

Abstract
Chronic exposure to intermittent hypoxia (CIH) increases carotid sinus nerve activity in normoxia and in response to acute hypoxia. We hypothesized that CIH augments basal and chemoreflex-stimulated sympathetic outflow through an angiotensin receptor-dependent mechanism. Rats were exposed to CIH for 28 days: a subset was treated with losartan. Then, lumbar sympathetic activity was recorded under anesthesia during 20-second apneas, isocapnic hypoxia, and potassium cyanide. We measured carotid body superoxide production and expression of angiotensin II type-1 receptor, neuronal nitric oxide synthase, and NADPH oxidase. Sympathetic activity was higher in CIH vs. control rats at baseline, during apneas and isocapnic hypoxia, but not cyanide. Carotid body superoxide production and expression of angiotensin II type 1 receptor and gp91phox subunit of NADPH oxidase were elevated in CIH rats, whereas expression of neuronal nitric oxide synthase was reduced. None of these differences were evident in animals treated with losartan. CIH-induced augmentation of chemoreflex sensitivity occurs, at least in part, via the renin-angiotensin system.
Keywords: chemoreceptors, angiotensin II, superoxide, angiotensin antagonist, oxidative stress
1. Introduction
Sympathetic nervous system overactivity is thought to contribute importantly to hypertension in patients with obstructive sleep apnea (OSA) (Hedner et al., 1988; Narkiewicz et al., 1998); however, the pathogenesis of this sympathoexcitation is incompletely understood. Chronic intermittent hypoxia (CIH) in rodents, an experimental model that mimics the episodic hypoxemia of OSA, elicits increases in arterial pressure that are critically dependent on the carotid chemoreceptor, the sympathetic nervous system, and signaling through the angiotensin II type 1 receptor (AT1R) (Fletcher et al., 1999; Fletcher et al., 1992; Fletcher et al., 1992); however, it is not clear how these systems interact to produce their hypertensive effect. The present findings advance the understanding of CIH-induced hypertension by demonstrating that this intervention sensitizes chemoreflex control of sympathetic outflow to the hindlimb, an effect that involves signaling through the AT1R.
Several lines of evidence indicate that recurrent exposure to brief hypoxic episodes, such as those that occur during OSA and CIH, alters chemoreflex function. In humans, inhibition of the carotid chemoreceptors with supplemental oxygen reduces sympathetic outflow and blood pressure in patients with OSA, but not control subjects (Narkiewicz, K. et al., 1998). In addition, hypoxia-induced sympathetic activation is enhanced in OSA patients vs. controls (Imadojemu et al., 2007; Narkiewicz, K. et al., 1998). Taken together these findings indicate OSA causes sensitization of the carotid chemoreflex.
In concordance with these findings, CIH in experimental animals produces increases in carotid sinus nerve discharge that outlast the period of exposure (Peng et al., 2003). Moreover, acute hypoxia elicits greater increases in carotid sinus nerve activity in CIH-exposed vs. control animals (Peng, Y. J. et al., 2003; Peng et al., 2006). This augmentation of carotid body sensory function appears to be redox-sensitive and dependent on increases in NADPH oxidase-derived superoxide (Peng et al., 2009; Peng, Y. J. et al., 2003). It is not known how CIH increases superoxide production; however, angiotensin II (Ang II), a well known activator of NADPH oxidase, may play a role, as it does in a rabbit model of heart failure (Li et al., 2006).
In addition to upregulation of excitatory influences, removal of inhibitory influences also enhances carotid chemoreflex sensitivity. Nitric oxide constrains carotid body afferent activity in vitro (Prabhakar et al., 1993). Also, carotid body expression of neuronal NO synthase (nNOS) and NO production are reduced and chemoreflex sensitivity is enhanced in rabbits with heart failure (Ding et al., 2008). To our knowledge, the effect of CIH on nNOS expression in the carotid body has not been investigated.
Additional evidence for CIH-induced carotid body sensitization comes from observations of the efferent limb of the carotid chemoreflex. Hypoxia- and hypercapnia-induced increases in preganglionic cervical sympathetic outflow were greater in CIH vs. control rats (Greenberg et al., 1999). Whether CIH also alters basal and chemoreflex-stimulated postganglionic sympathetic outflow to the hindlimb, a vascular bed that contributes importantly to blood pressure regulation, has not to our knowledge been studied.
The goal of the present study was to determine, in anesthetized rats, whether CIH augments basal and chemoreflex-stimulated sympathetic outflow to the hindlimb (lumbar sympathetic nerve activity; LSNA), and to investigate the role of the renin-angiotensin system in any observed changes. We hypothesized that CIH would increase basal LSNA, augment chemoreflex sensitivity, and increase Ang II-dependent superoxide production via signaling through the AT1R. To further investigate potential mechanisms of chemoreflex sensitization, we measured carotid body expression of AT1R and NADPH oxidase, proteins known to be associated with alterations in chemoreflex function in an animal model of heart failure (Ding, Y. et al., 2008; Li et al., 2007). Because impaired production of nitric oxide also contributes to chemoreflex hypersensitivity in this model (Sun et al., 1999), we measured carotid body expression of nNOS.
2. Methods
2.1 Animals
Adult male Sprague-Dawley rats (Harlan, Madison, WI) were used for all experiments. They had ad libitum access to water and standard chow (Purina) during exposure to either CIH or normoxia (NORM). Room temperature and relative humidity were maintained at 24±1° C and 20–70%. Rats were housed in accordance with recommendations set forth in the National Institutes of Health Guide for the Care of Laboratory Animals (NIH Pub. No. 85–23, Revised 1985). All protocols were approved by the University of Wisconsin-Madison School of Medicine and Public Health’s Institutional Animal Care and Use Committee.
In addition to CIH and NORM rats (n=9 per group) two additional groups of rats were treated with losartan in the drinking water (25–30 mg/kg/day) for 7 days prior to and during the 28-day exposure period (CIH-Los and NORM-Los, n=8 per group). A subset of rats from each group (n=4–5) was instrumented with indwelling femoral catheters to allow measurement of blood gases in the unanesthetized state on the 27th day of the exposure period. Body weight before exposure was not significantly different between groups (249±10, 239±4, 246±6, and 236±3 g, for NORM, CIH, NORM-Los, and CIH-Los respectively, p=0.463 by ANOVA).
2.2 Chronic intermittent hypoxia (CIH) and normoxia (NORM) exposures
The CIH protocol was identical to one we previously used to demonstrate impaired endothelium-dependent vasodilation (Phillips et al., 2004) and increased arterial pressure (10–15 mmHg) (Marcus et al., 2009). Briefly, rats in their home cages were placed into a Plexiglas chamber and exposed to intermittent hypoxia for 12 hours per day (from 18:00 hours to 06:00 hours) for 28 days. Oxygen concentration in the chamber was monitored using a heated zirconium sensor (Fujikura America, Pittsburgh, PA). A microprocessor-controlled timer was used to operate solenoid valves that controlled the flow of oxygen and nitrogen into the chamber to provide hypoxic exposures at 4-minute intervals. During the first minute of each cycle, nitrogen was introduced at a rate sufficient to achieve a fraction of inspired oxygen (FIO2) of 0.10 within 45 seconds and to maintain this level of FIO2 for an additional 60 seconds. Then, oxygen was introduced at a rate sufficient to achieve an FIO2 of 0.21 within 30 seconds and to maintain this level of FIO2 for the remainder of the 4-minute cycle. Control rats were housed under normoxic conditions adjacent to the hypoxia chamber for 28 days. There they were subjected to light, noise, and temperature stimuli similar to those experienced by the CIH rats.
2.3 Arterial blood gases in unanesthetized rats
After 14 days of CIH or NORM exposure, catheters were placed into the abdominal aorta distal to the renal arteries via the femoral artery. The catheter tubing was exteriorized by tunneling it beneath the skin to an opening between the scapulae. On day 27 of CIH or NORM exposure, rats were removed from their home cages and placed in a Plexiglas animal carrier for sampling of arterial blood under normoxic conditions. Rats were allowed to acclimatize to the surroundings for at least 30 minutes and care was taken to insure that rats were not sniffing or exploring when blood was drawn. Blood samples (0.2ml) were analyzed (Radiometer Copenhagen ABL505) and corrected for body temperature.
2.4 Surgical preparation
Within 4 hours of the end of the final exposure period on day 28, rats were anesthetized for study of chemoreflex function. Anesthesia was induced with isoflurane (5% in O2) and maintained with 2.5–3% isoflurane in O2. The right femoral artery and vein were cannulated for measurement of arterial pressure and blood gases and infusion of fluids and drugs. Rats were tracheotomized, paralyzed (pancuronium bromide, 3.5 mg/kg, i.v.), and mechanically ventilated (Rodent Ventilator 683, Harvard Apparatus, Holliston, MA, USA), and subsequently converted from isoflurane to urethane anesthesia (1.6–1.8 g/kg, i.v.) over the course of 20–25 minutes. An intravenous infusion of lactated Ringer’s solution (0.5–1 ml/hour) was maintained throughout the surgery and experiment. Rectal temperature was maintained at 37±1° C. using a heat lamp and heating pad.
2.5 Recording of sympathetic nerve activity
The lumbar sympathetic chain between the renal and inferior mesenteric arteries was exposed through a midline abdominal incision. The nerves were dissected free, placed on bipolar platinum electrodes, and covered with silicone elastomer (Kwik-Cast, World Precision Instruments, Sarasota, FL). The electrode wires were connected to a high impedance probe (Grass HIP511). Action potentials were amplified 20 thousand-fold by a Grass P511 AC Amplifier and bandpass filtered with a bandwidth of 100–1000 Hz. The filtered neurogram was routed through a storage oscilloscope (Gould Model 420, Cleveland, OH) and an amplitude discriminator to an audio amplifier. For permanent recording and analysis, the filtered neurogram was routed though a nerve-traffic analyzer (University of Iowa Bioengineering Model 66C-2), which counts nerve spikes exceeding a threshold voltage level (time constant, 0.5 sec), to a Grass Model 7 polygraph and Windaq computer data acquisition system (Dataq Instruments, Akron, OH). At the beginning of each experiment the threshold voltage level was set using a window discriminator to a level just above the background noise as determined during phenylephrine-induced sympathoinhibition. The window remained constant throughout the experiment.
We used the scaling method to quantify LSNA (Guild et al., 2009). At the end of each experiment, the 0% LSNA voltage level was measured during ganglionic blockade with pentolinium tartrate (10 mg/kg, i.v.). The maximal (or 100%) calibration voltage was determined in each rat by passing ammonia vapor over the nasal mucosa (Dorward et al., 1985). Absolute values for maximal voltage elicited by this stimulus were comparable in all rats (0.17±0.02, 0.18±0.03, 0.18±0.02, and 0.17±0.05 arbitrary units for NORM, CIH, NORM-Los, and CIH-Los respectively). For all analyses, LSNA was expressed as a percentage of the maximal LSNA during the nasopharyngeal response.
2.6 Physiological measurements
Blood pressure, arterial oxygen saturation (SaO2; MouseOx, Starr Life Sciences, Oakmont, PA), and end-tidal CO2 (PETCO2; Capnogard, Respironics Novametrix, Wallingford, CT) were measured continuously during the experiment. Arterial blood gases were measured periodically from 0.2 ml samples (Radiometer Copenhagen ABL505). We monitored depth of anesthesia by assessing changes in blood pressure in response to a foot pinch. Urethane was supplemented as necessary to maintain a surgical plane of anesthesia.
2.7 Chemoreflex testing protocol
Response to apneas
After surgical procedures were completed, arterial blood gases were measured. Using these measurements, ventilatory rate and tidal volume were adjusted until blood gases matched the mean values measured in unanesthetized rats from the same experimental group. After a stable baseline had been established, we began a series of six 20-second apneas produced by turning off the ventilator.
In the first set of three apneas, LSNA, mean arterial pressure (MAP), and arterial oxygen saturation were measured. The first three apneas were separated by 30 minutes each, because we were concerned that more frequent apneas would elicit long-term facilitation of LSNA as has been demonstrated for phrenic nerve discharge (Bach et al., 1999). During the second set of apneas, 0.2 ml of blood was drawn at end-apnea for determination of arterial blood gases. After each blood draw, 0.5 ml of lactated Ringer’s solution was infused to flush the catheter and replace volume. Because we considered it unlikely that long-term facilitation could affect arterial blood gases in paralyzed, mechanically ventilated rats, the second set of three apneas were separated by 10 minutes in the interest of time. At least 10 minutes after the final apnea, another arterial blood sample was taken to verify that the preparation remained stable during the experimental period.
Response to isocapnic hypoxia
We exposed a subset of rats from each group to three 2-minute periods of isocapnic hypoxia (FIO2, 0.12). The LSNA response to hypoxia was quantified by calculating the area under the LSNA curve (see below) for the first 30 seconds during hypoxia exposure and for the final 30 seconds of the pre-hypoxia baseline. We chose to analyze only the first 30 seconds of hypoxia exposure because longer periods of hypoxia caused decreases in blood pressure. The mean value for change in area under the curve (Δ AUC) from baseline to hypoxia in the three trials was used as an index of chemoreflex sensitivity.
Response to cyanide
Potassium cyanide was administered in three successive injections (30 μg/kg, i.v.) separated by 5 minutes Because of the rapid, transient LSNA responses to bolus injections of cyanide, we measured initial peak responses (averaged over 0.5 seconds) within 4 sec of injection. For each trial, the LSNA response was calculated by subtracting the peak LSNA value during cyanide (expressed as % of ammonia maximum) minus the value for the 60-sec baseline immediately prior to injection (also expressed as % of ammonia maximum). LSNA response values were averaged across the three trials.
2.8 Stimulation of cutaneous afferents
To determine the effect of CIH on sympathetic responses to non-chemoreflex stimuli, we immersed the rat’s tail in hot water (55o C.) for 60 seconds, an intervention that produced transient increases in sympathetic activity and blood pressure. To quantify this response we subtracted the peak LSNA value during stimulation (expressed as % of ammonia maximum) minus the value for the 60-sec baseline immediately prior to immersion (also expressed as % of ammonia maximum).
2.9 Pressor response to angiotensin II
Following chemoreflex testing, we determined the efficacy of AT1R blockade by measuring the peak increase in mean arterial pressure elicited by intravenous injection of Ang II (25 ng/kg). Pressor responses were averaged across the three trials.
2.10 Carotid body superoxide production
Both carotid bodies were removed, flash frozen in liquid nitrogen, and stored at −80° C. until analyzed. Superoxide anion production was measured using the lucigenin chemiluminescence method. Carotid body samples were homogenized and centrifuged (3000 rpm) at 4° C. for 5 minutes. The supernatant was placed in 0.5 ml microfuge containing dark adapted lucigenin (5 μM), then read in a TD-20/20 Luminometer (Turner Designs, Sunnyvale, CA). Light emission was recorded for 5 minutes and expressed as mean light units (MLU)/min/100 μg protein.
2.11 Western blot analysis for carotid body AT1R, nNOS, and NADPH oxidase content
Carotid bodies were harvested (as described above) and pooled (two carotid bodies from each rat) to yield 25–30 μg protein/rat. We performed 4–6 blots in each experimental group, for which protein samples were obtained from 4–6 rats. In addition, using a stripping buffer, we used one membrane to measure different target proteins. The procedures for Western blot analysis have previously been described (Ding, Y. et al., 2008) and are briefly summarized here. The protein was extracted with lysing buffer (10 mM PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1% SDS) plus protease inhibitor cocktail (100 μl/ml) and centrifuged at 12.000 g for 20 minutes at 4o C. Five μg of protein (as determined by BCA protein assay kit from Pierce Chemical, Rockford, IL) was mixed with loading buffer containing mercaptoethanol, heated at 100° C. for 5 minutes, separated in a 10% polyacrylamide gel along with molecular weight standards, and transferred to a PVDF membrane. For AT1R and nNOS determinations, the membrane was probed with a mouse anti-AT1R and mouse monoclonal anti-nNOS (Santa Cruz Biotechnology, Santa Cruz, CA) and a peroxidase-conjugated goat anti-mouse IgG (Pierce Chemical, Rockford, IL). For NADPH oxidase components the membrane was probed with goat anti gp91phox, p67phox, p47phox, p40phox and p22phox antibodies (Santa Cruz, CA, USA) and a peroxidase-conjugated rabbit anti-goat IgG (Santa Cruz, CA, USA). The signals were detected using enhanced chemiluminescence substrate (Pierce) and the bands were analyzed using UVP BioImaging Systems. Protein loading was controlled by probing all Western blots with mouse anti-GAPDH antibody (Santa Cruz) or β-actin (NADPH oxidase components) and normalizing AT1R, nNOS, gp91phox, p67phox, p47phox, p40phox and p22phox protein intensity to that of GAPDH or β-actin as appropriate.
2.12 Data analysis
During apneas, LSNA (% maximum) and arterial oxygen saturation were averaged in 2.5-second bins to produce stimulus response curves. For each apnea, the stimulus response curve was quantified using the following logistic expression: R = Rmin + (Rmax−Rmin)/(1+(D/EC50)^S), where R=LSNA response, Rmin=baseline LSNA, Rmax=peak LSNA, D=arterial oxygen saturation, and S= Hill slope coefficient determining the steepness of each curve. If the R2 value for the curve fitting was less than 0.7, the data were not used. Slope coefficients were used to compare chemoreflex sensitivity between groups.
To quantify LSNA responses to isocapnic hypoxia, the area under the curve was calculated by graphing all data points within the time periods of interest (Sigmaplot software, version 8.02, Systat software, Chicago IL) and using the ‘area below curves’ function to calculate an AUC for each exposure period and pre-exposure baseline.
2.13 Statistics
All data are presented as means±SEM. Animal weight and age at the beginning of the experimental protocol was analyzed by 1-way ANOVA. Within-group determinations of change in LSNA, PETCO2, and SaO2 relative to zero were made using t-tests. Unpaired t-tests were used to compare all other variables between exposure group (i.e. CIH vs. NORM and CIH-Los vs. NORM-Los). We did not make comparisons across treatment groups (losartan-treated vs. untreated) in order to guard against the possibility that the drug would affect our tests of chemoreflex responsiveness in a non-specific way (e.g. via blood pressure lowering or its effect on central sympathetic outflow, plasma renin activity, plasma or tissue Ang II) (Gradman et al., 2008; Li et al., 1996). Statistical significance was accepted when p<0.05.
3. Results
3.1 General characteristics of experimental animals
During the 28-day exposure period, CIH-exposed animals tended to gain less weight than their normoxia-exposed counterparts; however, the differences were not statistically significant (+87±8, +105±12, +85±10, and +111±7 grams in CIH, NORM, CIH-Los, and NORM-Los, respectively; p=0.173). There were no between-group differences in the age of animals on the day of study (88±2, 90±1, 90±2, and 91±1 days, p=0.600).
Baseline MAP, measured under anesthesia on the day of study, was significantly higher in CIH vs. NORM (127±6 vs. 105±10 mmHg; p=0.039), but not different between the two losartan-treated groups and (96±3 vs. 87±9 mmHg in CIH-Los and NORM-Los; p=0.161).
3.2 Arterial blood gases
Unanesthetized rats
Under normoxic conditions on the 27th day of exposure, PaCO2 was lower in CIH vs. NORM, but only reached statistical significance for CIH-Los vs. NORM-Los (Table 1). In CIH-Los, HCO3− was significantly lower, and PaO2 was significantly higher than NORM-Los, whereas pH was not different in CIH-exposed animals and their respective controls. Taken together these findings indicate that CIH caused a mild respiratory alkalosis. HCO3− dropped by more than we would predict necessary to compensate for the level of hyperventilation; therefore, CIH may have increased production of fixed acid (e.g. via increased anaerobic glycolysis).
Table 1.
Arterial blood gases and core temperature measured under normoxic conditions in unanesthetized rats after 27 days of CIH or normoxia exposure.
| PaCO2 (Torr) | PaO2 (Torr) | pH (units) | HCO3− (mEq/l) | Temperature (° C.) | |
|---|---|---|---|---|---|
| NORM | 37.4±0.8 | 86.4±1.4 | 7.49±0.02 | 27.0±1.0 | 37.1±0.1 |
| CIH | 34.8±0.8 | 88.1±1.9 | 7.46±0.02 | 24.9±1.4 | 36.7±0.3 |
| P-value | 0.053 | 0.566 | 0.309 | 0.363 | 0.404 |
| NORM-Los | 37.3±0.7 | 87.1±2.0 | 7.46±0.01 | 26.2±0.1 | 37.5±0.1 |
| CIH-Los | 33.0±0.9* | 94.5±1.5* | 7.46±0.01 | 23.1±0.3* | 37.4±0.1 |
| P-value | 0.006 | 0.019 | 0.924 | <0.001 | 0.337 |
p<0.05 vs. respective control condition.
Response to apneas
Apneas resulted in significant increases in PETCO2 and decreases in SaO2 (p<0.001 in all groups; Table 2). No significant between-group differences in were observed in CIH vs. NORM or CIH-Los vs. NORM-Los. The decreases in SaO2 were smaller in both groups of losartan-treated rats vs. those with no losartan; however, values for PaO2 at end-apnea were comparable in the 4 groups (34.1±1.7, 36.2±1.6, 39.6±1.7, and 38.1±2.5 for CIH, NORM, CIH-Los, and NORM-Los, respectively).
Table 2.
Changes in end-tidal CO2 (PETCO2) and arterial oxygen saturation (SaO2) caused by 20-second apneas. There were no differences between CIH-exposed rats and their respective control groups.
| ΔPETCO2 (Torr) | ΔSaO2 (%) | |
|---|---|---|
| NORM | +5.4±0.5 | −32±4 |
| CIH | +5.9±0.6 | −31±3 |
| P-value | 0.505 | 0.566 |
| NORM-Los | +4.5±0.2 | −26±4 |
| CIH-Los | +4.7±0.4 | −23±3 |
| P-value | 0.591 | 0.543 |
3.3 Pressor response to angiotensin II
Intravenous injection of Ang II (25 ng/kg) caused greater increases in mean arterial pressure in CIH vs. NORM (+14±3 vs. +9±1 mmHg, p=0.030). As expected the increases were attenuated in losartan-treated rats; however, there was no between-group difference in CIH-Los and NORM-Los (+4±1 and +4±1 mmHg, p=0.980).
3.4 Effect of CIH on baseline LSNA
After the 28-day exposure period, LSNA was measured under normoxic conditions prior to chemoreflex testing. Baseline LSNA was significantly higher in animals exposed to CIH vs. NORM (38±8 vs. 17±3% maximal; p=0.023). In contrast, baseline LSNA was not significantly different between groups of animals treated with losartan (28±3 vs. 23±4% maximal in CIH-Los and NORM-Los; p=0.361).
3.5 Effect of CIH on chemoreflex function
Apnea
Apneas increased LSNA in all groups. Typical LSNA and MAP responses to an apnea are shown in Figure 1 and stimulus-response curves are shown in Figure 2. LSNA was higher in CIH vs. NORM at all levels of SaO2. In contrast, there were no differences in LSNA in CIH-Los vs. NORM-Los. Figure 3 shows the slope coefficients of these stimulus response curves. Sympathetic activation during apnea was greater in CIH vs. NORM; whereas it was comparable in CIH-Los vs. NORM-Los. As mentioned above, 20-second apneas resulted in smaller desaturations in losartan-treated vs. untreated rats. For this reason, we repeated the slope coefficient computations using the same range of SaO2 values in all groups (from baseline to ~68–70%). In CIH vs. NORM, the statistically significant difference in slope coefficient remained (35±7 vs. 13±6; p=0.028), whereas there was no difference in CIH-Los vs. NORM-Los (20±6 vs. 16±3; p=0.286). The blood pressure responses to 20-second apneas were not different between CIH-exposed and normoxic control animals (+26±3 vs. +28±5 mmHg in CIH vs. NORM; p=0.781), nor were they different in groups treated with losartan (+16±3 vs. +18±5 mmHg in CIH-Los vs. NORM-Los; p=0.728).
Figure 1.

Representative response to a 20-second ventilator-induced apnea. This intervention produced marked increases in lumbar sympathetic nerve activity (LSNA) and mean arterial pressure (MAP). Pt, tracheal pressure.
Figure 2.

SaO2-LSNA relationship during 20-second apnea. LSNA was higher at baseline and at all levels of SaO2 in CIH vs. NORM (left panel). This difference in chemoreflex sensitivity was not evident in rats treated with losartan (right panel).
Figure 3.

Slope coefficients for the data shown in Figure 2. Slope coefficients were greater in CIH than NORM (left panel), whereas slope coefficients were comparable in CIH-Los and NORM-Los. *p<0.05 vs. the respective control condition.
Isocapnic Hypoxia
Acute exposure to isocapnic hypoxia (FIO2, 0.12) significantly increased LSNA in all rats (p<0.02 for all groups) (Figure, 4, top). The Δ AUC was greater in CIH (n=7) vs. NORM (n=6) (+405±115 vs. +185±34 units), but this difference was of borderline statistical significance (p=0.055). In animals treated with losartan, the LSNA responses to hypoxia were comparable in the two groups (+221±58 vs. +276±62 units for CIH-Los (n=6) and NORM-Los (n=7), respectively; p=0.532).
Figure 4.

Physiological records showing typical responses to isocapnic hypoxia (upper panel) and cyanide injection (lower panel). Insets in the upper panel show 4-second segments of neurogram with expanded timebase acquired during baseline (a) and peak response to hypoxia (b). The arrow in the lower panel indicates the timing of the cyanide injection.
Cyanide
Potassium cyanide (30 μg/kg, i.v.) produced significant transient increases in LSNA in all rats (p<0.03 for all groups) (Figure 4, bottom). In contrast to other tests of chemoreflex function, between-group differences in cyanide-induced sympathoexcitation were not statistically significant (+132±18 vs. +113±15% in CIH vs. NORM; p=0.227). Likewise, there was no between-group difference in the response to cyanide in losartan-treated animals (+115±19 vs. +89±9% in CIH-Los vs. NORM-Los; p=0.246).
3.6 Effect of CIH on heat-induced sympathetic activation
Stimulation of cutaneous afferents with hot water significantly increased LSNA in all rats (p<0.05 in all groups). Peak changes in LSNA were comparable in CIH vs. NORM (+37±5 vs. +33±4%, p=0.600). Likewise, there was no between-group difference in LSNA responses to this stimulus in rats treated with losartan (+40±4% in CIH-Los vs. +39±7% in NORM-Los, p=0.690).
3.7 Effect of CIH on carotid body superoxide production
Carotid body superoxide production was greater in CIH vs. NORM (1.40±0.10 vs. 0.47±0.04 relative light units/100 μg protein; p<0.001) (Figure 5). A statistically significant effect of CIH was also observed in animals treated with losartan (0.57±0.04 relative light units/100 μg protein in CIH-Los vs. 0.40±0.03 relative light units/100 μg protein in NORM-Los; p=0.003).
Figure 5.

Carotid body superoxide production. Superoxide production was greater in CIH than NORM (left panel). Carotid bodies from losartan-treated rats also showed a significant effect of CIH on superoxide production (right panel). RLU, relative light units. *p<0.05 vs. respective control condition.
3.8 Effect of CIH on protein expression in carotid bodies
AT1R
Expression of AT1R was increased in rats exposed to CIH relative to NORM (p=0.020; Figure 6). There was no between-group difference in rats treated with losartan (p=0.156).
Figure 6.
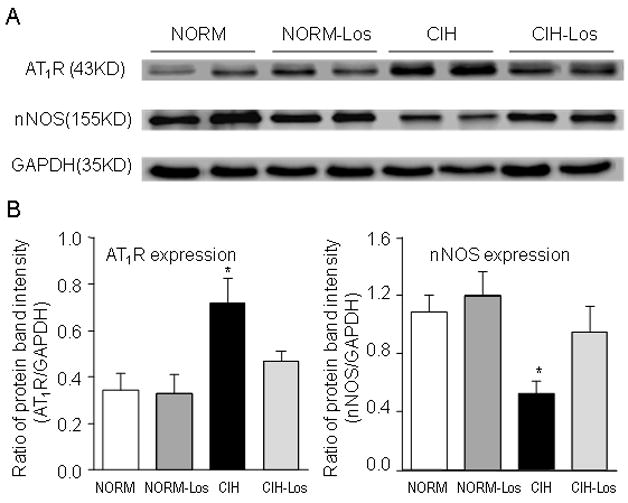
A. Representative blots showing carotid body angiotensin type I receptor (AT1R) and neuronal nitric oxide synthase (nNOS) expression. B. Summary data for AT1R and nNOS expression. AT1R expression was increased in CIH vs. NORM; this difference was not evident in rats treated with losartan (left panel). nNOS expression was decreased in CIH vs. NORM; this decrease was not evident in losartan-treated rats (right panel). *p<0.05 vs. respective control condition.
nNOS
Expression of nNOS was decreased in rats exposed to CIH relative to NORM (p=0.005; Figure 6). There was no between-group difference in rats treated with losartan (p=0.338).
NADPH oxidase
Expression of the gp91phox subunit was increased in CIH relative to NORM (p=0.001) (Figure 7). CIH did not alter expression of P22 phox, P47 phox, or P67 phox subunits (P40phox could not be detected in the rat carotid bodies). Expression of gp91phox was lower in CIH-Los vs. NORM-Los (p=0.003)
Figure 7.
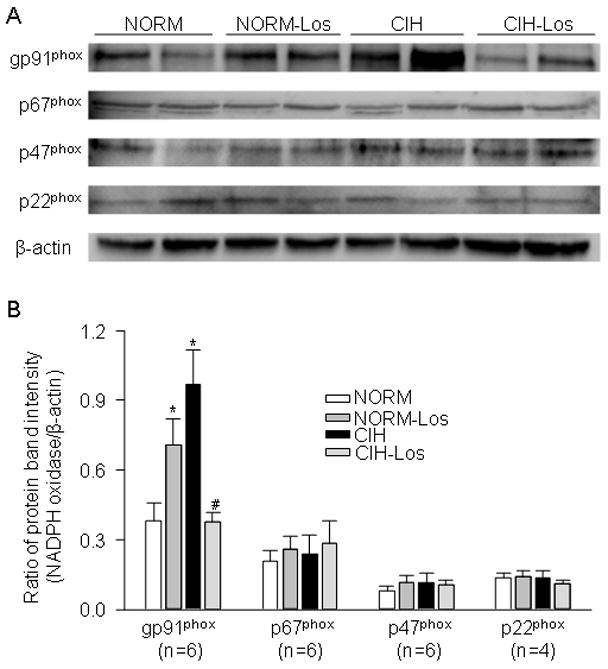
NADPH oxidase subunit expression in carotid bodies. A. Representative blots for gp91phox, p22 phox, p67 phox, and p47 phox expression. B. Summary data for NADPH subunits. Expression of the gp91 phox subunit was increased in CIH vs. NORM; CIH did not affect expression of other subunits (left panel). Expression of gp91phox in CIH-Los was reduced relative to NORM-Los (right panel). *p<0.05 vs. respective control condition.
4. Discussion
We sought to determine whether repetitive, brief exposures to hypoxia, administered 12 hours per day for 28 days, alter chemoreflex control of postganglionic sympathetic outflow to the hindlimb. The major finding was that CIH caused an increase in chemoreflex sensitivity that was dependent on signaling through the AT1R. This alteration in chemoreflex control of sympathetic outflow was accompanied by: 1) an increase in the baseline (normoxic) level of LSNA, 2) increases in the expression of AT1R and the gp91phox subunit of NADPH oxidase and decreases in the expression of nNOS in the carotid body, and 3) an increase in carotid body superoxide production. All of these effects were also dependent on activation of the renin-angiotensin system. These data provide supporting evidence for carotid chemoreflex sensitization in mediating CIH-induced increases in sympathetic activity under normoxic conditions and in response to acute hypoxic challenge. In addition, the data suggest that an Ang II-NADPH oxidase-superoxide signaling pathway is responsible for augmenting chemoreflex sensitivity in this experimental model.
4.1 CIH-induced sensitization of the carotid chemoreflex
Our conclusion that CIH augments carotid chemoreflex control of sympathetic outflow is based on several lines of evidence. First, the asphyxia produced by 20-second apnea elicited greater sympathoexcitation in rats previously exposed to CIH than in normoxic control animals. Second, acute exposure to isocapnic hypoxia produced larger increases in LSNA in CIH vs. NORM. Finally, our CIH model caused the same alterations in carotid body protein expression and superoxide production that are associated with sensitization of the carotid chemoreflex in an experimental model of heart failure (Schultz et al., 2007). Our findings are consistent with those of previous investigators who have shown, in isolated and intact carotid body preparations, that CIH causes sensory long-term facilitation of carotid sinus nerve activity (Peng, Y. J. et al., 2009; Peng, Y. J. et al., 2003; Peng, Y. J. et al., 2006; Rey et al., 2004). It is unlikely that the observed increase in chemoreflex sensitivity represents a non-specific effect on sympathetic responsiveness, because the increase in nerve traffic elicited by cutaneous afferent stimulation was not altered by CIH.
Previous investigators have shown that exposure to CIH increases the amount of activation in cervical preganglionic sympathetic nerves elicited by subsequent acute chemoreflex stimulation (Greenberg, H. E. et al., 1999). More recently, CIH has been shown to enhance renal sympathetic nerve responses to acute hypoxia and hypercapnia (Huang et al., 2009). Our results confirm and extend these findings by demonstrating that CIH augments carotid chemoreflex control of postganglionic lumbar sympathetic nerve activity. This adaptation results in heightened sympathetic vasoconstrictor outflow to the hindlimb, a vascular bed that contributes importantly to blood pressure regulation, during normoxia and also during chemoreflex stimulation with hypoxia and asphyxia. Furthermore, our data provide insight into the mediators of CIH-induced chemoreflex sensitization by describing a putative Ang II-NADPH oxidase-superoxide signaling pathway.
4.2 Mechanisms of CIH-induced increases in chemoreflex sensitivity
Ang II is capable of stimulating the carotid chemoreceptor directly in high doses (Allen, 1998), and modifying the chemoreceptor sensitivity to hypoxia at more physiological concentrations (Li, Y. L. et al., 2006). In rabbits, intravenous infusion of Ang II results in increased afferent discharge from the carotid chemoreceptors and increased renal sympathetic nerve activity during exposure to hypoxia (Li, Y. L. et al., 2006). These effects are blocked by co-infusion of an AT1R antagonist (Li, Y. L. et al., 2006), a superoxide dismutase mimetic (Li, Y. L. et al., 2007), or an inhibitor of NADPH oxidase (Li, Y. L. et al., 2007). Taken together these findings indicate that the stimulatory effect of Ang II on carotid chemoreflex sensitivity occurs via AT1R binding and activation of an NADPH oxidase- and superoxide-dependent signaling pathway.
In the present study, CIH exposure caused an increase in carotid body AT1R expression in control rats, but not in those treated with losartan. This finding is consistent with previous work showing that chronic continuous hypoxia increases AT1R expression in the carotid body (Leung et al., 2000), and that infusion of Ang II into the cerebral ventricles elicits increases in AT1R expression in the rostral ventrolateral medulla that are prevented by losartan (Gao et al., 2005). A potential reason for this Ang II-induced increase in AT1R expression is downregulation of nNOS (Campese et al., 2002; Qadri et al., 2001; Usui et al., 1998). In support of this idea, we observed that CIH caused decreases in carotid body nNOS expression. We have previously shown that decreased expression of nNOS in the carotid body and decreased nitric oxide production contribute to chemoreflex hypersensitivity in a rabbit heart failure model (Ding, Y. et al., 2008; Li et al., 2005; Sun, S. Y. et al., 1999). In the present study, we did not directly investigate the role of nitric oxide in CIH-induced chemoreflex hypersensitivity, however our data suggest the possibility that renin-angiotensin system-mediated changes in nNOS enzyme expression could contribute to changes in chemoreflex function associated with CIH.
The present data confirm previous findings that CIH increases production of reactive oxygen species in the carotid body (Peng, Y. J. et al., 2006) and that this change is attended by increased NADPH oxidase (NOX) activity and protein expression (Peng, Y. J. et al., 2009). We extend these observations with our finding that CIH-induced increases in superoxide production and gp91phox expression are not evident in the presence of AT1R blockade, suggesting that Ang II signaling plays an important role in CIH-induced changes in carotid body redox balance. Although the sources of CIH-induced increases in superoxide production have not been definitively identified, inhibition of NOX, a known producer of superoxide in the carotid body (Li, Y. L. et al., 2007), prevents CIH-induced changes in chemoreflex function (Peng, Y. J. et al., 2009). Interestingly, we observed that gp91phox expression was greater in NORM-Los than CIH-Los animals. The reason for this unexpected observation is unclear.
In addition to NOX, other enzyme systems may contribute to increases in superoxide production associated with CIH. Xanthine oxidase (XO) is a potential source of superoxide that is stimulated by hypoxia (Kelley et al., 2006) and that may interact with Ang II-NADPH oxidase pathways to increase superoxide production (Landmesser et al., 2007). Nothing is known at the present time about the effects of CIH on XO activity or expression. Our results indicate that AT1R blockade greatly reduced but did not eliminate increased superoxide production associated with CIH, suggesting that other sources of superoxide are upregulated by CIH. The contribution of XO to CIH-induced changes in carotid body redox state merits further study.
The present findings suggest that CIH alters, via Ang II/AT1R-dependent pathways, carotid chemoreceptor function under baseline, normoxic conditions and in response to acute hypoxia. Nevertheless, it is also possible that these sympathoexcitatory effects of CIH may arise from oxygen sensitive neurons in the central nervous system, especially those responsible for relaying or modulating carotid chemoreceptor input (e.g. presympathetic neurons in the rostral ventrolateral medulla) (Guyenet, 2000; Sun et al., 1994). Furthermore, prevention of CIH-induced sympathetic activation by AT1R blockade could also occur via a central nervous system action, because losartan is able to cross the blood brain barrier (Pediconi et al., 2005). In a rabbit model of heart failure which produces sustained sympathetic activation and sensitization of the carotid chemoreflex, central AT1R blockade reduces sympathetic tone (Zucker et al., 2009).
4.3 Methodological considerations
By measuring LSNA responses to cyanide injection, a stimulus thought to be specific for the carotid chemoreceptor (Barros et al., 2002), we sought to differentiate the peripheral from the central effects of CIH. In our study, LSNA responses to cyanide were somewhat larger, but not statistically different, in CIH-exposed vs. normoxic control rats; however, these results are not conclusive due to inadequate statistical power. Previous investigations of carotid body sensory responses to hypoxia and cyanide in CIH-exposed vs. normoxic control animals have yielded inconsistent results. In adults cats (Rey, S. et al., 2004), rat pups (Peng and Prabhakar 2004), and adult mice (Peng, Y. J. et al., 2006), CIH produced an augmentation in carotid sinus nerve activation during acute hypoxia, but not cyanide injection. In contrast, in adult rats, carotid sinus nerve responses to both hypoxia and cyanide were augmented in CIH-exposed animals relative to controls (Peng et al., 2004). The reasons for this inconsistency are unknown; however, it is clear that CIH-induced increases in carotid body hypoxic sensitivity are not always accompanied by increases in response to cyanide.
In this study, the oxygen desaturation produced by 20-second apnea was attenuated in the CIH-Los and NORM-Los groups relative to the untreated groups. Although PaO2 at end-apnea was somewhat higher in the losartan-treated vs. untreated rats, the attenuated desaturation may imply a shift in the oxyhemoglobin dissociation curve. Losartan produces mild decreases in hemoglobin content (Mohanram et al., 2008); however, it is unclear how this could affect oxyhemoglobin dissociation. Perhaps losartan could alter the affinity of hemoglobin for oxygen by increasing production of NO and NO derivatives (Campese, V. M. et al., 2002; Olson et al., 2004; Qadri, F. et al., 2001; Ritter et al., 2003; Stepuro et al., 2006; Su et al., 2009).
Our control animals were housed in cages adjacent to the hypoxia chamber but were not exposed to cyclic changes in airflow. Although both groups of rats were exposed to transient noises associated with solenoid valve operation and airflow change, we considered the possibility that the rats inside the chamber experienced stress induced by alterations in airflow and temperature. It is unlikely that our results were influenced by these factors because: 1) the inflow port of the hypoxia chamber contained baffles that minimized air jets, 2) the hypoxia-exposed rats resided in individual plastic cages placed inside the chamber; thus, they were protected from direct exposure to changes in airflow, and 3) the thermal environments were the same in the chamber and adjacent areas of the room where the NORM rats were housed (mean temperatures 23.9±0.1 vs. 24.1±0.2° C.).
Although our CIH paradigm models the intermittent hypoxemia associated with sleep apnea in humans, it fails to mimic the intermittent hypercapnia, airway obstructions, and sleep disruption that accompany this clinical condition. We chose to study the effects of intermittent hypoxia per se because previous investigators have shown that hypercapnia and sleep disruption are not primary contributors to blood pressure elevation in this setting (Bao et al., 1999; Brooks et al., 1997). It is impossible to faithfully reproduce OSA-induced sleep fragmentation and sleep limitation in rodents, even when intermittent hypoxia is applied during the light cycle, because the animals typically sleep during only 60% of this period (Franken et al., 1999). In addition, when rodents are exposed to intermittent hypoxia during the light cycle they shift their diurnal sleep-wake patterns and exhibit more sleep time in the dark cycle when they are unperturbed (Polotsky et al., 2006). We would like to emphasize that our CIH paradigm elicits increases in arterial pressure that are comparable to those observed by other investigators who administered intermittent hypoxia during the animals’ light cycles (Fletcher, E. C. et al., 1992; Lai et al., 2006; Marcus, N. J. et al., 2009; Troncoso Brindeiro et al., 2007).
4.4 Conclusions
This study demonstrates that alterations in chemoreflex function associated with CIH are largely mediated by Ang II signaling through the AT1R. The potential mechanisms underlying these alterations in function are CIH-induced increases in Ang II production resulting in increased AT1R and NADPH oxidase subunit expression and increased superoxide production in the carotid body.
Acknowledgments
The authors acknowledge the technical assistance of Dr. Safraaz Mahamed, Kristin Sorenson, Scott Smith, and Jenni Ballweg. We are indebted to Drs. E. Burt Olson, Jr. and Steven Polishinski for their assistance with design and operation of the hypoxia chamber. This work was supported by grants from the National Heart Lung and Blood Institute (grant numbers HL 074072 to BJM, T32 HL07654 to NJM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen AM. Angiotensin AT1 receptor-mediated excitation of rat carotid body chemoreceptor afferent activity. J Physiol. 1998;510 (Pt 3):773–781. doi: 10.1111/j.1469-7793.1998.773bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach KB, Kinkead R, Mitchell GS. Post-hypoxia frequency decline in rats: sensitivity to repeated hypoxia and alpha2-adrenoreceptor antagonism. Brain Res. 1999;817:25–33. doi: 10.1016/s0006-8993(98)01181-0. [DOI] [PubMed] [Google Scholar]
- Bao G, Metreveli N, Fletcher EC. Acute and chronic blood pressure response to recurrent acoustic arousal in rats. Am J Hypertens. 1999;12:504–510. doi: 10.1016/s0895-7061(99)00032-1. [DOI] [PubMed] [Google Scholar]
- Barros RC, Bonagamba LG, Okamoto-Canesin R, de Oliveira M, Branco LG, Machado BH. Cardiovascular responses to chemoreflex activation with potassium cyanide or hypoxic hypoxia in awake rats. Auton Neurosci. 2002;97:110–115. doi: 10.1016/s1566-0702(02)00050-4. [DOI] [PubMed] [Google Scholar]
- Brooks D, Horner RL, Kozar LF, Render-Teixeira CL, Phillipson EA. Obstructive sleep apnea as a cause of systemic hypertension. Evidence from a canine model. J Clin Invest. 1997;99:106–109. doi: 10.1172/JCI119120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campese VM, Ye S, Zhong H. Downregulation of neuronal nitric oxide synthase and interleukin-1beta mediates angiotensin II-dependent stimulation of sympathetic nerve activity. Hypertension. 2002;39:519–524. doi: 10.1161/hy0202.102815. [DOI] [PubMed] [Google Scholar]
- Ding Y, Li YL, Schultz HD. Downregulation of carbon monoxide as well as nitric oxide contributes to peripheral chemoreflex hypersensitivity in heart failure rabbits. J Appl Physiol. 2008;105:14–23. doi: 10.1152/japplphysiol.01345.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorward PK, Riedel W, Burke SL, Gipps J, Korner PI. The renal sympathetic baroreflex in the rabbit. Arterial and cardiac baroreceptor influences, resetting, and effect of anesthesia. Circ Res. 1985;57:618–633. doi: 10.1161/01.res.57.4.618. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Bao G, Li R. Renin activity and blood pressure in response to chronic episodic hypoxia. Hypertension. 1999;34:309–314. doi: 10.1161/01.hyp.34.2.309. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Behm R, Miller CC, III, Stauss H, Unger T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol. 1992;72:1978–1984. doi: 10.1152/jappl.1992.72.5.1978. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Culman J, Miller CC, Unger T. Sympathetic denervation blocks blood pressure elevation in episodic hypoxia. Hypertension. 1992;20:612–619. doi: 10.1161/01.hyp.20.5.612. [DOI] [PubMed] [Google Scholar]
- Franken P, Malafosse A, Tafti M. Genetic determinants of sleep regulation in inbred mice. Sleep. 1999;22:155–169. [PubMed] [Google Scholar]
- Gao L, Wang W, Li YL, Schultz HD, Liu D, Cornish KG, Zucker IH. Sympathoexcitation by central ANG II: roles for AT1 receptor upregulation and NAD(P)H oxidase in RVLM. Am J Physiol Heart Circ Physiol. 2005;288:H2271–H2279. doi: 10.1152/ajpheart.00949.2004. [DOI] [PubMed] [Google Scholar]
- Gradman AH, Pinto R, Kad R. Current concepts: renin inhibition in the treatment of hypertension. Curr Opin Pharmacol. 2008;8:120–126. doi: 10.1016/j.coph.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Greenberg HE, Sica A, Batson D, Scharf SM. Chronic intermittent hypoxia increases sympathetic responsiveness to hypoxia and hypercapnia. J Appl Physiol. 1999;86:298–305. doi: 10.1152/jappl.1999.86.1.298. [DOI] [PubMed] [Google Scholar]
- Guild SJ, Barrett CJ, McBryde FD, Van Vliet BN, Head GA, Burke SL, Malpas SC. Quantifying sympathetic nerve activity; problems and pitfalls, the need for standardization. Exp Physiol. 2009 doi: 10.1113/expphysiol.2008.046300. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. Neural structures that mediate sympathoexcitation during hypoxia. Respir Physiol. 2000;121:147–162. doi: 10.1016/s0034-5687(00)00125-0. [DOI] [PubMed] [Google Scholar]
- Hedner J, Ejnell H, Sellgren J, Hedner T, Wallin G. Is high and fluctuating muscle nerve sympathetic activity in the sleep apnoea syndrome of pathogenetic importance for the development of hypertension? J Hypertens Suppl. 1988;6:S529–S531. doi: 10.1097/00004872-198812040-00166. [DOI] [PubMed] [Google Scholar]
- Huang J, Lusina S, Xie T, Ji E, Xiang S, Liu Y, Weiss JW. Sympathetic response to chemostimulation in conscious rats exposed to chronic intermittent hypoxia. Respir Physiol Neurobiol. 2009;166:102–106. doi: 10.1016/j.resp.2009.02.010. [DOI] [PubMed] [Google Scholar]
- Imadojemu VA, Mawji Z, Kunselman A, Gray KS, Hogeman CS, Leuenberger UA. Sympathetic chemoreflex responses in obstructive sleep apnea and effects of continuous positive airway pressure therapy. Chest. 2007;131:1406–1413. doi: 10.1378/chest.06-2580. [DOI] [PubMed] [Google Scholar]
- Kelley EE, Hock T, Khoo NK, Richardson GR, Johnson KK, Powell PC, Giles GI, Agarwal A, Lancaster JR, Jr, Tarpey MM. Moderate hypoxia induces xanthine oxidoreductase activity in arterial endothelial cells. Free Radic Biol Med. 2006;40:952–959. doi: 10.1016/j.freeradbiomed.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Lai CJ, Yang CC, Hsu YY, Lin YN, Kuo TB. Enhanced sympathetic outflow and decreased baroreflex sensitivity are associated with intermittent hypoxia-induced systemic hypertension in conscious rats. J Appl Physiol. 2006;100:1974–1982. doi: 10.1152/japplphysiol.01051.2005. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Spiekermann S, Preuss C, Sorrentino S, Fischer D, Manes C, Mueller M, Drexler H. Angiotensin II induces endothelial xanthine oxidase activation: role for endothelial dysfunction in patients with coronary disease. Arterioscler Thromb Vasc Biol. 2007;27:943–948. doi: 10.1161/01.ATV.0000258415.32883.bf. [DOI] [PubMed] [Google Scholar]
- Leung PS, Lam SY, Fung ML. Chronic hypoxia upregulates the expression and function of AT(1) receptor in rat carotid body. J Endocrinol. 2000;167:517–524. doi: 10.1677/joe.0.1670517. [DOI] [PubMed] [Google Scholar]
- Li Q, Dale WE, Hasser EM, Blaine EH. Acute and chronic angiotensin hypertension: neural and nonneural components, time course, and dose dependency. Am J Physiol. 1996;271:R200–R207. doi: 10.1152/ajpregu.1996.271.1.R200. [DOI] [PubMed] [Google Scholar]
- Li YL, Gao L, Zucker IH, Schultz HD. NADPH oxidase-derived superoxide anion mediates angiotensin II-enhanced carotid body chemoreceptor sensitivity in heart failure rabbits. Cardiovasc Res. 2007;75:546–554. doi: 10.1016/j.cardiores.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YL, Li YF, Liu D, Cornish KG, Patel KP, Zucker IH, Channon KM, Schultz HD. Gene transfer of neuronal nitric oxide synthase to carotid body reverses enhanced chemoreceptor function in heart failure rabbits. Circ Res. 2005;97:260–267. doi: 10.1161/01.RES.0000175722.21555.55. [DOI] [PubMed] [Google Scholar]
- Li YL, Xia XH, Zheng H, Gao L, Li YF, Liu D, Patel KP, Wang W, Schultz HD. Angiotensin II enhances carotid body chemoreflex control of sympathetic outflow in chronic heart failure rabbits. Cardiovasc Res. 2006;71:129–138. doi: 10.1016/j.cardiores.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Marcus NJ, Olson EB, Jr, Bird CE, Philippi NR, Morgan BJ. Time-dependent adaptation in the hemodynamic response to hypoxia. Respir Physiol Neurobiol. 2009;165:90–96. doi: 10.1016/j.resp.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanram A, Zhang Z, Shahinfar S, Lyle PA, Toto RD. The effect of losartan on hemoglobin concentration and renal outcome in diabetic nephropathy of type 2 diabetes. Kidney Int. 2008;73:630–636. doi: 10.1038/sj.ki.5002746. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJ, Montano N, Dyken ME, Phillips BG, Somers VK. Contribution of tonic chemoreflex activation to sympathetic activity and blood pressure in patients with obstructive sleep apnea. Circulation. 1998;97:943–945. doi: 10.1161/01.cir.97.10.943. [DOI] [PubMed] [Google Scholar]
- Olson S, Oeckler R, Li X, Du L, Traganos F, Zhao X, Burke-Wolin T. Angiotensin II stimulates nitric oxide production in pulmonary artery endothelium via the type 2 receptor. Am J Physiol Lung Cell Mol Physiol. 2004;287:L559–L568. doi: 10.1152/ajplung.00312.2003. [DOI] [PubMed] [Google Scholar]
- Pediconi D, Martarelli D, Fontanazza A, Pompei P. Effects of losartan and irbesartan administration on brain angiotensinogen mRNA levels. Eur J Pharmacol. 2005;528:79–87. doi: 10.1016/j.ejphar.2005.10.062. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Yuan G, Wang N, Deneris E, Pendyala S, Natarajan V, Kumar GK, Prabhakar NR. NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J Neurosci. 2009;29:4903–4910. doi: 10.1523/JNEUROSCI.4768-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci U S A. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol. 2004;96:1236–1242. doi: 10.1152/japplphysiol.00820.2003. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marce M, Kumar GK, Semenza GL, Prabhakar NR. Heterozygous HIF-1alpha deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol. 2006;577:705–716. doi: 10.1113/jphysiol.2006.114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips SA, Olson EB, Morgan BJ, Lombard JH. Chronic intermittent hypoxia impairs endothelium-dependent dilation in rat cerebral and skeletal muscle resistance arteries. Am J Physiol Heart Circ Physiol. 2004;286:H388–H393. doi: 10.1152/ajpheart.00683.2003. [DOI] [PubMed] [Google Scholar]
- Polotsky VY, Rubin AE, Balbir A, Dean T, Smith PL, Schwartz AR, O’Donnell CP. Intermittent hypoxia causes REM sleep deficits and decreases EEG delta power in NREM sleep in the C57BL/6J mouse. Sleep Med. 2006;7:7–16. doi: 10.1016/j.sleep.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Kumar GK, Chang CH, Agani FH, Haxhiu MA. Nitric oxide in the sensory function of the carotid body. Brain Res. 1993;625:16–22. doi: 10.1016/0006-8993(93)90132-7. [DOI] [PubMed] [Google Scholar]
- Qadri F, Arens T, Schwartz EC, Hauser W, Dominiak P. Angiotensin-converting enzyme inhibitors and AT1-receptor antagonist restore nitric oxide synthase (NOS) activity and neuronal NOS expression in the adrenal glands of spontaneously hypertensive rats. Jpn J Pharmacol. 2001;85:365–369. doi: 10.1254/jjp.85.365. [DOI] [PubMed] [Google Scholar]
- Rey S, Del Rio R, Alcayaga J, Iturriaga R. Chronic intermittent hypoxia enhances cat chemosensory and ventilatory responses to hypoxia. J Physiol. 2004;560:577–586. doi: 10.1113/jphysiol.2004.072033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter O, Schuh K, Brede M, Rothlein N, Burkard N, Hein L, Neyses L. AT2 receptor activation regulates myocardial eNOS expression via the calcineurin-NF-AT pathway. FASEB J. 2003;17:283–285. doi: 10.1096/fj.02-0321fje. [DOI] [PubMed] [Google Scholar]
- Schultz HD, Li YL, Ding Y. Arterial chemoreceptors and sympathetic nerve activity: implications for hypertension and heart failure. Hypertension. 2007;50:6–13. doi: 10.1161/HYPERTENSIONAHA.106.076083. [DOI] [PubMed] [Google Scholar]
- Stepuro TL, Zinchuk VV. Nitric oxide effect on the hemoglobin-oxygen affinity. J Physiol Pharmacol. 2006;57:29–38. [PubMed] [Google Scholar]
- Su KH, Tsai JY, Kou YR, Chiang AN, Hsiao SH, Wu YL, Hou HH, Pan CC, Shyue SK, Lee TS. Valsartan regulates the interaction of angiotensin II type 1 receptor and endothelial nitric oxide synthase via Src/PI3K/Akt signalling. Cardiovasc Res. 2009;82:468–475. doi: 10.1093/cvr/cvp091. [DOI] [PubMed] [Google Scholar]
- Sun MK, Reis DJ. Central neural mechanisms mediating excitation of sympathetic neurons by hypoxia. Prog Neurobiol. 1994;44:197–219. doi: 10.1016/0301-0082(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Sun SY, Wang W, Zucker IH, Schultz HD. Enhanced activity of carotid body chemoreceptors in rabbits with heart failure: role of nitric oxide. J Appl Physiol. 1999;86:1273–1282. doi: 10.1152/jappl.1999.86.4.1273. [DOI] [PubMed] [Google Scholar]
- Troncoso Brindeiro CM, da Silva AQ, Allahdadi KJ, Youngblood V, Kanagy NL. Reactive oxygen species contribute to sleep apnea-induced hypertension in rats. Am J Physiol Heart Circ Physiol. 2007;293:H2971–H2976. doi: 10.1152/ajpheart.00219.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui M, Ichiki T, Katoh M, Egashira K, Takeshita A. Regulation of angiotensin II receptor expression by nitric oxide in rat adrenal gland. Hypertension. 1998;32:527–533. doi: 10.1161/01.hyp.32.3.527. [DOI] [PubMed] [Google Scholar]
- Zucker IH, Schultz HD, Patel KP, Wang W, Gao L. Regulation of central angiotensin type 1 receptors and sympathetic outflow in heart failure. Am J Physiol Heart Circ Physiol. 2009;297:H1557–H1566. doi: 10.1152/ajpheart.00073.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]