

Abstract
As a promiscuous redox partner, the biological role of cytochrome P450 reductase (CPR) depends significantly on protein-protein interactions. We tested a hypothesized CPR docking site by mutating D113, E115, and E116 to alanine and assaying activity toward various electron acceptors as a function of ionic strength. Steady-state cytochrome c studies demonstrated the mutations improved catalytic efficiency and decreased the impact of ionic strength on catalytic parameters when compared to wild type. Based on activity toward 7-ethoxy-4-trifluoro-methylcoumarin, CYP2B1 and CPR favored formation of an active CYP2B1·CPR complex and inactive (CYP2B1)2·CPR complex until higher ionic strength whereby only the binary complex was observed. The mutations increased dissociation constants only for the binary complex and suppressed the ionic strength effect. Studies with a non-binding substrate, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) suggest changes in activity toward cytochrome c and CYP2B1 reflect alterations in the route of electron transfer caused by the mutations. Electrostatic modeling of catalytic and binding parameters confirmed the importance of D113 and especially the double mutant E115 and E116 as mediators in forming charge-charge interactions between CPR and complex partners.
Keywords: cytochrome P450 reductase, docking, site-directed mutagenesis, CYP2B1, protein-protein interactions, electrostatic interactions
1. Introduction
Despite its name, mammalian cytochrome P450 reductase (CPR)1 is a promiscuous electron donor for many proteins, and consequently, protein-protein interactions play a critical role in the biological functions of this enzyme. Horecker in 1950 [1] first identified the enzyme as an NADPH-dependent cytochrome c3+ (cyt c) reductase; however, later studies showed that this flavoprotein was localized to the endoplasmic reticulum (microsomes) [2, 3] and not the mitochondrion, where cyt c is found. A critical biological role for CPR activity was demonstrated by Lu and Coon when reconstitution of a microsomal fatty acid hydroxylase was shown to require cytochrome P450 (P450 or CYP for a specific isoform), CPR, and phospholipid for activity [4]. Although studies have predominantly focused on the role of CPR in P450 catalysis, CPR also transfers electrons to heme oxygenase in heme degradation [5], squalene monooxygenase [6] and 7-dehydro-cholesterol reductase [7] in the synthesis of sterols, and cytochrome b5 (cyt b5) in fatty acid desaturation and elongation [8]. Interestingly, mammals express only one form of CPR to participate in these various complexes. How CPR structure enables this catalytic promiscuity is unclear, but undoubtedly relevant to the activities of the resultant complexes in the cell.
The formation and stabilization of CPR complexes reflects the contribution of negatively-charged CPR residues and positively-charged residues on complex partners (reviewed in [9]). Increases in ionic strength (I) mask these charged residues, thereby disrupting CPR complexes including those with cyt c and multiple P450s. More specifically, the CPR docking site may consist of a cluster of acidic residues in the α helix F-β sheet 5 loop as shown by site-directed mutagenesis [10] and cross-linking studies [11] (Fig. 1, blue loop). Nevertheless, chemical labeling experiments [12] and antibody inhibition studies [13] implicated another site consisting of the β sheet 2-α helix C loop (Fig. 1, yellow loop). Within this region and especially the loop are a number of negatively-charged residues that participate in docking to redox partners. Surprisingly, these loops reside on opposite faces of CPR suggesting the enzyme binds protein partners through multiple docking sites and/or sub-sites as part of one large docking site. Further validation of these docking sites is clearly needed.
Fig. 1.

Proposed docking sites for protein redox partners and CPR based on the X-ray crystallographic structure [49]. Loops potentially mediating protein-protein interactions are shown relative to the electron donating FMN moiety. FMN, red; α helix F-β sheet 5 loop, blue; β sheet 2-α helix C loop, yellow; mutated acidic residues, orange. Molecular graphics were generated by PYMOL (http://pymol.sourceforge.net).
Although not identified in those original studies, we hypothesize that D113, E115, and E116 within the β sheet 2-α helix C loop of rat CPR contribute at a minimum to a second, spatially-distinct docking site for binding protein partners (Fig. 1, orange). These residues are conserved among all mammalian CPRs including humans (Table 1). Even yeast CPR retains the corresponding acidic residues at positions D113 and E115. The occupancy of the site created by this loop would impact the number and functional properties of potential CPR complexes, and hence the contribution of P450s to the metabolism of foreign, biologically active (xenobiotic) compounds.
Table 1.
Alignment of yeast, rat, and human CPR amino acid sequences for the β sheet 2-α helix C loop using LALIGN [45].
To test our hypothesized docking site, we mutated D113, E115, and E1 16 to alanine and assayed activity toward various electron acceptors as a function of ionic strength. Unlike carboxamide analog substitutions for these residues, alanine eliminates electrostatic and hydrogen-bonding contributions to complexation. In these studies, we employed CPR-K56Q as a surrogate for the “wild type” form of the reductase. This mutation generates an enzyme resistant to proteolytic degradation without altering its activity [14, 15]. To avoid compensation between residues 115 and 116, we made only a double mutant, E115/6A. Because the proposed mutations may affect CPR properties unrelated to docking, we assayed activity toward 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), a small organic molecule. This substrate undergoes one electron reduction only through the transient formation of a collision complex rather than associating to a specific binding site [16]. Due to the extensive use cyt c as a model CPR partner, we measured cyt c reduction under steady-state conditions.
More importantly, we determined the impact of the mutations on the ability to reconstitute rat CYP2B1 activity toward 7-ethoxy-4-trifluoromethylcoumarin (7-EFC) through a high throughput method. Initial studies revealed non-hyperbolic (non-binary) relationship between the observed CYP2B1 activity and protein concentration. We interpreted these results by identifying the most statistically favorable mechanisms among several possibilities using the software program DynaFit [17]. For all catalytic studies, we varied ionic strength to disrupt electrostatic interactions and assess the impact on catalytic efficiencies (kcat/Km) for MTT and cyt c and binding interactions, i.e. complex dissociation constants, for CYP2B1. All data sets were then analyzed by electrostatic modeling to assess the effect of mutations on the relative charge at the docking site [18].
2. Materials and methods
2.1. Materials
MTT, cyt c (horse heart), components of NADPH regenerating system (NADP+, glucose 6-phosphate, torula yeast glucose 6-phosphate dehydrogenase) for catalytic assay were purchased from Sigma-Aldrich, as were dilauroyl-L-α-phosphatidylcholine, bovine erythrocyte superoxide dismutase, and catalase. 7-EFC and reference standard 7-hydroxy-4-trifluoromethylcoumarine (7-HFC) were obtained from Molecular Probes, Inc. (Eugene, OR). HPLC grade acetonitrile and other basic chemicals were purchased from Fisher Scientific (Houston, TX). Rat CYP2B1 was prepared as described previously [19].
2.2. Construction and expression of rat CPR enzymes
D113, E115, and E116 were substituted with alanine, and the corresponding CPR proteins prepared using established protocols. The expression plasmid for rat CPR-K56Q was used as a template for site-directed mutagenesis. This plasmid included the substitution of Lys56 to Gln to suppress proteolytic cleavage of the N-terminus [14, 15]. For simplicity, this form of the enzyme is referred to as “wild type”. The Stratagene QuikChange kit was used to introduce the desired substitutions as per manufacturer protocol. The corresponding primers were designed primers to incorporate alanine codons optimized for protein expression in Escherichia coli. The primers for these reactions are shown below, whereby the underlined regions are sites for mutations:
D113A:
Forward 5′-GCATGTCCGCAGCGCCTGAAGAGTATGACTTGGCC
Reverse 5′-CATACTCTTCAGGCGCTGCGGACATGCCCCGC
E115/6A:
Forward 5′-GTCCGCAGACCCTGCGGCGTATGACTTGGCCGACC
Reverse 5′-GGCCAAGTCATACGCCGCAGGGTCTGCGGACATGC
The wild type and resulting mutant reductase proteins were expressed in Topp3 strain of E. coli cells (Stratagene) and subsequently purified as described by others [20–23] with some modifications [15]. In brief, after solubilizing the protein, CPR was purified by affinity chromatography using 2′,5′-ADP agarose affinity column. Following elution from the affinity column, CPR was loaded onto DEAE resin to facilitate the removal of detergents from solubilization steps. High salt eluted the protein, which was then applied to Zeba desalting spin column (Pierce Biotechnology, Rockford, IL) pre-equilibrated with 50 mM potassium phosphate pH 7.4, 1 mM EDTA and 20 % glycerol, the storage buffer. Final samples were prepared as high concentration stocks such that dilutions during experimental studies minimized any effects from the storage buffer. These stocks were stored at −80 °C. Protein concentrations were quantitated spectrally based on absorbance properties of CPR flavins (FAD and FMN) as reported by French and Coon [24]. As a measure of purity, total protein levels were determined using the bicinchoninic acid (BCA) assay as described by the manufacturer (Thermo Scientific/Pierce). We also analyzed the purity of samples by SDS-PAGE.
2.3. Quantitation of CPR flavins
Based on a published method by Lopez-Anaya and Mayersohn [25], an HPLC procedure was developed by our group to assess the incorporation of FAD and FMN in CPR proteins. Protein samples were diluted in a brown screw cap vial to 100 nM in 10 mM potassium phosphate pH 7.4, 1 mM EDTA containing 100 nM riboflavin as an internal standard. Samples were boiled 5 min, cooled 3 min on ice, and then microfuged 10 min. The resulting supernatant was then transferred to an amber HPLC vial. Each sample was injected on a Waters Symmetry C18 3.5 μm 4.6 × 75 mm column with a 90:10 25 mM potassium phosphate pH 2.6/H2O:CH3CN mobile phase at a flow rate of 1.5 ml min−1. The elution of the flavins was monitored by fluorescence (excitation: 450 nm, emission: 535 nm). All three compounds eluted within 6 min. The corresponding concentrations of the samples were then determined relative to standards.
2.4. MTT reduction assay
Reduction of MTT by CPR was determined spectrophotometrically by measuring the increase of 610 nm at 25 °C [16]. Reaction mixtures included 5 nM CPR, 100 μM NADPH, 25 g/L Emulgen 909, 20 to 300 mM KCl in 20 mM HEPES buffer (pH 7.5), and varying concentration of MTT (1 to 100 μM) in a total volume of 1 ml. Emulgen 909 was included to ensure CPR remained in a monomeric state [26]. Reactions were initiated by the addition of CPR, and the increase in absorbance at 610 nm was monitored for 60 s to calculate initial velocities. These data were plotted against substrate concentration and then fit to a non-linear regression described by the Michaelis-Menten equation using Graph-Pad Prism software (San Diego, USA), which yielded the corresponding kinetic parameters, Km and kcat.
2.5. Cyt c3+ reduction assay
The kinetic parameters of cyt c reduction by CPR were determined by monitoring the formation of reduced cyt c (cyt c 2+) at 550 nm at 25 °C using a modification of a previously published method by Yasukochi and Masters[20]. The reaction mixtures contained 1 nM CPR, 100 μM NADPH, 25g/L Emulgen 909 in 20 mM HEPES (pH 7.5), and varying concentrations of cyt c (0.25 to 100 μM) in a total volume of 1 ml as described previously. It also contained KCl to achieve the desired ionic strength. CPR can aggregate, which can affect results from catalytic studies. As a solution, Emulgen 909 was included to ensure CPR remained in a monomeric state as shown by light scattering studies [26]. The initial velocities were determined continuously at 550 nm using a Jasco V-550 spectrophotometer. These data were plotted against substrate concentration and then fit to a non-linear regression described by the Michaelis-Menten equation using Graph-Pad Prism software (San Diego, USA), which yielded the corresponding kinetic parameters, Km and kcat.
2.6. CYP2B1 activity assay
Initial reaction rates for CYP2B1-mediated 7-EFC O-deethylation were determined using reconstituted systems by modifying a published protocol [27]. Reactions included 20 mM HEPES (pH 7.5), 20 μM DLPC, 50 μM 7-EFC, CPR mutants (0.025 μM), and an NADPH regenerating system with increasing KCl concentration to achieve the desired ionic strength. The NADPH regenerating system ensures the reductase remains under saturating conditions of cofactor during the reaction. Reactions were at eight CYP2B1 concentrations ranging from 15 to 400 nM to correspond to the eight wells for each column of the microplate. Following the addition of all components except NADP+, the reactions in the assay block were incubated at 37 °C for 5 min. The reactions were initiated upon addition of NADP+ and then generally incubated for 30 min at 37 °C. At three time points, an aliquot was taken from the reaction, quenched with acetonitrile, and further analyzed by PerkinElmer Victor3V 1420 Multi-label Counter (Boston, MA) possessing 410 nm excitation and 510 nm emission bandpass filters. 7-HFC was used for generation of standard curves.
Initial titration studies were clearly non-hyperbolic indicating that CYP2B1 and CPR associated through a more complex mechanism than a simple binary complex. As described for CYP1A2 [28] and CYP2E1 [29], CYP2B1 and CPR in this study were hypothesized to form the expected functional binary (CYP2B1·CPR) complex and possibly a higher order ternary ((CYP2B1)2·CPR) complex, which may or may not be active as described by Kpr and Kppr, respectively in Fig. 2. The prospect of CYP2B1 self-association into dimers (Kpp), which is commonly observed for some, but not all P450s (discussed in [30]), was also introduced as a potential step in the mechanism. To limit the number of possibilities, every potential mechanism was required to include the formation of a binary complex resulting in six different complexation mechanisms.
Fig. 2.

Possible modes of interaction between CYP2B1 and CPR. The traditional binary complex between CYP2B1 and CPR is simplest mechanism of interaction. Other possible complexes may form including a higher order complex, (CYP2B1)2·CPR, and a CYP2B1 dimer, (CYP2B1)2. The contributions of these steps to the interaction between the proteins were assessed using DynaFit software [17, 29].
The most probable mechanism and corresponding parameters for the mode of interaction between CYP2B1 and CPR was identified using the advanced tools of numerical analysis and applied statistics, as implemented in the software DynaFit [17, 29]. A representative input file (script) for this analysis is included in Supplemental Data.
2.7. Electrostatic modeling
The contribution of charged groups was estimated by the decrease in catalytic efficiency for the cyt c reaction (kcat/Km) or the reciprocal of the increase in dissociation constant for complex between CYP2B1 and CPR (1/Kpr). This effort was made possible through the use of a semi-empirical relationship for the electrostatic energy of interaction between complex partners based on studies between cyt c and its oxidation-reduction partners [18]. For this model, specific complementary charge-pair interactions between positively-charged residues for cyt c and CYP2B1 and negatively-charged acidic groups for CPR are assumed to dominate the model of interaction. The natural log of the respective parameters (lnk) was plotted as a function of the square root of ionic strength (I1/2) and then fit to Equation 1. In this equation, “n” is the relative number of charge pairs in the interaction and influences the slope of the curve. The variable “a” is the effective radius of the region of contact. The average distance between the contacts is reflected in “r”, which influences the amount of curvature. The lnkinf term is the respective catalytic efficiency for cyt c reduction or dissociation constant for CYP2B1·CPR at infinite ionic strength and sets the vertical limit for the equation. To simplify the analysis, we assumed the size of the site of interaction and the distance between contacts were the same for CPR and complex partners. Based on initial fitting results, we assigned as constants the “a” value to 1.7 and “r” to 2.3, which is similar to values reported previously by others [18].
| (1) |
2.8. Confidence intervals for model parameters
Nonsymmetrical confidence intervals for all model parameters at stated probability levels were computed by using the profile-t method of Bates and Watts [31].
3. Results
3.1. Analysis CPR protein preparations
The homogeneity and integrity of CPR samples are critical for assessing the interactions between complex partners. After quantitating CPR spectrally [24], we analyzed the incorporation of flavins (FAD and FMN). As shown in Table 2, all three enzymes incorporated the correct 1:1 stoichiometry of FAD and FMN. Both concentrations were within error of the CPR concentration used in the experiment (100 nM). The SDS-PAGE analysis of the proteins revealed a single band corresponding to the approximate size of CPR (78 kDa) for each preparation, and thus demonstrated the homogeneity of the samples for these studies (Fig. 3). These observations were validated by determinations of the total protein concentration for the samples relative to a bovine serum albumin standard (Table 2). Minimal variations in those values suggest common purity among the samples.
Table 2.
Analysis of CPR protein preparationsa
| CPR | [CPR]b(nM) | Flavins (nM)c | [Protein]totald (μg/mL) | |
|---|---|---|---|---|
| [FAD] | [FMN] | |||
| “Wild type”e | 100 | 102 ± 2 | 114 ± 8 | 7.5 ± 0.2 |
| D113A | 100 | 113 ± 7 | 100 ± 5 | 7.4 ± 0.1 |
| E115/6A | 100 | 105 ± 3 | 98 ± 6 | 7.6 ± 0.1 |
Standard deviation shown for the average value from 3 to 5 experiments.
CPR concentrations were determined spectrally based on absorbance properties of the flavins (FAD and FMN) as reported by others [24]. A 100 nM CPR solution was then used for other studies.
Flavin concentrations were determined by HPLC in Methods section 2.3.
Total protein concentration was determined by the bicinchoninic acid (BCA) assay as described by the manufacturer (Thermo Scientific/Pierce).
“Wild type” is CPR-K56Q
Fig. 3.
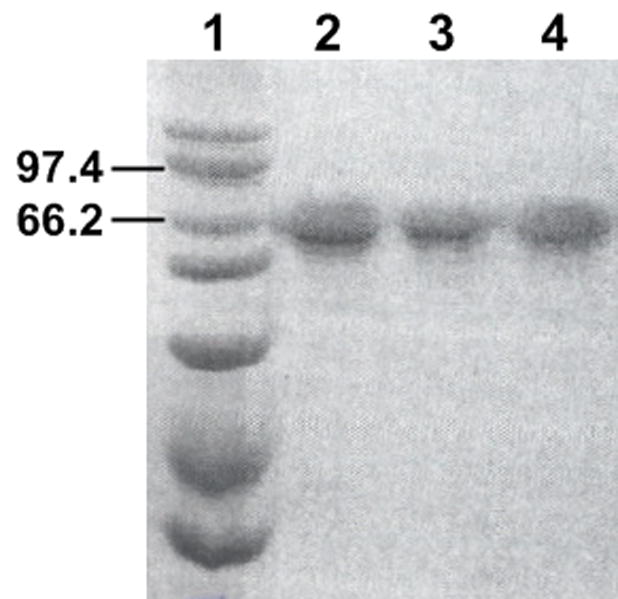
SDS-PAGE analysis of CPR preparations. In lane 1, a Bio-Rad high molecular weight standard was included to assess the size of proteins resolved by the gel. The most relevant standards at 97.4 and 66.2 kDa are indicated. A total of 10 mg protein from each CPR preparation was loaded in lanes 2 (“wild type”), 3 (D113A), and 4 (E115/6A). All samples were separated using a 10 % acrylamide gel with a length of 10 cm and subsequently stained with coommasie brilliant blue dye.
3.2. The effect of mutations on CPR catalytic properties
To determine the effects of the mutations on CPR properties independent of binding, we measured reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) for all CPR enzymes as a negative control (Table 3, kFig. 4). Compared to wild type, both mutations increased cat approximately two-fold, but did not affect Km at the lowest ionic strength (10 mM). As the ionic strength increased, wild type kcat and Km increased 4.8-fold and 4.5-fold, respectively, such that the catalytic efficiency (kcat/Km) was essentially unchanged. Similarly, both kinetic parameters increased approximately four-fold for D113A and consequently no change in catalytic efficiency. For E115/6A, there was a slight decrease in catalytic efficiency at higher ionic strengths due to a larger increase in Km than observed for kcat. At the highest ionic strength, doubling the NADPH concentration did not affect the kinetic parameters indicating NADPH was still saturating.
Table 3.
Effect of CPR mutation on MTT reduction as a function of ionic strengtha
| “Wild type”b | D113A | E115/6A | |||||||
|---|---|---|---|---|---|---|---|---|---|
| I (mM) | kcat (min−1) | Km (μM) | kcat/Km (min−1μM−1) | kcat (min−1) | Km (μM) | kcat/Km (min−1μM−1) | kcat (min−1) | Km (μM) | kcat/Km (min−1μM−1) |
| 10 | 350 (330–370) | 2.2 (1.4–3.0) | 160 | 630 (610–650) | 2.1 (1.7–2.5) | 300 | 780 (740–820) | 1.8 (1.3–2.3) | 430 |
| 30 | 510 (470–550) | 3.1 (2.0–4.2) | 160 | 830 (800–870) | 2.6 (2.1–3.1) | 320 | 1100 (990–1200) | 2.6 (1.6–3.6) | 410 |
| 60 | 700 (660–740) | 3.9 (3.0–4.8) | 180 | 1200 (1100–1200) | 4.1 (3.6–4.8) | 290 | 1500 (1400–1600) | 3.8 (2.9–4.6) | 390 |
| 110 | 1100 (1000–1200) | 6.2 (4.6–7.7) | 180 | 1800 (1700–1900) | 6.3 (5.2–7.4) | 290 | 2300 (2100–2400) | 6.5 (5.3–7.6) | 350 |
| 210 | 1700 (1600–1900) | 10 (7.6–13) | 170 | 2400 (2300–2500) | 8.7 (7.2–10) | 280 | 3400 (3200–3600) | 11 (9.3–13) | 300 |
Kinetic parameters were determined as described under Materials and Methods. Values in parentheses denote 95 % confidence intervals for respective parameters.
“Wild type” is CPR-K56Q, the proteolytically resistant form of the enzyme, used to generate the mutants.
Fig. 4.

Effect of CPR mutation on MTT reduction as function of ionic strength. MTT reduction by wild type (A), D113A (B), and E115/6A (C) was measured with varying concentrations of MTT (1 to 100 μM) as a function of ionic strength for 60 s. Color intensity reflects ionic strength from 10 to 210 mM. The reported values reflect the results from three experiments performed in duplicate and include the standard deviation from mean. The parameters from the fits of the kinetic profiles are summarized in Table 3.
3.3. The effect of CPR mutations on cyt c reduction
As a model protein complex dominated by electrostatic interactions, we measured cyt c reduction by CPR as a function of ionic strength (Table 4, kFig. 5). Compared to wild type, both mutations increased cat by 50 % and Km by 25 %, respectively, at the lowest ionic strength (30 mM). As the ionic strength increased, wild type kcat and Km increased 2.1-fold and 11-fold, respectively, resulting in an overall decrease in catalytic efficiency (kcat/Km). While the kcat increased by the same magnitude for the mutants, the corresponding Km value increased only 9.2-fold for D113A and 6.4-fold for E115/6A. These trends indicate a suppression of catalytic efficiency as a result of the mutations. Doubling the NADPH concentration did not alter the kinetic parameters indicating NADPH remained saturating at the highest ionic strength.
Table 4.
Effect of CPR mutation on cyt c reduction as a function of ionic strengtha
| “Wild type”b | D113A | E115/6A | |||||||
|---|---|---|---|---|---|---|---|---|---|
| I (mM) | kcat (min−1) | Km (μM) | kcat/Km (min−1 μM−1) | kcat (min−1) | Km (μM) | kcat/Km (min−1 μM−1) | kcat (min−1) | Km (μM) | kcat/Km (min−1 μM−1) |
| 30 | 1000 (980–1100) | 2.0 (1.7–2.3) | 500 | 1500 (1400–1600) | 2.5 (1.9–3.0) | 600 | 1500 (1400–1600) | 2.4 (1.4–3.3) | 630 |
| 60 | 880 (820–940) | 4.0 (3.7–4.2) | 220 | 1400 (1300–1500) | 5.2 (4.0–6.3) | 270 | 1500 (1400–1600) | 3.6 (3.1–5.1) | 420 |
| 110 | 1200 (1100–1300) | 8.5 (5.5–12) | 140 | 2200 (2000–2400) | 11 (8.2–15) | 200 | 2200 (2000–2300) | 6.0 (4.2–7.7) | 360 |
| 210 | 2000 (1900~2000) | 20 (18–21) | 100 | 3900 (3700~4000) | 26 (23~28) | 150 | 3500 (3400~3600) | 13 (12~15) | 270 |
| 310 | 2100 (2100–2200) | 23 (21–26) | 91 | 3300 (3200–3500) | 25 (23–28) | 130 | 3500 (3400–3700) | 16 (14–18) | 220 |
Kinetic parameters were determined as described under Materials and Methods. Values in parentheses denote 95 % confidence intervals for respective parameters.
“Wild type” is CPR-K56Q, the proteolytically resistant form of the enzyme, used to generate the mutants.
Fig. 5.

Effect of CPR mutation on cyt c reduction as a function of ionic strength. Cyt c reduction by wild type (A), D113A (B), and E115/6A (C) is shown with varying ionic strength. Experiments were performed with 1 nM CPR, 100 μM NADPH, 0.25g/L Emulgen 909 in 20 mM HEPES (pH 7.5), and varying concentrations of cyt c (0.25 to 100 μM). Color intensity reflects ionic strength from 30 to 310 mM. The reported values reflect the results from three experiments performed in duplicate and include the standard deviation from mean. The parameters from the fits of the kinetic profiles are summarized in Table 4.
3.4. The effect of CPR mutation on CPR-CYP2B1 complexation
For more biologically relevant studies with a CPR redox partner, we measured the ability of reconstituted CYP2B1 to oxidize 7-EFC as a function of ionic strength (48 to 189 mM). Unlike the MTT and cyt c studies, the measured rates as a function of titrant (CYP2B1) concentration were not hyperbolic. At lower ionic strengths, the increase in CYP2B1 concentration led to an increase in the observed rate followed a decline, indicating a simple binary mechanism of association was not sufficient to describe the mode of interaction between CYP2B1 and CPR (Fig. 6). As described for CYP1A2 [28] and CYP2E1 [29], we hypothesized that CYP2B1 and CPR may interact to form the expected functional binary (CYP2B1·CPR) complex and a higher order ternary ((CYP2B1)2·CPR) complex in the potential presence of a CYP2B1 dimers (Fig. 2). We then globally fit the data for CYP2B1 and the reductases to the resulting potential complexation mechanisms and statistically analyzed the quality of the fits, to select the most probable model.
Fig. 6.

Effect of CPR mutation on CYP2B1 mediated 7-EFC activity as a function of ionic strength. CYP2B1 mediated 7-EFC activity is shown for wild type (A), D113A (B), E115A/E116A (C) with increasing ionic strength. Experiments were performed with 20 mM HEPES (pH 7.5), 20 μM DLPC, 50 μM 7-EFC, CPR mutants (0.025 μM), varying concentrations of CYP2B1 (15 to 400 nM), and an NADPH regenerating system with increasing KCl concentration to achieve the desired ionic strength. Color intensity reflects ionic strength from 48 to 189 mM. The reported values reflect the results from three experiments performed in duplicate and include the standard deviation from mean. The corresponding fits of the kinetic profiles reflect the statistically most probable mechanism as determined by DynaFit software (see Results and Supplemental Data). The parameters from the fits are summarized in Table 5.
After globally fitting reactions to these models, we generated a corresponding Akaike Information Criterion (AICc) to identify statistically the most plausible model based on the quality of the fits. Due to the volume of these data, we included the raw results in Supplemental Data. The significance rules as outlined by Burnham and Anderson ([32], p. 70) provided a metric to rank models such that the lower AICc values indicated comparatively high support for the mechanism. When comparing the AAICc, values between 0 and 2 indicate substantial support, while values between 4 and 7 signify considerably less support. A value for AAICc greater than 10 indicates essentially no support for the model.
For wild type and mutants, the mechanism of interaction between CYP2B1 and CPR favored formation of the functional CYP2B1·CPR complex and an inactive (CYP2B1)2·CPR complex until higher ionic strength whereby only the binary complex was observed (Fig. 2). The statistical analyses for each sample set are included in Supplemental Data. In some cases, models including a P450 dimer or active (CYP2B1)2·CPR complex were statistically possible; however, an examination of the parameters for those models revealed extremely large uncertainty among most of the parameters. Moreover, the best fit Vmax value for an active (CYP2B1)2·CPR complex was always essentially zero (0.000001), which reduces the model down to the one with an inactive ternary complex. The fit of the data to the preferred mechanisms are shown in Fig. 6, and the parameters summarized in Table 5. Compared to wild type, the D113 mutation led to a 50 % increase for Kpr and no change in catalytic activity (Vmax) at the lowest ionic strength (48 mM). By contrast, these values increased 2.7- and 4.1-fold, respectively, for E115/6A. At higher ionic strengths, wild type Kpr increased 2.5-fold, while Vmax did not begin a steady decrease until 88 mM ionic strength. A similar trend for the binary complex parameters was observed for D113A. For E115/6A, the increase in ionic strength led to an immediate decrease in both parameters. Interestingly, the magnitude of the ionic strength effect on Kpr and Vmax for all CPRs coincided with the disappearance of the inactive ternary complex.
Table 5.
Effect of CPR mutation on CYP2B1 7-EFC activity as a function of ionic strengtha
| “Wild type”b | D113A | E115/6A | |||||||
|---|---|---|---|---|---|---|---|---|---|
| I (mM) | Vmax (nM min−1) | Kpr (nM) 7.9 | Kppr (nM) | Vmax (nM min−1) | Kpr (nM) 12 | Kppr (nM) | Vmax (nM min−1) | Kpr (nM) | Kppr (nM) |
| 48 | 1.1 (0.8–1.7) | (1.0–30) | 570 (200–2500) | 1.3 (1.0–1.9) | (3.5–34) | 740 (270–3600) | 4.5 (4.0–5.2) | 21 (14–30) | 710 (470–1200) |
| 53 | 1.2 (0.9–2.0) | 11 (2.2–40) | 630 (200–4100) | 1.4 (1.1–2.1) | 15 (5.1–42) | 800 (280–5700) | 4.0 (3.3–5.3) | 25 (13–47) | 870 (390–2900) |
| 59 | 1.1 (0.8–1.8) | 14 (3.3–47) | 620 (200–3600) | 1.4 (1.1–1.8) | 20 (9.9–39) | 1100 (460–8300) | 3.6 (3.0–4.7) | 23 (12–44) | 1600 (590~>90000)d |
| 88 | 0.7 (0.5–1.3) | 19 (5.4–63) | 770 (220–23000) | 1.0 (0.8–1.5) | 19 (7.7–46) | 1700 (470~>510000)c | 2.2 (2.0–2.4) | 21 (13–32) | |
| 137 | 0.3 (0.2–0.3) | 22 (11–40) | 0.5 (0.4–0.6) | 25 (10–53) | 1.6 (1.5–1.7) | 31 (24–42) | |||
| 189 | 0.2 (0.2–0.3) | 16 (5.1–38) | 0.3 (0.3–0.4) | 37 (18–71) | 1.1 (0.9–1.1) | 28 (18–43) | |||
“Wild type” is CPR-K56Q, the proteolytically resistant form of the enzyme, used to generate the mutants. Kinetic parameters were determined as described under Materials and Methods. Values in parentheses denote 95 % confidence intervals for respective parameters unless otherwise indicated.
“Wild type” is CPR-K56Q, the proteolytically resistant form of the enzyme, used to generate the mutants.
Confidence intervals at 80%.
Confidence intervals at 93%.
3.5 Electrostatic modeling
In all cases, catalytic efficiency for MTT reduction displayed no curvature as a function of ionic strength (Fig. 7, Panel A), which obviated the ability to fit the data to the electrostatic modeling equation. To emphasize the lack of curvature, the data were fit effectively by a linear regression. Despite the lack of curvature, the data from CPR-E115/6A studies did demonstrate a gradual decrease in efficiency as a function of ionic strength.
Fig. 7.

Electrostatic modeling of CPR complexes. (A) For MTT studies, the natural log of catalytic efficiencies taken from Table 3 were plotted against the square root of ionic strength ranging from 10 to 210 mM and fit to Equation 1. (B) For cyt c studies, the natural log of catalytic efficiencies taken from Table 4 were plotted against the square root of ionic strength ranging from 30 to 310 mM and fit to Equation 1. (C) For CYP2B1 studies, the natural log of 1/Kpr values taken from Table 5 were plotted against the square root of ionic strength ranging from 48 to 189 mM and fit to Equation 1. Symbols: wild type, circles (●); D113A, triangles (▲); E115/6A, inverted triangles (▼;). Color intensity reflects increasing ionic strength. The reported values reflect the results from three to eight experiments and include the standard deviation from mean. The parameters from the fits of the kinetic profiles are summarized in Table 6.
As expected, the catalytic efficiency for cyt c reduction was dependent on the formation of electrostatic interactions between cyt c and CPR (Fig. 7, Panel B). Higher ionic strength led to a decrease in catalytic efficiency. The impact of this effect correlated with the predicted roles for residues targeted for mutagenesis as shown by the electrostatic modeling parameters in Table 6. The “n” value is a relative measure of electrostatic interactions for the Michaelis complex during turnover in these experiments. This parameter decreased 1.3-fold for D113A and 2.5-fold for E115/6A, respectively. These mutations also increased the respective catalytic efficiencies for the enzymes at infinite ionic strength. As observed for the “n” values, the mutation of E115/6 produced a more significant effect than the D113 mutation.
Table 6.
Electrostatic modeling for cyt c and CYP2B1a
| Cyt c reduction | Reconstituted CYP2B1 activity | |||
|---|---|---|---|---|
| CPR | n | (kcat/Km)∞ | n | Kpr,∞ |
| “Wild type”b | 8.5 (7.0–9.9) | 79 (65–94) | 7.4 (5.0–9.8) | 32 (25–44) |
| D113A | 6.5 (5.3–7.7) | 120 (100–140) | 7.4 (4.6–10) | 42 (33–57) |
| E115/6A | 3.9 (2.6–5.3) | 240 (200–280) | 6.6 (4.4–8.8) | 54 (45–69) |
Modeling parameters were determined as described under Materials and Methods. Values in parentheses denote 95 % confidence intervals for respective parameters.
“Wild type” is CPR-K56Q, the proteolytically resistant form of the enzyme, used to generate the mutants.
Similarly, the mutations decreased binding between CYP2B1 and CPR presumably from a loss in electrostatic interactions (Table 6). When compared to wild type Kpr at infinite ionic strength, the mutant dissociation constant for the binary complex increased 1.3-fold for D113A and 1.7-fold for E115/6A, respectively. Unlike the data for cyt c reduction, the mutations had a less severe effect on the relative number of contacts between the proteins. The “n” values for CYP2B1 complexes with all reductases were within the same confidence intervals.
4. Discussion
4.1. Non-docking mutational effects
Although charged residues are known to contribute to protein-protein interactions, we identified acidic residues of the β sheet 2-α helix C loop participating in binding and unexpectedly, catalysis for complexes between CPR and redox partners. Control experiments with MTT revealed alanine mutations of D113 and E115/6 increased substrate turnover (kcat) at a given ionic strength, but did not impact catalytic efficiencies as a function of ionic strength. There are two possible explanations for the mutational effects on turnover; the mutations improved the redox properties of CPR to favor direct MTT reduction and/or increased superoxide anion generation by the enzyme as an indirect pathway to MTT reduction [33]. CPR reduction of molecular oxygen to superoxide anion occurs slowly, if at all [2], and thus it is more likely that mutations changed redox properties of the flavins. Unlike cyt c and CYP2B1, the electrostatic modeling efforts revealed no curvature for MTT, which is consistent with the premise that no electrostatically-stabilized complex forms between CPR and the small organic molecule.
In the absence of binding, mutations must alter the ability of CPR to reduce electron acceptors by changing FMN redox properties and/or the electron transfer pathway. While proximal charges can influence redox properties of molecules, this possibility does not seem likely given targeted residues are greater than 20 Å from the FMN moiety (Fig. 1). An alternative and more plausible explanation is that mutation of negatively-charged residues alters the mode of interaction between CPR and electron acceptors thereby changing the pathway for electron transfer. The sensitivity of this step to structure would then impact electron transfer rates and consequently the ability to reduce MTT, cyt c, and reconstitute CYP2B1 activity. In the latter two cases, catalysis and binding to both proteins were sensitive to ionic strength indicating a link between binding and electron transfer events through the targeted residues.
4.2. Generation of nonfunctional complexes between cyt c and CPR
Unlike previous chemical labeling studies [12] suggesting otherwise, we report the β sheet 2-α helix C loop mediates cyt c reduction; these contrasting results may reflect the design of the respective experimental studies. In the former study, incomplete labeling of multiple acid residues by chemical labeling of CPR would dilute the impact of the loss of those contacts between CPR and cyt c, whereas our site-directed mutagenesis studies targeted a specific site and completely eliminate the negative charge. The previous study measured cyt c reduction activity in the presence of 300 mM potassium phosphate pH 7.4. Under such high ionic strength conditions (666 mM), electrostatic interactions would contribute little to catalysis. Our use of a range of lower ionic strengths favored these types of protein-protein contacts, and thus revealed the significance of D113 and E115/6 in this reaction.
The catalytic roles for D113 and E115/6 interacting with the positively-charged surface of cyt c were manifest in both catalytic parameters. The significant kcat effect demonstrated the respective residues played a role in step(s) limiting turnover of cyt c. Under low ionic strength conditions (< 500 mM), release of products, NADP+ and/or cyt c2+, was shown to contribute, if not determine, kcat for this reaction [34]. Nevertheless, those studies were performed in phosphate buffer. Phosphate and cyt c form complexes that impact the interaction and electron-transfer properties between cyt c and redox partners [35], and thus the findings from the previous kinetic studies are not likely applicable to our efforts with an HEPES/KCl system. Based on the MTT experiments, we speculate that electron transfer or a step related to this event contributes to cyt c reduction such that mutation of D113 and E115/6 alters this process to increase the turnover rate for CPR. Similarly, loss of E115/6 facilitated formation of the Michaelis complex at higher ionic strengths as evidenced by decreases in Km values. Taken together, catalytic efficiency (kcat/Km) improved greatly as a consequence of the loss of the acidic residues. These findings suggest that the β sheet 2-α helix C loop facilitates generation of inactive or less functional complexes between CPR and cyt c. Because cyt c is not a biological redox partner for CPR, it is plausible that cyt c recognition of the CPR docking site is not highly evolved and thus not specific.
4.3. Complex interactions between CYP2B1 and CPR
Like CYP1A2 [28] and CYP2E1 [29], CYP2B1 forms multiple complexes with CPR that may or may not be functional. From our study, CYP2B1 and CPR initially form a high affinity functional CYP2B1·CPR complex. At higher CYP2B1 concentrations, association of another CYP2B1 molecule creates an inactive higher order complex, which we assumed to be (CYP2B1)2·CPR. While further study may reveal the stoichiometry of this complex, it is possible that this complex is a different higher order complex or a nonspecific complex, such as an aggregate. Regardless, the contributions of this complex are relatively limited toward the mechanism of CYP2B1-CPR interactions. The formation of (CYP2B1)2·CPR is much less favored than the binary complex as evidenced by dissociation constants more than 40-fold greater than those observed for the binary complex. The sensitivity of dissociation constants for both complexes to ionic strength demonstrates a common stabilizing role for electrostatic interactions.
Our proposed mechanism of complexation between CYP2B1 and CPR contrasts with the binary mechanism suggested by others [36]. A symmetrical Job plot resulted when CYP2B1 and CPR were varied but the total protein concentration held constant [36]. This type of curve supports a 1:1 stoichiometry for the complex between the proteins. Similarly, the titration of CYP2B1 with CPR yielded a hyperbolic curve when P450 activity was measured, which would be expected for binary complex formation. However, subsequent publications by the authors [37, 38] demonstrated asymmetrical Job plots, which the authors speculated was the result of detergent contamination or protein aggregation. It is also possible that CYP2B1 was forming a higher order complex as reported for CYP1A2 [28] and CYP2E1 [29], and suggested by our CYP2B1 work. Moreover, we titrated CPR with CYP2B1 to reflect the known excess of P450 relative to CPR in cells [39], which contrasts with the use of CPR as the titrant in the earlier CYP2B1 studies. The choice of titrant can impact the mechanism of interaction observed between the P450s and CPR [29]. Lastly, differences in buffer systems and ionic strength between the respective studies could account for variations in the apparent number and stoichiometry of complexes as indicated by our experiments with CYP2B1 and CPR.
4.4. Alterations of the functional CYP2B1·CPR complex
The presence of D113 and E115/6 primarily stabilized the functional CYP2B1·CPR complex. Mutation of those residues increased Kpr, although the effect was far more significant for E115/6A. These effects on binding between CYP2B1 and CPR correlated with the relative decrease in electrostatic interactions for the corresponding binary complex. While these residues are important in complex interactions, the respective Kpr values for all CPRs did not change until an ionic strength of 137 mM. Presumably, D113 and E115/6 are among multiple contacts between CYP2B1 and CPR. The docking surface between these enzymes may include other electrostatic interactions as well as hydrogen bonding or hydrophobic contacts that further stabilize the complex against the changes in ionic strength.
In addition to stabilizing CYP2B1·CPR, the E115/6 residues determine the functional properties of the binary complex. Alanine mutations led to a significant increase in the ability of CYP2B1 to O-demethylate EFC. Although not studied for this system, the rate-limiting step for P450 catalysis is typically the second electron transfer during the catalytic cycle [40]. As suggested for the increase in MTT and cyt c activities, E115/6A may alter the pathway for electron transfer to yield more productive interaction between CPR and redox partners. While our goal was to demonstrate a role for this residue in binding, this novel catalytic role will provide an important focal point for future studies.
These observations raise the question as to why would loss of electrostatic interactions consistently improve catalysis. A simple explanation would be the role of the residues in forming non-specific interactions. If this were true, then why are these residues conserved among CPRs? In addition, higher ionic strength would presumably disperse non-specific interactions; however the effect of the mutation on catalysis is independent of ionic strength. Alternatively, the conservation of the residues may reflect contributions to different protein-protein interactions formed and broken during catalysis rather than a single set of contacts for a static complex. Stabilizing interactions revealed by binding constants may not support efficient electron transfer as shown through effects on catalysis. In support of this dynamic model for protein-protein interactions, other groups have produced several lines of evidence indicating CPR undergoes conformational changes during electron transfer events [41–43]. Lastly, it is possible that these residues are important for CPR complexes with other partners, such as heme oxygenase [5], squalene monooxygenase [6], and cyt b5 [44]. Taken together, we have confirmed that the role of E115/6 as important contact residues in complex formation and revealed an unexpected contribution to catalysis.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health NCRR COBRE Grant 1 P20 RR015569-06 and the Chonnam National University, Second Stage BK21 Project from the Ministry of Education, Sciences, & Technology of the Republic of Korea.
We thank Haoming Zhang (PFH) for preparation of rat CYP2B1 enzyme. We thank Dmitri Davydov (University of California – San Diego) for helpful advice regarding a buffer system for cyt c studies and Frank Millet (University of Arkansas) for discussions on the electrostatic modeling efforts. Drew R. Jones (UAMS) was also helpful in the preparation of this manuscript.
Footnotes
The abbreviations used are: CYP2B1, cytochrome P450 2B1; CPR, cytochrome P450 reductase; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; cyt c, cytochrome c (horse heart); cyt b5, cytochrome b5; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; 7-ethoxy-4-trifluoromethylcoumarin, 7-EFC; 7-hydroxy-4-trifluoromethylcoumarin, 7-HFC; NADP+, nicotinamide adenosine dinucleotide phosphate (oxidized); I, ionic strength.
The file includes a sample script (input data) for the DynaFit software [17] and table of subsequent model discrimination results for interactions between CPR (wild type and mutant) and CYP2B1.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Horecker BL. Triphosphopyridine nucleotide-cytochrome c reductase in liver. J Biol Chem. 1950;183:593–605. [Google Scholar]
- 2.Phillips AH, Langdon RG. Hepatic triphosphopyridine nucleotide-cytochrome c reductase: isolation, characterization, and kinetic studies. J Biol Chem. 1962;237:2652–2660. [PubMed] [Google Scholar]
- 3.Williams CH, Kamin H. Microsomal triphosphopyridine nucleotide-cytochrome c reductase of liver. J Biol Chem. 1962;237:587–595. [PubMed] [Google Scholar]
- 4.Lu AY, Coon MJ. Role of hemoprotein P-450 in fatty acid omega-hydroxylation in a soluble enzyme system from liver microsomes. J Biol Chem. 1968;243:1331–1332. [PubMed] [Google Scholar]
- 5.Schacter BA, Nelson EB, Marver HS, Masters BS. Immunochemical evidence for an association of heme oxygenase with the microsomal electron transport system. J Biol Chem. 1972;247:3601–3607. [PubMed] [Google Scholar]
- 6.Ono T, Bloch K. Solubilization and partial characterization of rat liver squalene epoxidase. J Biol Chem. 1975;250:1571–1579. [PubMed] [Google Scholar]
- 7.Nishino H, Ishibashi T. Evidence for requirement of NADPH-cytochrome P450 oxidoreductase in the microsomal NADPH-sterol Delta7-reductase system. Arch Biochem Biophys. 2000;374:293–298. doi: 10.1006/abbi.1999.1602. [DOI] [PubMed] [Google Scholar]
- 8.Oshino N, Imai Y, Sato R. A function of cytochrome b5 in fatty acid desaturation by rat liver microsomes. J Biochem. 1971;69:155–167. doi: 10.1093/oxfordjournals.jbchem.a129444. [DOI] [PubMed] [Google Scholar]
- 9.Strobel HW, Hodgson AV, Shen SJ. NADPH cytochrome P450 reductase and its structural and functional domains. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. 2. Plenum Press; New York: 1995. pp. 225–244. [Google Scholar]
- 10.Shen AL, Kasper CB. Role of acidic residues in the interaction of NADPH-cytochrome P450 oxidoreductase with cytochrome P450 and cytochrome c. J Biol Chem. 1995;270:27475–27480. doi: 10.1074/jbc.270.46.27475. [DOI] [PubMed] [Google Scholar]
- 11.Nisimoto Y. Localization of cytochrome c-binding domain on NADPH-cytochrome P-450 reductase. J Biol Chem. 1986;261:14232–14239. [PubMed] [Google Scholar]
- 12.Nadler SG, Strobel HW. Role of electrostatic interactions in the reaction of NADPH-cytochrome P-450 reductase with cytochromes P-450. Arch Biochem Biophys. 1988;261:418–429. doi: 10.1016/0003-9861(88)90358-x. [DOI] [PubMed] [Google Scholar]
- 13.Nadler SG, Strobel HW. Identification and characterization of an NADPH-cytochrome P450 reductase derived peptide involved in binding to cytochrome P450. Arch Biochem Biophys. 1991;290:277–284. doi: 10.1016/0003-9861(91)90542-q. [DOI] [PubMed] [Google Scholar]
- 14.Bonina T, Gilep AA, Estabrook RW, Usanov SA. Engineering of proteolytically stable NADPH-cytochrome P450 reductase. Biochemistry (Mosc) 2005;70:357–65. doi: 10.1007/s10541-005-0122-3. [DOI] [PubMed] [Google Scholar]
- 15.Collom SL, Jamakhandi AP, Tackett AJ, Radominska-Pandya A, Miller GP. CYP2E1 active site residues in substrate recognition sequence 5 identified by photoaffinity labeling and homology modeling. Arch Biochem Biophys. 2007;459:59–69. doi: 10.1016/j.abb.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yim S, Yun CH, Ahn T, Jung HC, Pan JG. A continuous spectrophotometric assay for NADPH-cytochrome P450 reductase activity using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. J Biochem Mol Biol. 2005;38:366–9. doi: 10.5483/bmbrep.2005.38.3.366. [DOI] [PubMed] [Google Scholar]
- 17.Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 18.Smith H, Ahmed A, Millett F. Electrostatic interaction of cytochrome c with cytochrome c1 and cytochrome oxidase. J Biol Chem. 1981;256:4984–4990. [PubMed] [Google Scholar]
- 19.Von Weymarn LB, Sridar C, Hollenberg PF. Identification of amino acid residues involved in the inactivation of cytochrome P450 2B1 by two acetylenic compounds: the role of three residues in nonsubstrate recognition Sites. J Pharmacol Exp Ther. 2004;311:71–79. doi: 10.1124/jpet.104.069757. [DOI] [PubMed] [Google Scholar]
- 20.Yasukochi Y, Masters BS. Some properties of a detergent-solubilized NADPH-cytochrome c(cytochrome P-450) reductase purified by biospecific affinity chromatography. J Biol Chem. 1976;251:5337–5344. [PubMed] [Google Scholar]
- 21.Shen AL, Porter TD, Wilson TE, Kasper CB. Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J Biol Chem. 1989;264:7584–7589. [PubMed] [Google Scholar]
- 22.Li HY, Darwish K, Poulos TL. Characterization of recombinant Bacillus megaterium cytochrome P-450 BM-3 and its two functional domains. J Biol Chem. 1991;266:11909–11914. [PubMed] [Google Scholar]
- 23.Sevrioukova I, Truan G, Peterson JA. The flavoprotein domain of P450BM-3: expression, purification, and properties of the flavin adenine dinucleotide- and flavin mononucleotide-binding subdomains. Biochemistry. 1996;35:7528–7535. doi: 10.1021/bi960330p. [DOI] [PubMed] [Google Scholar]
- 24.French JS, Coon MJ. Properties of NADPH-cytochrome P-450 reductase purified from rabbit liver microsomes. Arch Biochem Biophys. 1979;195:565–577. doi: 10.1016/0003-9861(79)90383-7. [DOI] [PubMed] [Google Scholar]
- 25.Lopez-Anaya A, Mayersohn M. Quantification of riboflavin, riboflavin 5′-phosphate and flavin adenine dinucleotide in plasma and urine by high-performance liquid chromatography. J Chromatogr. 1987;423:105–13. doi: 10.1016/0378-4347(87)80332-8. [DOI] [PubMed] [Google Scholar]
- 26.Kanaeva IP, Skotselyas ED, Turkina IF, Petrochenko EV, Davydov DR, Kondrashin SK, Dzuzenova CS, Bachmanova GI, Archakov AI. Reduction and catalytic properties of cytochrome P-450 LM2 in reconstituted system containing monomeric carriers. Biochem Biophys Res Commun. 1987;147:1295–1299. doi: 10.1016/s0006-291x(87)80211-5. [DOI] [PubMed] [Google Scholar]
- 27.Hanna IH, Reed JR, Guengerich FP, Hollenberg PF. Expression of human cytochrome P450 2B6 in Escherichia coli: characterization of catalytic activity and expression levels in human liver. Arch Biochem Biophys. 2000;376:206–216. doi: 10.1006/abbi.2000.1708. [DOI] [PubMed] [Google Scholar]
- 28.Kelley RW, Reed JR, Backes WL. Effects of ionic strength on the functional interactions between CYP2B4 and CYP1A2. Biochemistry. 2005;44:2632–2641. doi: 10.1021/bi0477900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jamakhandi A, Kuzmic P, Sanders DE, Miller GP. Global analysis of protein-protein interactions reveals multiple CYP2E1-reductase complexes. Biochemistry. 2007;46:10192–201. doi: 10.1021/bi7003476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hazai E, Bikadi Z, Simonyi M, Kupfer DD. Association of cytochrome P450 enzymes is a determining factor in their catalytic activity. J Comput Aided Mol Des. 2005;19:271–85. doi: 10.1007/s10822-005-4995-4. [DOI] [PubMed] [Google Scholar]
- 31.Bates D, Watts DG. Nonlinear Regression Analysis and its Applications. Wiley; New York: 1988. [Google Scholar]
- 32.Burnham KP, Anderson DR. A Practical Information-Theoretic Approach. 2. Springer; New York: 2002. Model Selection and Multimodel Inference. [Google Scholar]
- 33.Sutherland M, Learmonth BA. The tetrazolium dyes MTS and XTT provide new quantitative assays for superoxide and superoxide dismutase. Free Radic Res. 1997;27:283–9. doi: 10.3109/10715769709065766. [DOI] [PubMed] [Google Scholar]
- 34.Sem DS, Kasper CB. Effect of ionic strength on the kinetic mechanism and relative rate limitation of steps in the model NADPH-cytochrome P450 oxidoreductase reaction with cytochrome c. Biochemistry. 1995;34:12768–12774. doi: 10.1021/bi00039a037. [DOI] [PubMed] [Google Scholar]
- 35.Pettigrew G, Moore GR. Cytochromes c. Biological aspects. Springer-Verlag; Berlin: 1987. [Google Scholar]
- 36.Miwa GT, West SB, Huang MT, Lu AYH. Studies on the association of cytochrome P-450 and NADPH-cytochrome c reductase during catalysis in a reconstituted hydroxylating system. J Biol Chem. 1979;254:5695–5700. [PubMed] [Google Scholar]
- 37.Miwa GT, Lu AYH. Studies on the stimulation of cytochrome P-450-dependent monooxygenase activity by dilauroylphosphatidylcholine. Arch Biochem Biophys. 1981;211:454–458. doi: 10.1016/0003-9861(81)90477-x. [DOI] [PubMed] [Google Scholar]
- 38.Miwa GT, Lu AYH. The association of cytochrome P-450 and NADPH-cytochrome P-450 reductase in phospholipid membranes. Arch Biochem Biophys. 1984;234:161–166. doi: 10.1016/0003-9861(84)90337-0. [DOI] [PubMed] [Google Scholar]
- 39.Estabrook RW, Hildebrandt AG, Baron J, Netter KJ, Leibman K. A new spectral intermediate associated with cytochrome P-450 function in liver microsomes. Biochem Biophys Res Commun. 1971;42:132–139. doi: 10.1016/0006-291x(71)90372-x. [DOI] [PubMed] [Google Scholar]
- 40.Guengerich F. Rate-limiting steps in cytochrome P450 catalysis. Biol Chem. 2002;383:1553–64. doi: 10.1515/BC.2002.175. [DOI] [PubMed] [Google Scholar]
- 41.Reed JR, Hollenberg PF. New perspectives on the conformational equilibrium regulating multi-phasic reduction of cytochrome P450 2B4 by cytochrome P450 reductase. J Inorg Biochem. 2003;97:276–286. doi: 10.1016/s0162-0134(03)00310-6. [DOI] [PubMed] [Google Scholar]
- 42.Grunau A, Paine MJ, Ladbury JE, Gutierrez A. Global effects of the energetics of coenzyme binding: NADPH controls the protein interaction properties of human cytochrome P450 reductase. Biochemistry. 2006;45:1421–1434. doi: 10.1021/bi052115r. [DOI] [PubMed] [Google Scholar]
- 43.Grunau A, Geraki K, Grossmann JG, Gutierrez A. Conformational dynamics and the energetics of protein--ligand interactions: role of interdomain loop in human cytochrome P450 reductase. Biochemistry. 2007;46:8244–8255. doi: 10.1021/bi700596s. [DOI] [PubMed] [Google Scholar]
- 44.Iyanagi T, Makino N, Mason HS. Redox properties of the reduced nicotinamide adenine dinucleotide phosphate-cytochrome P-450 and reduced nicotinamide adenine dinucleotide-cytochrome b5 reductases. Biochemistry. 1974;13:1701–1710. doi: 10.1021/bi00705a023. [DOI] [PubMed] [Google Scholar]
- 45.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yabusaki Y, Murakami H, Ohkawa H. Primary structure of Saccharomyces cerevisiae NADPH-cytochrome P450 reductase deduced from nucleotide sequence of its cloned gene. J Biochem. 1988;103:1004–1010. doi: 10.1093/oxfordjournals.jbchem.a122370. [DOI] [PubMed] [Google Scholar]
- 47.Murakami H, Yabusaki Y, Ohkawa H. Expression of rat NADPH-cytochrome P-450 reductase cDNA in Saccharomyces cerevisiae. DNA. 1986;5:1–10. doi: 10.1089/dna.1986.5.1. [DOI] [PubMed] [Google Scholar]
- 48.Shephard EA, Palmer CNA, Segall HJ, Phillips IR. Quantification of cytochrome P450 reductase gene expression in human tissues. Arch Biochem Biophys. 1992;294:168–172. doi: 10.1016/0003-9861(92)90152-m. [DOI] [PubMed] [Google Scholar]
- 49.Wang M, Roberts DL, Paschke R, Shea TM, Masters BSS, Kim JJP. Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc Natl Acad Sci USA. 1997;94:8411–8416. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.