

Abstract
Adenosine is released from injured or hypoxic tissues where it exerts numerous anti-inflammatory effects including suppression of neutrophil functions. Although most previous work has implicated the A2AAR, we have recently shown that selective activation of the abundantly expressed A3AR inhibits neutrophil superoxide production and chemotaxis providing a potential mechanistic explanation for the efficacy of A3AR agonists in experimental animal models of inflammation. In this study, we hypothesized that the A3AR suppresses neutrophil functions by inhibiting the monomeric GTPase Rac, a central regulator of chemokine-directed neutrophil migration and superoxide production. We found that pre-treating neutrophils with the highly selective A3AR agonist CP-532,903 reduced fMLP-induced Rac activation using an ELISA-based assay that detects all three Rac isoforms. CP-532,903 also inhibited fMLP-induced F-actin formation, a downstream effector function of Rac relevant to neutrophil migration, but not activation of ERK1/2 or p38. Pre-treating neutrophils with CP-532,903 did not stimulate cAMP production or alter fMLP-induced calcium transients, implicating that A3AR stimulation does not inhibit Rac activation or neutrophil activities by suppressing Ca2+ signaling, elevating the intracellular concentration of cAMP, or by cross-desensitizing fMLP receptors. Our results suggest that activation of the A3AR signals to suppress neutrophil functions by interfering with the monomeric GTPase Rac, thus contributing to the ant-inflammatory actions of adenosine.
Keywords: adenosine, adenosine receptors, neutrophils, inflammation, Rac, cell signaling
1. Introduction
Neutrophils are attracted to inflamed tissues through the production of chemoattractant mediators including chemokines, lipid molecules (platelet-activating factor, leukotrienes), complement components (C5a), and bacterial proteins including N-formylated peptides [1, 2]. Most of these molecules bind to cell surface G protein-coupled receptors that stimulate neutrophils to migrate, and ultimately to phagocytose microorganisms and cell debris, secrete granule contents containing degradative enzymes, activate the NADPH oxidase to generate reactive oxygen species, and stimulate the synthesis of other pro-inflammatory molecules that help to recruit additional immune cell populations [1, 2]. While these actions of neutrophils are critical for normal wound healing, the excessive release of toxic mediators can damage host tissue contributing to the pathogenesis of numerous acute and chronic inflammatory diseases.
Adenosine is formed in inflamed tissues from the enzymatic degradation of ATP released from activated or injured cells, which serves to dampen the inflammatory reaction and promote inflammation resolution by suppressing the activity of most cells of the immune system including the neutrophil [3, 4]. Adenosine potently inhibits neutrophil adhesion to endothelial cells, degranulation, superoxide production, and pro-inflammatory mediator production [3, 4]. Among the four adenosine receptor (AR) subtypes (A1, A2A, A2B, and A3), most previous studies have implicated the A2AAR in mediating the inhibitory effects of adenosine on neutrophils via the cAMP/protein kinase A (PKA) signaling axis and/or through cAMP-independent activation of a protein phosphatase [3, 4]. However, we have recently discovered that activating the Gi protein-coupled A3AR also functions in murine neutrophils to inhibit superoxide production and chemotaxis [5]. Suppression of neutrophil activity represents a potential mechanism by which A3AR agonists provide benefit in experimental animal models of inflammation [6-11].
The small GTPase Rac plays a central role in regulating responses to inflammatory signals in neutrophils. Rac2 is a necessary component of the NADPH oxidase complex that is assembled in endosomes and at the plasma surface upon chemoattractant receptor stimulation [12]. Rac2 also regulates rearrangement of the cytoskeleton and neutrophil migration [13-15]. Requirement of Rac2 in chemotaxis and the formation of reactive oxygen species has been demonstrated in studies using neutrophils from Rac2-null mice [14] and from patients that carry key Rac2 mutations [16, 17]. Although Rac2 is the primary isoform found in human hematopoietic cells, Rac1 is equally expressed in murine neutrophils where it also regulates both superoxide production and direction sensing during chemotaxis [18, 19].
In this study, we provide evidence that the A3AR inhibits fMLP-induced Rac activation in murine neutrophils. This occurs by mechanisms that do not involve alterations in Ca2+ signaling, cAMP elevation, or cross-desensitization of the fMLP receptor. Suppression of chemokine receptor-induced activation of Rac represents a potential intracellular signaling mechanism by which the A3AR suppresses neutrophil activities.
2. Materials and methods
2.1. Materials
Cell culture reagents were purchased from Invitrogen (Carlsbad, CA). Fura2-AM and pluronic F-127 were purchased from Molecular Probes-Invitrogen (Eugene, OR). Anti phospho-ERK1/2 (extracellular signal-regulated kinase 1/2), anti phospho-p38, anti ERK1/2, anti p38 and horse radish peroxidase-linked anti-rabbit IgG were purchased from Cell Signaling Technology, Inc. (Danvers, MA). CL-XPosure™ Film and Restore Western Blot stripping buffer were from Pierce Biotechnology, Inc. (Rockford, IL). cAMP assay kits and Hybond™-C nitrocellulose membrane were obtained from GE Healthcare Bio-Sciences Corp. (Piscataway, NJ). Ro 20-1724 was obtained from BIOMOL International L.P. (Plymouth Meeting, PA). Western Lighting™ Chemiluminescence Reagent Plus was from PerkinElmer LAS, Inc. (Boston, MA). G-LISA Kit was purchased from Cytoskeleton, Inc. (Denver, CO). Percoll was purchased from Amersham Biosciences (Piscataway, NJ). CP-532,903 was a gift from Dr. W. Ross Tracey (Pfizer Global Research and Development, Groton, CT), adenosine deaminase (ADA) was purchased from Roche Applied Science (Indianapolis, IN), and all remaining drugs and reagents were purchased from Sigma-Aldrich (St. Louis, MO).
2.2. Mice
C57BL/6 wild-type (WT) mice were purchased from Taconic Farms from Dr. (Germantown, NY). Congenic C57BL/6 A3KO mice were a kind gift Marlene Jacobson (Merck Research Labs, West Point, PA; [20]). All animal experiments were conducted with approval of the Institutional Animal Care and Use Committee at the Medical College of Wisconsin.
2.3. Isolation of Mouse Bone Marrow Neutrophils
Morphologically mature neutrophils were purified from mouse bone marrow by isotonic Percoll gradient centrifugation, as previously described [5, 21]. Briefly, mice were euthanized by anoxia with carbon dioxide. Tibias and femurs of mice were flushed with neutrophil isolation buffer (1 x HBSS without Ca2+ and Mg2+, and containing 0.4% sodium citrate) and layered on a 3-step Percoll gradient (72%, 64%, 52%). Following centrifugation at 1,060 x g for 30 min, cells at the 72%:64% interface, were removed and washed once with isolation buffer before use in experiments.
2.4. Rac activation assays
Freshly isolated neutrophils were re-suspended in HBSS containing 1 unit/mL ADA and then aliquoted into Eppendorf tubes (2.5 × 106 cells/250 μL). After incubating at 37°C for 30 min in the presence of vehicle or CP-532,903, the cells were stimulated with fMLP (1 μM) for the times indicated. The reactions were terminated by the addition of two volumes of ice-cold neutrophil isolation buffer with simultaneous quick chilling in a dry ice/ethanol bath. Total activated Rac (isoforms 1, 2 and 3) in the cell lysates (1 μg/μL) was quantified using the G-LISA kit that utilizes 96-well plates coated with a Rac-GTP binding domain-containing effector protein and a non-specific Rac antibody. In control assays, the cells were first subjected to one freeze-thaw cycle after which the lysates were incubated for 10 min at 37°C in the reaction pH 7.4) buffer (50 mM HEPES, 100 mM NaCl, 2 mM EDTA, and 6 mM MgCl2, containing 200 μM GTPγS, prior to assay for activated Rac.
2.5. F-actin measurements
Freshly isolated neutrophils were resuspended in HBSS buffer containing 1 unit/mL ADA and aliquoted into Eppendorf tubes (2 × 106 cells/250 μL). After incubating for 30 min at 37°C in the presence of vehicle or CP-532,903, the cells were stimulated with fMLP (1 μM) for 30 sec, after which the cells were fixed with 3.7% paraformaldehyde (in HBSS without Ca2+/Mg2+) for 30 min on ice. The cells were washed and then incubated for another 30 min on ice with FITC-phalloidin (2.5 μM) in PBS/2%FBS supplemented with 100 μg/mL L-α-palmitoyl-lysophophatidylcholine to permeabilize and stain the cells. F-actin content was quantified by flow cytometry using a Bectin Dickinson FACSCaliber flow cyctometer by measuring a total of at least 10,000 cells per sample.
2.6. Western blot analysis for phosphorylation of ERK1/2 and p38
Freshly isolated neutrophils were resuspended in HBSS buffer containing 1 unit/mL ADA and aliquoted into Eppendorf tubes (~2.5 × 106 cells/250 μL). After incubating for 30 min at 37°C with vehicle or CP-532,903, the cells were stimulated with fMLP (1 μM) for the times indicated. The assays were terminated by the addition of two-fold volume of ice cold neutrophil isolation buffer followed by rapid chilling in dry ice/ethanol. Cell lysates were prepared by adding 50 μL of Triton-X lysis buffer consisting of 25 mM Tris-HCl, pH 7.5, 100 mM NaCl, 50 mM NaF, 5 mM EDTA, 1% v/v Triton X-100, 40 mM β-glycerophosphate, 40 mM paranitrophenylphosphate, 1 mM sodium orthovanadate, 1 mM phenyl ethylsulfonyl fluoride (PMSF), 10 μg/mL leupeptin, 10 μg/mL pepstatin A, and 10 μg/mL aprotinin. The lysates were clarified by centrifugation at 16,000 X g for 5 min at 4°C and the protein concentrations of the soluble extracts were quantified using the BioRad (Bradford) protein assay. The extracts (20 μg protein) were denatured with 5 X Laemmli sample buffer (62.5 mM Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, 20% β-mercaptoethanol, and 0.0025% bromophenol blue) and boiled for 5 min prior to separation by standard 10% SDS-polyacrylamide gel electrophoresis. Proteins were wet blotted onto nitrocellulose membranes and probed with anti-phospho-ERK1/2 (1:2000) or anti-phospho-p38 (1:2000) antibodies. The blots were stripped with Restore PLUS Western blot stripping buffer (Pierce) and reprobed with antibodies against the respective total proteins (anti-ERK1/2, 1:2000; anti-p38, 1:2000). Horseradish peroxidase-conjugated secondary antibodies (1:1000) were visualized by enhanced chemiluminescence detection (Western Lightning™ Reagent Plus, PerkinElmer). Densitometry analysis was performed using Scion Image software (from the National Institutes of Health).
2.7. Intracellular Ca2+ Assays
Freshly isolated neutrophils were loaded with the Ca2+-specific fluorescent probe Fura-2 AM (5 μM) in neutrophil isolation buffer for 30 min at 37°C. Cells were washed and resuspended in HBSS containing 1 unit/mL ADA at a concentration of 1×106 cells/mL. Fluorescence at baseline and after the addition of various activating agents was continuously measured with a spectrofluorimeter (Becton Dickenson) in a stirred thermostable cuvette (37°C) using an emission wavelength of 510 nm and excitation wavelengths of 340 and 380 nm. The intracellular concentration of calcium ([Ca2+]i) was calculated using the Grynkiewicz equation [22]:
Where [Ca2+]i is given in nM units, Kd is the dissociation constant of Fura-2 AM (224 nM under standard conditions), R represents the ratio of fluorescence (F) emission at 510 nm following excitation at 340 and 380 nm (F340/F380) and Q is the ratio of minimal and maximal fluorescence following excitation at 380 nm. Rmax was measured after the addition of 20 μM digitonin to release all of the intracellular FURA-2 AM. Rmin was subsequently measured after the addition of 50 mM EGTA to chelate the free calcium. All results are plotted as [Ca2+]i (nM) versus time.
2.8. Quantification of intracellular cAMP accumulation
Freshly isolated neutrophils were resuspended in HBSS containing 1 unit/mL ADA and 20 μM Ro 20-1724 (phosphodiesterase inhibitor) and then transferred to polypropylene tubes (1×105 cells/200 μL). After equilibrating at 37°C for 5 min, vehicle or agonists were added at the concentrations indicated for 15 min. The assays were terminated by adding 500 μl of 0.15 N HCl. cAMP in the acid extract was determined by radioimmunoassay according to the manufacturer’s protocol (GE Healthcare; Piscastaway, NJ).
3. Results
3.1. Activation of the A3AR reduces fMLP-induced Rac activation
We have previously observed that activating the A3AR inhibits superoxide production and chemotaxis of mouse bone marrow neutrophils in response to fMLP as well as a panel of other chemoattractive agents [5]. Considering that Rac plays an important role in regulating both of these responses in murine neutrophils, we examined whether stimulating the A3AR interferes with fMLP-induced Rac activation using an ELISA-based assay that quantifies the active guanosine-5′-triphosphate (GTP)-bound form of all three Rac isoforms. Like other small GTPases, Rac cycles between a GDP-bound inactive state and a GTP-bound active state. Initially, we examined the time-course of Rac activation in response to 1 μM fMLP. As shown in Figure 1, the level of active Rac was increased as early as 5 s after exposure to fMLP and returned to baseline levels by 1 min. In positive control assays, active Rac was increased nearly 4-fold when cell lysates were treated with the non-hydrolyzable GTP analog GTPγS (Figure 1A). Previous studies using human and murine neutrophils have similarly reported that Rac is rapidly and transiently activated in response to fMLP [23, 24]. The magnitude and time-course of fMLP-induced activation was similar in assays using neutrophils isolated from A3KO mice (Figure 1A).
Figure 1.

Effect of CP-532,903 on fMLP-induced Rac activation in mouse bone marrow neutrophils obtained from wild-type mice and from A3ARKO mice. Rac activity in whole-cell lysates was quantified using an ELISA-based assay kit (G-LISA kit, Cytoskeleton, Inc), as described under Materials and Methods. (A) The time-course of Rac activation following stimulation with fMLP (1 μM). (B & C) Rac activity 15 sec after the addition of 1 μM fMLP to wild-type (B) and A3KO (C) cells pretreated for 30 min with 100 nM CP-532,903 (CP). The data are presented as the percent increase over baseline activity. In control assays, cell lysates were incubated with 200 μM GTPγS. All assays were conducted in the presence of 1 unit/ml ADA. Mean ± SEM. *, p < 0.05 versus the fMLP-treated group by one-way ANOVA and Bonferroni’s t test, n = 3-7.
We subsequently examined whether treating neutrophils for 30 min with vehicle or the A3AR agonist CP-532,903 (100 nM) reduces fMLP-induced Rac activation. A 30-min pretreatment protocol was used since we observed previously that maximal inhibition of fMLP-induced superoxide production is achieved when the cells are exposed to CP-532,903 for at least 18 min [5]. Although treatment with CP-532,903 did not alter the basal level of active Rac, it markedly reduced the degree of activation produced by fMLP (Figure 1B). This result indicates that stimulating the A3AR in murine neutrophils inhibits the ability of fMLP receptors to couple to Rac signaling. To confirm that CP-532,903 functions specifically through activation of the A3AR, parallel studies were conducted using neutrophils isolated from A3KO mice [20]. As shown in Figure 1C, the inhibitory effect of CP-532,903 on fMLP-induced Rac activation was not apparent in assays using A3KO neutrophils.
3.2. Activation of the A3AR inhibits fMLP-induced F-actin generation but not fMLP-induced phosphorylation of ERK or p38
Both Rac1 and Rac2 participate in chemoattractant-induced neutrophil migration by promoting actin polymerization at the leading edge [15]. Rac2 has also been reported to be upstream of ERK1/2 and p38 mitogen-activated protein (MAP) kinase activation by chemoattractants [15], which importantly regulate neutrophil superoxide production and chemotaxis. We therefore examined whether activation of the A3AR interferes with these two downstream effector functions of Rac. For F-actin assays, neutrophils in suspension were pre-treated with either vehicle or CP-532,903 (100 nM) for 30 min and then stimulated with fMLP (1 μM) for 30 s before staining the cells with FITC-conjugated phalloidin. For the MAP kinase assays, mouse bone marrow neutrophils were pretreated with vehicle or CP-532,903 for 30 min and then stimulated with fMLP for up to 10 min after which phosphorylated ERK1/2 and p38 were quantified by Western immunoblotting. As shown in Figure 2A, exposure to fMLP significantly increased F-actin content over 2-fold in vehicle-treated control cells; this increase was nearly abolished in cells pretreated with CP-532,903. Treatment with fMLP also induced rapid but transient phosphorylation of both ERK1/2 and p38 (Figure 3). In contrast to the results of the F-actin assays, treatment with CP-532,903 did not inhibit fMLP-induced phosphorylation of either ERK1/2 or p38 kinases.
Figure 2.

Effect of CP-532,903 on fMLP-induced F-actin formation in mouse bone marrow neutrophils. Neutrophils were pretreated with vehicle (A) or 100 nM CP-532,903 (B) for 30 min at 37°C in the presence of 1 unit/ml ADA, and then stimulated with 1 μM fMLP for 30 sec. Cells were stained with FITC-conjugated phalloidin and intracellular fluorescence was quantified by flow cytometry assessing a total of 10,000 cells per sample. Mean ± SEM. *, p < 0.05 versus the vehicle-treated group by Student’s t test, n = 8.
Figure 3.

Effect of CP-532,903 on fMLP-induced activation of ERK1/2 and p38 in mouse bone marrow neutrophils. (A) Representative Western immunoblots showing phosphorylated and total levels of ERK1/2 and p38. (B) and (C) show the results of densitometric analysis of the Western immunoblots for ERK1/2 and p38, respectively. Ratios of phosphorylated to total protein were normalized to baseline levels. Mean ± SEM. n = 3-4.
3.3. Activation of the A3AR does not alter fMLP-induced intracellular Ca2+ transients or stimulate cAMP elevation
Activation of chemoattractant receptors including fMLP receptors induces transient elevations in intracellular Ca2+ [25, 26], which is prerequisite for the pro-inflammatory activities of neutrophils including superoxide production and degranulation as well as adhesion required for cell migration [25, 26]. It has previously been suggested that intracellular Ca2+ signaling may contribute to Rac activation in response to stimulation by G protein-coupled receptors [27]. Moreover, one potential mechanism by which A2AAR activation inhibits the pro-inflammatory activity of neutrophils is by accelerating the sequestration of intracellular Ca2+ through elevation of cAMP and activation of protein kinase A (PKA; [26, 28]). We therefore examined whether activation of the A3AR influences fMLP-induced Ca2+ transients in murine neutrophils or stimulates cAMP production. As shown, treating cells with CP-532,903 had no effect on fMLP-induced Ca2+ transients (Figure 4). In addition, treatment with CP-532,903 did not promote cAMP accumulation in neutrophils at concentrations as high as 10 μM (Figure 5). In contrast, in control studies stimulating the cells with the A2AAR agonist CGS 21680 or forskolin produced a significant increase in cAMP accumulation (Figure 5). These results therefore indicate that activation of the A3AR does not alter Ca2+ signaling responses in murine neutrophils. By inference, these results also demonstrate that activation of the A3AR does not produce heterologous desensitization of fMLP receptors [29-33].
Figure 4.

Changes in intracellular [Ca2+] in mouse bone marrow neutrophils in response to fMLP. The cells were pretreated for 30 min with vehicle or CP-532,903 (100 nM) prior to measurement of intracellular [Ca2+] during stimulation with fMLP (1 μM). The intracellular [Ca2+] was measured in suspended cells loaded with FURA-2/AM in HBSS containing 1 unit/ml ADA, as described in Materials and Methods. The data shown are representative of 3-4 independent experiments.
Figure 5.

CP-532,903 does not stimulate cAMP production in mouse bone marrow neutrophils. Neutrophils suspended in HBSS containing 1 unit/ml ADA and 20 μM Ro 20-1724 were stimulated with vehicle or increasing concentrations of CP-532,903 for 15 min. In control experiments, the cells were stimulated with the A2AAR agonist CGS 21680 (1 μM) or forskolin (10 μM). The assays were terminated by adding 0.15 N HCl. cAMP in the acid extract was determined by radioimmunoassay. Mean ± SEM. *, p < 0.05 versus the vehicle-treated group by one-way ANOVA and Dunnett’s t test, n = 3.
4. Discussion
We recently identified that the A3AR is abundantly expressed in murine neutrophils and that activation of this AR subtype, along with the A2AAR, inhibits fMLP-induced superoxide production and also slows chemotaxis [5]. Considering that it couples to Gi inhibitory proteins similar to most chemoattractant receptors, the intracellular mechanisms by which the A3AR suppresses neutrophil activities was not readily apparent. In this study, we provide evidence that activating the A3AR signals to inhibit activation of Rac, a small GTPase intimately involved in regulating both neutrophil superoxide production and chemotaxis.
Among the three Rac isoforms (Rac1, 2, and 3), it is generally viewed that Rac2 is the major isoform expressed in neutrophils, and that Rac2 regulates chemoattractant-induced neutrophil functions, including chemotaxis and superoxide production [15, 17, 18]. However, defects in chemotaxis and superoxide production of neutrophils from Rac2 null mice are further augmented by the additional loss of Rac1 [18]. In addition, Rac1 deficiency alone results in an inability of neutrophils to detect and orient within a chemotactic gradient [19]. Thus, both Rac1 and Rac2 appear to play contributing roles in regulating the pro-inflammatory actions of neutrophils. In the present investigation, Rac activity was not completely inhibited in CP-532,903-pretreated cells. Since the assay used in our studies did not discriminate between individual Rac isoforms, it remains possible that A3AR activation resulted in specific inhibition of one of the two Rac isoforms expressed in neutrophils. Such a scenario could explain our previous observations that A3AR activation with CP-532,903 produces only a 50% inhibition of stimulated superoxide production and only a modest reduction in chemotaxis [5].
We examined whether activation of the A3AR influences F-actin formation and MAPK activation, two downstream effector system regulated by Rac. In neutrophils and multiple other cell types, Rac regulates actin polymerization involved in cell migration, through effects on Pak1-LIM kinase 1 that inhibits actin depolymerization and gelsolin that regulates actin polymerization [34-36]. Both ERK and p38 signaling are also known to be involved in chemoattractant-induced superoxide generation and migration, although the exact mechanisms remain unclear [37, 38]. Using phalloidan staining coupled with fluorescence detection, we found that pretreating neutrophils with CP-532,903 markedly attenuated fMLP-induced F-actin formation, providing further support for the hypothesis that activating the A3AR signals to inhibit Rac activation thereby slowing migration. In contrast, we found that CP-532,903 treatment did not alter fMLP-induced activation of ERK or p38 kinases. This observation is consistent with previous findings suggesting that fMLP-induced activation of MAP kinases in neutrophils involves multiple different input signals and is only partially dependent upon Rac activity [15].
Our studies ruled out two potential mechanisms by which stimulating the A3AR may lead to inhibition of fMLP-stimulated Rac activation and neutrophil activities, including cAMP elevation and alterations in the magnitude and duration of fMLP-induced Ca2+ transients. Although traditionally thought to stimulate Gi proteins, it has previously been suggested that the A3AR increases cAMP production in eosinophils, reportedly through atypical coupling to Gsα or through the release of mediators that act upon other Gs protein-coupled receptors [39, 40]. Since Ca2+ transients were unaffected and MAP kinases continued to be activated, our results further oppose a potential mechanism related to A3AR-induced cross-desensitization of fMLP receptors, a mechanism that has been suggested to explain the ability of Gi protein-coupled μ and δ opioid receptors to inhibit neutrophil chemotaxis [31, 41, 42].
The question remains as to the precise mechanism by which stimulating the A3AR produces selective interference with fMLP-induced Rac activation. The A3AR has been reported to activate phosphitadylinositol-3-kinase (PI3K) via the release of βγ subunits from Gi/o in various cell types leading to activation of ERK 1/2 [43-47]. In our studies, we also observed that activation of the A3AR produced transient phosphorylation of Erk 1/2 without inducing an increase in the intracellular free Ca2+ concentration (Figure 6), suggesting that the A3AR might signal to regulate PI3K, phosphoinositide metabolism, and Erk1/2 in neutrophils selectively without promoting activation of phospholipase C, Ca2+ signaling, and activation of protein kinases C (PKC) isoenzymes. It is well established that activation of neutrophils in response to fMLP and other chemokines involves, in addition to mobilization of intracellular Ca2+ and PKC activation, localized generation of phosphoinositides by PI3K at the leading edge, which recruits guanine nucleotide exchange factors (GEFs) that functionally link fMLP receptors to Rac activation. Of particular interest is the major Rac activator in neutrophils P-Rex1 [48-53]. This Rac2-specific GEF, which translocates to the leading edge in areas of Rac2 activation and is synergistically activated by the binding of phosphatidylinositol(3,4,5)triphosphate (PIP3) and βγ subunits of heterotrimeric G proteins, has been implicated in regulating neutrophil NADPH oxidase activity and chemotaxis in response to a wide variety of extracellular stimuli including fMLP [48-53]. Accordingly, we speculate that pre-activation of the A3AR in neutrophils may sequester necessary signaling components (PI3K and Rac-specific GEFS such as P-Rex1) away from subsequently stimulated fMLP receptors localized to the leading edge, thereby interfering with Rac activation. Essentially, we predict that activating the A3AR alters the spatiotemporal signaling events required for fMLP-induced activation of Rac and subsequently Rac-dependent neutrophil functions.
Figure 6.
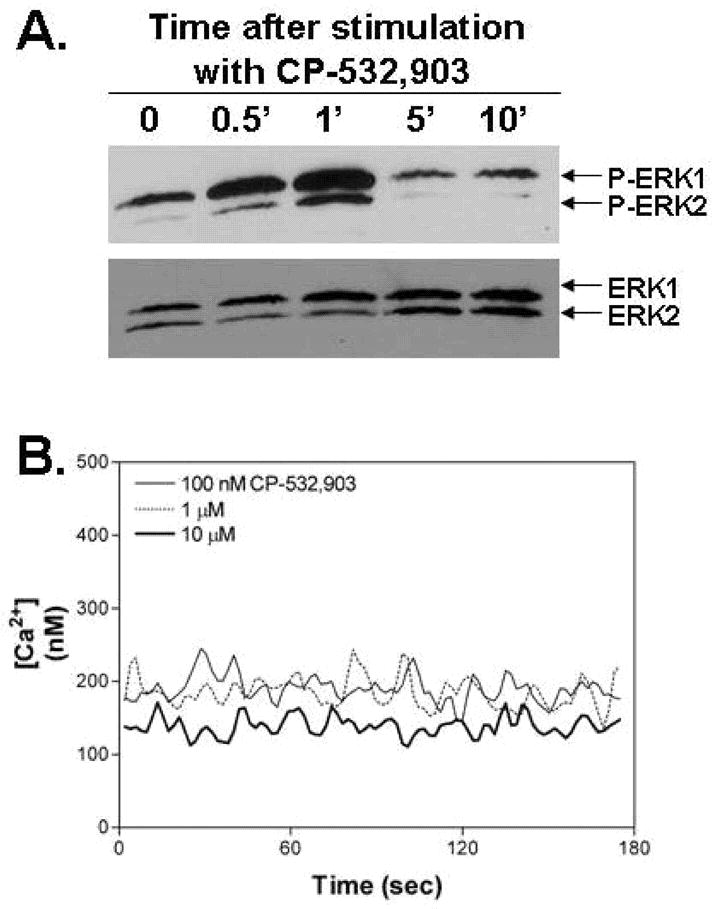
Treating neutrophils with CP-532,903 (100 nM) directly produced a transient increase in ERK1/2 phosphorylation (A) but did not induce changes in the intracellular [Ca2+] at concentrations as high as 10 μM (B). The data shown are representative of three independent experiments.
All of our studies in this report were conducted in the presence of adenosine deaminase to remove any effects of endogenous adenosine that might be produced in our assays. This was done in order to isolate the actions of the A3AR in neutrophils and to parallel our previously published study in which we showed that the A3AR signals to inhibit neutrophil activation [5]. It is important to note that chemoattractants stimulate the release of ATP and the subsequent production of adenosine that likely influences signaling by various purinergic receptors including the A3AR [54]. Indeed, it has been proposed by Chen and colleagues [54] that the release of purines at the leading edge of neutrophils coordinates signaling responses and drives directional migration of the cells.
In conclusion, this study provides evidence that the A3AR signals in murine neutrophils to inhibit Rac activation in response to the bacterial chemoattractant fMLP. This likely explains the inhibitory effect of A3AR activation on the pro-inflammatory activities of neutrophils. The precise mechanism by which the A3AR signals to inhibit Rac activation remains to be identified. However, three possibilities were excluded in this report, namely alterations in Ca2+ signaling, cAMP elevation, and receptor cross-desensitization.
Acknowledgments
We thank Dr. Jeffrey Woodliff (Department of Pediatrics, Medical College of Wisconsin) for his assistance with flow cytometry analyses and Dr. W. Ross Tracey (Pfizer Global Research and Development) for providing CP-532,903. This research was supported in part by the National Institutes of Health National Heart, Lung, and Blood Institute (Grant R01 HL077707) and by the American Heart Association (Grant 0615533Z).
Abbreviations
- ADA
adenosine deaminase
- AR
adenosine receptor
- C5a
complement component 5a
- CGS 21680
2-[p-(2-carboxyethyl)phenethylamino]-5′-N-ethylcarboxamidoadenosine
- CP-532, 903
(2S,3S,4R,5R)-3-amino-5-[6-(2,5-dichlorobenzylamino)purin-9-yl]-4-hydroxytetrahydrofuran-2-carboxylic acid methylamide
- fMLP
formylated-Methionine-Leucine-Phenylalanine
- GEF
guanine nucleotide exchange factor
- KO
knockout
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nathan C. Points of control in inflammation. Nature. 2002;420:846–52. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 2.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–82. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 3.Hasko G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 2004;25:33–9. doi: 10.1016/j.it.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nature Rev Drug Discov. 2008;7:759–70. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Hoeven D, Wan TC, Auchampach JA. Activation of the A3 adenosine receptor suppresses superoxide production and chemotaxis of mouse bone marrow neutrophils. Mol Pharmacol. 2008;74:685–96. doi: 10.1124/mol.108.048066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ge ZD, Peart JN, Kreckler LM, Wan TC, Jacobson MA, Gross GJ, et al. Cl-IB-MECA [2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor. J Pharmacol Exp Ther. 2006;319:1200–10. doi: 10.1124/jpet.106.111351. [DOI] [PubMed] [Google Scholar]
- 7.Jordan JE, Thourani VH, Auchampach JA, Robinson JA, Wang NP, Vinten-Johansen J. A3 adenosine receptor activation attenuates neutrophil function and neutrophil-mediated reperfusion injury. Am J Physiol. 1999;277:H1895–905. doi: 10.1152/ajpheart.1999.277.5.H1895. [DOI] [PubMed] [Google Scholar]
- 8.Lee HT, Kim M, Joo JD, Gallos G, Chen JF, Emala CW. A3 adenosine receptor activation decreases mortality and renal and hepatic injury in murine septic peritonitis. Am J Physiol Regul Integr Comp Physiol. 2006;291:R959–69. doi: 10.1152/ajpregu.00034.2006. [DOI] [PubMed] [Google Scholar]
- 9.Montesinos MC, Desai A, Delano D, Chen JF, Fink JS, Jacobson MA, et al. Adenosine A2A or A3 receptors are required for inhibition of inflammation by methotrexate and its analog MX-68. Arthritis Rheum. 2003;48:240–7. doi: 10.1002/art.10712. [DOI] [PubMed] [Google Scholar]
- 10.Montesinos MC, Yap JS, Desai A, Posadas I, McCrary CT, Cronstein BN. Reversal of the antiinflammatory effects of methotrexate by the nonselective adenosine receptor antagonists theophylline and caffeine: evidence that the antiinflammatory effects of methotrexate are mediated via multiple adenosine receptors in rat adjuvant arthritis. Arthritis Rheum. 2000;43:656–63. doi: 10.1002/1529-0131(200003)43:3<656::AID-ANR23>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 11.Wan TC, Ge ZD, Tampo A, Mio Y, Bienengraeber MW, Tracey WR, et al. The A3 adenosine receptor agonist CP-532,903 [N6-(2,5-dichlorobenzyl)-3′-aminoadenosine-5′-N-methylcarboxamide] protects against myocardial ischemia/reperfusion injury via the sarcolemmal ATP-sensitive potassium channel. J Pharmacol Exp Ther. 2008;324:234–43. doi: 10.1124/jpet.107.127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bokoch GM, Zhao T. Regulation of the phagocyte NADPH oxidase by Rac GTPase. Antiox Redox Signal. 2006;8:1533–48. doi: 10.1089/ars.2006.8.1533. [DOI] [PubMed] [Google Scholar]
- 13.Carstanjen D, Yamauchi A, Koornneef A, Zang H, Filippi MD, Harris C, et al. Rac2 regulates neutrophil chemotaxis, superoxide production, and myeloid colony formation through multiple distinct effector pathways. J Immunol. 2005;174:4613–20. doi: 10.4049/jimmunol.174.8.4613. [DOI] [PubMed] [Google Scholar]
- 14.Kim C, Dinauer MC. Rac2 is an essential regulator of neutrophil nicotinamide adenine dinucleotide phosphate oxidase activation in response to specific signaling pathways. J Immunol. 2001;166:1223–32. doi: 10.4049/jimmunol.166.2.1223. [DOI] [PubMed] [Google Scholar]
- 15.Roberts AW, Kim C, Zhen L, Lowe JB, Kapur R, Petryniak B, et al. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity. 1999;10:183–96. doi: 10.1016/s1074-7613(00)80019-9. [DOI] [PubMed] [Google Scholar]
- 16.Ambruso DR, Knall C, Abell AN, Panepinto J, Kurkchubasche A, Thurman G, et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Nat Acad Sci. 2000;97:4654–9. doi: 10.1073/pnas.080074897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams DA, Tao W, Yang F, Kim C, Gu Y, Mansfield P, et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood. 2000;96:1646–54. [PubMed] [Google Scholar]
- 18.Gu Y, Filippi MD, Cancelas JA, Siefring JE, Williams EP, Jasti AC, et al. Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science. 2003;302:445–9. doi: 10.1126/science.1088485. [DOI] [PubMed] [Google Scholar]
- 19.Sun CX, Downey GP, Zhu F, Koh AL, Thang H, Glogauer M. Rac1 is the small GTPase responsible for regulating the neutrophil chemotaxis compass. Blood. 2004;104:3758–65. doi: 10.1182/blood-2004-03-0781. [DOI] [PubMed] [Google Scholar]
- 20.Salvatore CA, Tilley SL, Latour AM, Fletcher DS, Koller BH, Jacobson MA. Disruption of the A3 adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J Bio Chem. 2000;275:4429–34. doi: 10.1074/jbc.275.6.4429. [DOI] [PubMed] [Google Scholar]
- 21.Lieber JG, Webb S, Suratt BT, Young SK, Johnson GL, Keller GM, et al. The in vitro production and characterization of neutrophils from embryonic stem cells. Blood. 2004;103:852–9. doi: 10.1182/blood-2003-04-1030. [DOI] [PubMed] [Google Scholar]
- 22.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Bio Chem. 1985;260:3440–50. [PubMed] [Google Scholar]
- 23.Geijsen N, van Delft S, Raaijmakers JA, Lammers JW, Collard JG, Koenderman L, et al. Regulation of p21rac activation in human neutrophils. Blood. 1999;94:1121–30. [PubMed] [Google Scholar]
- 24.Li S, Yamauchi A, Marchal CC, Molitoris JK, Quilliam LA, Dinauer MC. Chemoattractant-stimulated Rac activation in wild-type and Rac2-deficient murine neutrophils: preferential activation of Rac2 and Rac2 gene dosage effect on neutrophil functions. J Immunol. 2002;169:5043–51. doi: 10.4049/jimmunol.169.9.5043. [DOI] [PubMed] [Google Scholar]
- 25.Bokoch GM. Chemoattractant signaling and leukocyte activation. Blood. 1995;86:1649–60. [PubMed] [Google Scholar]
- 26.Tintinger G, Steel HC, Anderson R. Taming the neutrophil: calcium clearance and influx mechanisms as novel targets for pharmacological control. Clin Exp Immunol. 2005;141:191–200. doi: 10.1111/j.1365-2249.2005.02800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price LS, Langeslag M, ten Klooster JP, Hordijk PL, Jalink K, Collard JG. Calcium signaling regulates translocation and activation of Rac. J Bio Chem. 2003;278:39413–21. doi: 10.1074/jbc.M302083200. [DOI] [PubMed] [Google Scholar]
- 28.Anderson R, Visser SS, Ramafi G, Theron AJ. Accelerated resequestration of cytosolic calcium and suppression of the pro-inflammatory activities of human neutrophils by CGS 21680 in vitro. Br J Pharmacol. 2000;130:717–24. doi: 10.1038/sj.bjp.0703344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ali H, Richardson RM, Haribabu B, Snyderman R. Chemoattractant receptor cross-desensitization. J Bio Chem. 1999;274:6027–30. doi: 10.1074/jbc.274.10.6027. [DOI] [PubMed] [Google Scholar]
- 30.Didsbury JR, Uhing RJ, Tomhave E, Gerard C, Gerard N, Snyderman R. Receptor class desensitization of leukocyte chemoattractant receptors. Proc Nat Acad Sci. 1991;88:11564–8. doi: 10.1073/pnas.88.24.11564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grimm MC, Ben-Baruch A, Taub DD, Howard OM, Resau JH, Wang JM, et al. Opiates transdeactivate chemokine receptors: δ and μ opiate receptor-mediated heterologous desensitization. J Exp Med. 1998;188:317–25. doi: 10.1084/jem.188.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomhave ED, Richardson RM, Didsbury JR, Menard L, Snyderman R, Ali H. Cross-desensitization of receptors for peptide chemoattractants. Characterization of a new form of leukocyte regulation. J Immunol. 1994;153:3267–75. [PubMed] [Google Scholar]
- 33.Zhang N, Yang D, Dong H, Chen Q, Dimitrova DI, Rogers TJ, et al. Adenosine A2A receptors induce heterologous desensitization of chemokine receptors. Blood. 2006;108:38–44. doi: 10.1182/blood-2005-06-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arcaro A. The small GTP-binding protein Rac promotes the dissociation of gelsolin from actin filaments in neutrophils. J Bio Chem. 1998;273:805–13. doi: 10.1074/jbc.273.2.805. [DOI] [PubMed] [Google Scholar]
- 35.Azuma T, Witke W, Stossel TP, Hartwig JH, Kwiatkowski DJ. Gelsolin is a downstream effector of Rac for fibroblast motility. EMBO J. 1998;17:1362–70. doi: 10.1093/emboj/17.5.1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nature Cell Biol. 1999;1:253–9. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- 37.Dewas C, Fay M, Gougerot-Pocidalo MA, El-Benna J. The mitogen-activated protein kinase extracellular signal-regulated kinase 1/2 pathway is involved in formyl-methionyl-leucyl-phenylalanine-induced p47phox phosphorylation in human neutrophils. J Immunol. 2000;165:5238–44. doi: 10.4049/jimmunol.165.9.5238. [DOI] [PubMed] [Google Scholar]
- 38.Zu YL, Qi J, Gilchrist A, Fernandez GA, Vazquez-Abad D, Kreutzer DL, et al. p38 mitogen-activated protein kinase activation is required for human neutrophil function triggered by TNF-α or FMLP stimulation. J Immunol. 1998;160:1982–9. [PubMed] [Google Scholar]
- 39.Knight D, Zheng X, Rocchini C, Jacobson M, Bai T, Walker B. Adenosine A3 receptor stimulation inhibits migration of human eosinophils. J Leuk Bio. 1997;62:465–8. doi: 10.1002/jlb.62.4.465. [DOI] [PubMed] [Google Scholar]
- 40.Walker BA, Jacobson MA, Knight DA, Salvatore CA, Weir T, Zhou D, et al. Adenosine A3 receptor expression and function in eosinophils. Am J Respir Cell Mol Biol. 1997;16:531–7. doi: 10.1165/ajrcmb.16.5.9160835. [DOI] [PubMed] [Google Scholar]
- 41.Rogers TJ, Steele AD, Howard OM, Oppenheim JJ. Bidirectional heterologous desensitization of opioid and chemokine receptors. Ann N Y Acad Sci. 2000;917:19–28. doi: 10.1111/j.1749-6632.2000.tb05369.x. [DOI] [PubMed] [Google Scholar]
- 42.Zhang N, Hodge D, Rogers TJ, Oppenheim JJ. Ca2+-independent protein kinase Cσ mediate heterologous desensitization of leukocyte chemokine receptors by opioid receptors. J Bio Chem. 2003;278:12729–36. doi: 10.1074/jbc.M300430200. [DOI] [PubMed] [Google Scholar]
- 43.Hammarberg C, Fredholm BB, Schulte G. Adenosine A3 receptor- mediated regulation of p38 and extracellular-regulated kinase ERK1/2 via phosphatidylinositol-3′-kinase. Biochem Pharmacol. 2004;67:129–34. doi: 10.1016/j.bcp.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 44.Hammarberg C, Schulte G, Fredholm BB. Evidence for functional adenosine A3 receptors in microglia cells. J Neurochem. 2003;86:1051–4. doi: 10.1046/j.1471-4159.2003.01919.x. [DOI] [PubMed] [Google Scholar]
- 45.Schulte G, Fredholm BB. Human adenosine A1, A2A, A2B, and A3 receptors expressed in Chinese hamster ovary cells all mediate the phosphorylation of extracellular-regulated kinase 1/2. Mol Pharmacol. 2000;58:477–82. [PubMed] [Google Scholar]
- 46.Schulte G, Fredholm BB. Signaling pathway from the human adenosine A3 receptor expressed in Chinese hamster ovary cells to the extracellular signal-regulated kinase 1/2. Mol Pharmacol. 2002;62:1137–46. doi: 10.1124/mol.62.5.1137. [DOI] [PubMed] [Google Scholar]
- 47.Schulte G, Fredholm BB. Signalling from adenosine receptors to mitogen-activated protein kinases. Cell Signal. 2003;15:813–27. doi: 10.1016/s0898-6568(03)00058-5. [DOI] [PubMed] [Google Scholar]
- 48.Barber MA, Donald S, Thelen S, Anderson KE, Thelen M, Welch HC. Membrane translocation of P-Rex1 is mediated by G protein βγ subunits and phosphoinositide 3-kinase. J Bio Chem. 2007;282:29967–76. doi: 10.1074/jbc.M701877200. [DOI] [PubMed] [Google Scholar]
- 49.Dong X, Mo Z, Bokoch G, Guo C, Li Z, Wu D. P-Rex1 is a primary Rac2 guanine nucleotide exchange factor in mouse neutrophils. Curr Biol. 2005;15:1874–9. doi: 10.1016/j.cub.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 50.Mayeenuddin LH, Garrison JC. Phosphorylation of P-Rex1 by the cyclic AMP-dependent protein kinase inhibits the phosphatidylinositiol (3,4,5)-trisphosphate and G βγ-mediated regulation of its activity. J Bio Chem. 2006;281:1921–8. doi: 10.1074/jbc.M506035200. [DOI] [PubMed] [Google Scholar]
- 51.Welch HC, Coadwell WJ, Ellson CD, Ferguson GJ, Andrews SR, Erdjument-Bromage H, et al. P-Rex1, a PtdIns(3,4,5)P3- and G βγ regulated guanine-nucleotide exchange factor for Rac. Cell. 2002;108:809–21. doi: 10.1016/s0092-8674(02)00663-3. [DOI] [PubMed] [Google Scholar]
- 52.Welch HC, Condliffe AM, Milne LJ, Ferguson GJ, Hill K, Webb LM, et al. P-Rex1 regulates neutrophil function. Curr Biol. 2005;15:1867–73. doi: 10.1016/j.cub.2005.09.050. [DOI] [PubMed] [Google Scholar]
- 53.Zhao T, Nalbant P, Hoshino M, Dong X, Wu D, Bokoch GM. Signaling requirements for translocation of P-Rex1, a key Rac2 exchange factor involved in chemoattractant-stimulated human neutrophil function. J Leuk Biol. 2007;81:1127–36. doi: 10.1189/jlb.0406251. [DOI] [PubMed] [Google Scholar]
- 54.Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314:1792–5. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]