

Abstract
BACKGROUND AND AIMS
Mallory-Denk bodies (MDBs) are keratin (K)-rich cytoplasmic hepatocyte inclusions commonly associated with alcoholic steatohepatitis. Given the significant gender differences in predisposition to human alcohol-related liver injury, and the strain difference in mouse MDB formation, we hypothesized that gender affects MDB formation.
METHODS
MDBs were induced in male and female mice overexpressing K8, which are predisposed to MDB formation, and in non-transgenic mice by feeding the porphyrinogenic compound 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC). MDB presence was determined by histological, immunofluorescence and biochemical analyses and correlated to liver injury using serologic and pathological markers. Cytoskeletal and metabolic liver protein analysis, in vitro metabolism studies, and measurement of oxidative stress markers and protoporphyrin-IX were performed.
RESULTS
Male mice formed significantly more MDBs, which was modestly attenuated by estradiol. MDB formation was accompanied by elevated oxidative stress. Female mice had significantly fewer MDBs and oxidative stress-related changes, but had elevated ductular reaction and protoporphyrin-IX accumulation and MDB-preventive K18 induction. Evaluation of the microsomal cytochrome-P450 (CYP) enzymes revealed significant gender differences in protein expression and activity in untreated and DDC-fed mice, and showed that DDC is metabolized by CYP3A. The changes in CYPs account for the gender differences in porphyria and DDC metabolism. DDC metabolite formation and oxidative injury accumulate upon chronic DDC exposure in males, despite more efficient acute metabolism in females.
CONCLUSIONS
Gender dimorphic formation of MDBs and porphyria associate with differences in CYPs, oxidative injury and selective keratin induction. These findings may extend to human MDBs and other neuropathy- and myopathy-related inclusions.
Keywords: Liver inclusions, Cytochrome P450, keratins
Introduction
Mallory-Denk bodies (MDBs) are hepatocellular inclusions observed in multiple liver diseases, including alcoholic and non-alcoholic steatohepatitis (ASH and NASH, respectively), chronic cholestasis, hepatocellular carcinoma (HCC), and the copper-storage disorders Wilson disease and Indian childhood cirrhosis1, 2. MDBs are defined by their morphological appearance as large, irregularly-shaped perinuclear inclusions or small peripheral aggregates residing within ballooned hepatocytes that have a flocculent cytoplasm1.
The formation of MDBs is induced experimentally in mice by chronic feeding with griseofulvin, an antifungal agent, or 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC), an analog of the dihydropyridine calcium channel blockers3, 4. The established animal models have been essential in elucidating the major molecular components of MDBs and unraveling several mechanisms that lead to their formation. For example, the keratin (K) intermediate filament (IF) proteins K8 and K18 are the major constituents of MDBs5. Upon liver injury, such as the injury associated with MDB formation, K8 and K18 mRNA and protein levels are induced, but with a characteristic K8-to-K18 ratio of greater than 1, which is critical for MDB formation1, 5. For example, mice that are genetically altered to over-express K8 or mice that lack K18 develop MDBs spontaneously upon aging6, 7. Additionally, when challenged with an MDB-inducing agent, K8 over-expressing mice develop MDBs more rapidly than their wild-type littermates6. The bulk of keratins localized within MDBs are modified post-translationally by phosphorylation, ubiquitination, and, in the case of K8, transamidation via transglutaminase-2 (TG2)5. Similar to other inclusion-related disorders, such as neurodegeneration, and signifying cellular defects within the protein degradation machinery, MDBs also contain ubiquitin (Ub), heat shock proteins, and p62, which are induced in response to various stressful stimuli, in particular oxidative stress8, 9. The genetic background is critical in dictating the extent of mouse MDB formation, which parallels the observation that MDBs are seen only in some patients with disorders typically associated with the presence of MDBs10. Another facet of MDB-related liver disorders is the importance of gender in determining an individual’s susceptibility to, and progression of the disease. For example, females have an increased risk for developing advanced alcoholic liver disease (ALD) and cirrhosis11. However, the relationship between liver disease progression and MDB formation is currently unknown.
Since their inception over three decades ago, experimental models of MDB formation have traditionally utilized male mice, likely because of historical reasons pertaining to the use of these agents to induce hepatocellular carcinoma, which is more frequent in males. Bearing in mind the possible clinical impact of alcohol-related injury in males versus females, we tested the hypothesis that MDBs are formed in a gender-dependent manner, and that their formation correlates with the extent or type of liver injury. Several mechanisms were explored to understand the basis of a gender difference in DDC-induced MDB formation that we observed, including the role of microsomal metabolism, oxidative stress, protoporphyrin accumulation, and the expression of intermediate filaments (IFs) and other MDB component proteins.
Materials and Methods
Antibodies
The antibodies (Abs) we used were directed to: K8 (Troma) and K19 (Troma III) (Developmental Studies Hybridoma Bank); mouse/human K8-K18 Ab-8592 and mouse/human K18 Ab-466812; TG2, heat shock protein 60 (Hsp60) and GST-mu (Labvision); Ub and p62 (Santa Cruz Biotechnology); CYP2A6, CYP2C9, CYP7A1 and CYP3A1 (Abcam); and CYP2E1 (a kind gift from Dr. Harry Gelboin).
Animal experiments
Non-transgenic FVB/N mice and mouse K8-overexpressing transgenic mice (FVB/N background)6 were used (9 mice/group). Presence of the K8 transgene was verified by PCR screening of genomic mouse-tail DNA6. Mice were fed powdered chow (Formulab Diet 5008, Dean’s Animal Feeds or LabDiet 5001, PMI Nutrition International) containing 0.1% DDC (Sigma-Aldrich) for 6 weeks (K8-overexpressing mice). FVB/N mice were fed a 0.1% DDC-containing diet (10 or 90 days). Control mice were fed a standard mouse diet (Teklad Global Diet 2019, Harlan Teklad or LabDiet 5001). For estradiol (E2)-related experiments, E2 (10 μg/kg; Sigma-Aldrich) or vehicle (sesame oil) were injected subcutaneously into mice 3x/week. Mice were euthanized by CO2 inhalation, and blood was collected by intracardiac puncture for measurement of AST, ALT, and ALP. Livers were removed, weighed and sectioned into 1–2 mm slices then apportioned for: fixation in 10% formaldehyde followed by hematoxylin and eosin (H&E) staining, snap-freezing in liquid nitrogen for biochemical and PCR analyses, or embedded in optimal-cutting-temperature medium and frozen for immunofluorescence (FL) staining. Animal use was approved by and carried out in accordance with the Committee for the Use and Care of Animals at the University of Michigan.
Histological and immunofluorescence analysis
H&E-stained liver sections were assessed by an experienced clinical hepatopathologist (E.M.B.) for the parameters shown in Fig.1. Scoring was done in a blinded fashion with regard to treatment condition or gender. MDBs were counted using a 20× lens and 10 fields were analyzed per liver specimen. Liver injury was assessed semi-quantitatively using 0–4 scale (0, none; 1, rare; 2, moderate; 3, frequent; 4, abundant). Descriptive assessments were also noted for each liver. FL staining was done as described previously12. Sections were viewed using confocal microscopy (Zeiss 510-Meta). To quantify the extent of MDB formation, a 20x lens was used and the numbers of cells with K8/K18 and Ub-positive aggregates were counted (10 fields/liver section).
Fig. 1.

Histological evidence for gender dimorphic formation of MDBs in the livers of DDC-fed FVB/N mice. MDB detection by immunofluorescence (FL) staining of K8/K18 (red) and Ub (green) in male, female, and estradiol (E2)-treated male mice fed DDC for 90 days (top; scale bar represents 200 μm). Histological analysis demonstrating MDBs (middle) and ductular reaction (bottom), each denoted by the arrowheads (scale bars represent 5 μm).
Protein analysis
Total liver lysates were prepared using a homogenization buffer [0.187 M Tris-HCl (pH 6.8), 3% SDS and 5 mM EDTA] and samples were subsequently diluted to a desired protein concentration with 4X reducing Laemmli sample buffer. Equal amounts of proteins were separated by SDS-PAGE and stained by Coomassie blue, or were transferred to polyvinylidene difluoride membranes for immunoblotting12 and the resulting antigen-Ab complexes were detected by enhanced chemiluminescence (PerkinElmer Life Sciences). Specific Coomassie blue-stained bands were also characterized using routine mass spectrometry (Michigan Proteome Consortium, University of Michigan).
Quantitative real-time PCR (qPCR)
Total RNA was isolated using an RNeasy-Midi Kit (Qiagen) and then translated into cDNA using Taqman Reverse Transcription Reagents (Applied Biosystems). qPCR was performed using MyiQ real-time PCR detection system (Bio-Rad) and SYBR green for monitoring cDNA amplification (primers are listed in Supplementary Table 1). Samples were analyzed in triplicates and three individual mice were tested for each group. L7 ribosomal protein was used as an internal control and transcript levels relative to L7 were determined and reported as the mean ± s.d.
GST activity and lipid peroxidation assays
Total GST activity was determined by measuring the conjugation of the GST substrate 1-chloro-2,4-dinitrobenzene (CDNB) with reduced glutathione in the presence of liver lysates from various treatment groups (n=3 animals/group) and normalized to the amount of protein. Lipid hydroperoxides were measured using a kit (Cayman Chemical, Ann Arbor, MI) and 4 livers/condition.
DDC metabolism
Pooled liver microsomes (3 mice/condition) were prepared following established procedures13. The metabolic reactions contained 75 μg microsomal protein and 25 μM DDC in 0.1 M potassium phosphate buffer (pH 7.4) and were incubated (45 minutes, 37°C) in the presence/absence of NADPH and ketoconazole. Reaction mixtures were extracted (4 volumes of ethyl acetate), dried and dissolved in methanol for analysis by electrospray ionization-liquid chromatography/mass spectrometry (ESI-LC/MS). For ESI-LC/MS, samples (10 μl) were injected onto a Hypersil ODS column (Thermo Electron Corporation, Waltham, MA) that had been equilibrated with 50% solvent B (0.1% acetic acid in methanol) and 50% solvent A (0.1% acetic acid in water). The analytes were resolved (0.3 ml/min flow rate) using a gradient (0–5 min, 50% B; 5–20 min, 50 to 100% B; 20–25 min, 100% B; 25–26 min, 100 to 50% B; and 26–30 min, 75% B), followed by LCQ mass analysis (Thermo Electron Corporation, Waltham, MA). Data were acquired in positive ion mode using the Xcalibur software package (Thermo Electron Corporation).
Quantification of DDC and its metabolite
Liver tissue (160 mg) from 3 animals/group was homogenized (0.1 M potassium phosphate buffer, pH 7.4) and spiked with 200 nM 4-ethyl DDC, a structural analog of DDC, as an internal standard. Samples were extracted and analyzed by ESI-LC/MS. DDC and metabolite amounts were calculated based on a standard curve generated by injecting various known amounts of DDC and internal standard.
CYP content, catalytic activity and inactivation by DDC
Total CYP content was determined by the reduced CO spectral method14 and quantified using the CYP extinction coefficient of 91 mM−1-cm−1. The microsomal CYP catalytic activity was determined by the metabolism of probe substrates [7-benzyloxy-, 7-ethoxy-, and 7-methoxy- 4-(trifluoromethyl) coumarin (7-BFC, 7-EFC, 7-MFC)] yielding a fluorescent product after 15 minutes of reaction time. Inactivation assays were carried out as previously described15. To determine the effect of DDC metabolism by CYP3A4 on CYP2C9 inactivation, reconstituted CYP3A4 was incubated with 100 μM DDC in the absence/presence of NADPH, after which an equimolar and equal volume of CYP2C9 was added. The catalytic activity of CYP2C9 was measured immediately or 5 min after addition to the CYP3A4 reaction. Inactivation was expressed as percent activity loss after 5 min, relative to 0 min of incubation time.
PPIX accumulation
Liver tissue homogenates from the designated treatment groups were extracted with 20 volumes of 0.9 N perchloric acid/ethanol (1:1 mixture). PPIX levels in the extracts were determined using a fluorescence-based method (400 nm excitation and 605 nm emission) and a standard curve generated by measuring the fluorescence of authentic PPIX standard and normalized to liver tissue weight. The control livers had undetectable levels of PPIX (not shown).
Statistics
The data were presented and analyzed with GraphPad Prism 5 Software, as described in the Figure Legends.
Results
Gender difference in MDB formation in transgenic K8 over-expressing mice
We initially performed a pilot experiment to determine the role of gender on MDB formation using the previously-characterized mouse K8 over-expressing mice, which form MDBs after only 6 weeks of DDC feeding, in contrast to the 12–20 week feeding regimen required for non-transgenic models6. The presence of MDBs, assessed by immunofluorescence staining for K8/K18 and Ub, routine histopathology and biochemically by detection of high molecular weight protein aggregates containing K8 and Ub16, was significantly more prominent in male mice (Supplementary Fig. 1).
Gender difference in MDB formation in non-transgenic FVB/N mice
To confirm the results of the pilot study in non-transgenic mice and to also test the effect of estradiol administration on MDB formation in male mice, we fed FVB/N mice DDC for 90 days and administered either vehicle or E2 (10μg/kg, 3 days/week). Blinded histological and immunofluorescence analyses showed a striking difference in MDB formation (Fig. 1 and Supplementary Fig. 2), in that males had 5–9 times more MDBs than females (Table 1). Biochemical analysis for the presence of K8/K18-Ub complexes and p62 induction supported the MDB detection analysis (Fig. 2A). Estradiol produced a modest decrease in MDB formation (Fig. 1,2). There was similar K8 mRNA expression between genders, but a greater K18 induction in the female mice, resulting in a lower K8:K18 ratio which, together with the lower induction of p62 (Fig. 2A-B), protects from MDB formation as demonstrated previously using genetic mouse models16.
Fig. 2.
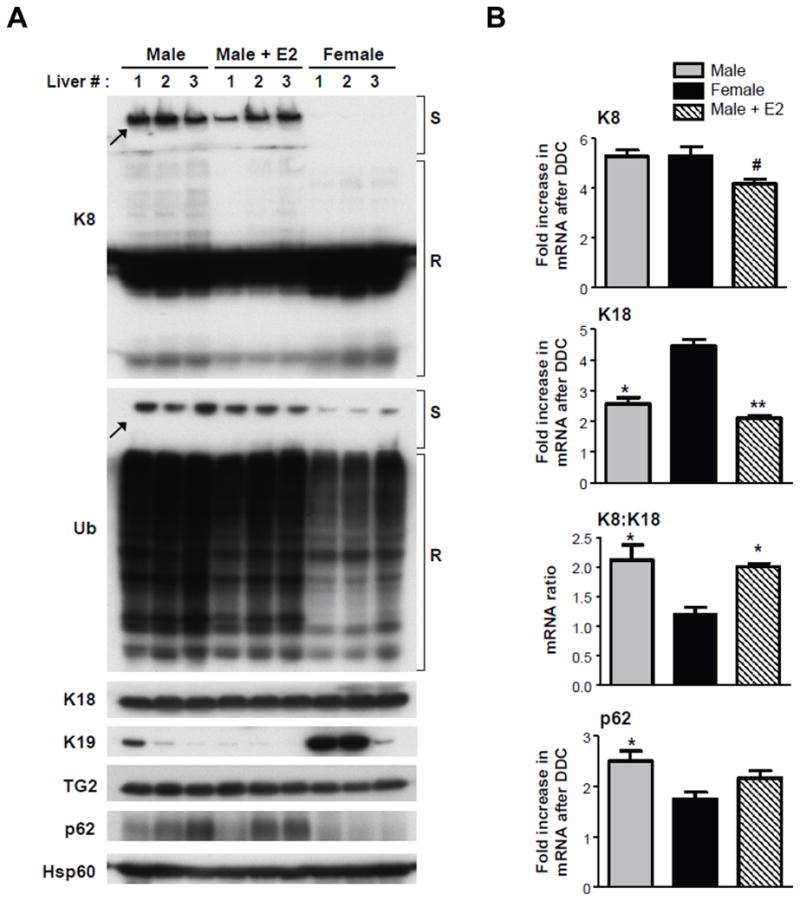
Biochemical evidence for gender dimorphic formation of MDBs in the livers of DDC-fed FVB/N mice. (A) Immunoblot analysis for the presence of high molecular weight K8 and Ub-containing complexes (arrows) in stacking gels (S), K8 monomer and Ub conjugates in SDS-PAGE resolving gels (R), and expression of K18, K19, TG2, p62 and Hsp60 (used as a loading control) in DDC-fed male, female and E2-treated male mice. (B) Quantitative real-time (PCR) analysis for mRNA expression of K8, K18 and p62. Values represent the mean ±s.e.m. (n=3) *P<0.05, **P<0.01 compared to female group; #P<0.05 compared to male group, one way ANOVA.
Differential correlation between the presence of MDBs and various markers of liver injury
Serological evaluation revealed elevated levels of the liver injury markers aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels, which were greater in the male DDC-fed mice, regardless of E2 administration (Table 1). Histological evaluation further showed increased acidophil bodies (representing apoptosis) and fat globules in male mouse hepatocytes. Conversely, female mice displayed increased ductular reaction (Fig. 1, Table 1), as well as an increased expression of K19 protein (Fig. 2) which if found in cholangiocytes and hepatic progenitor cells, but not adult hepatocytes. Thus, MDB formation correlated positively with AST and ALT levels, apoptosis, and fat accumulation and negatively with ductular reaction (Table 1).
Gender differences in DDC-induced oxidative stress in mouse liver
To assess other contributions to the MDB gender dimorphism, major differences in protein expression were evaluated in livers from control and DDC-fed mice. The presence of a 25 kDa protein was found to be dramatically elevated in the DDC-fed mice, implying its potential involvement in MDB formation (Fig. 3A). Mass spectrometry analysis identified this protein as glutathione-S-transferase-mu-1 (GSTM1), which is upregulated during oxidative stress and catalyzes the conjugation of the antioxidant glutathione to reactive intermediates that can elicit cellular toxicity17. There was greater GSTM1 induction and total GST activity in male DDC-fed mice (Fig. 3B,C). One hallmark of oxidative stress is the formation of highly reactive lipid hydroperoxides (LOOHs). Quantification of liver LOOHs revealed dramatic increases of approximately 60- and 100-fold in female and male DDC-fed mice, respectively (Fig. 3D). This prompted us to investigate DDC metabolism by hepatic enzymes as a potential source of the chemically-reactive intermediates.
Fig. 3.

Gender differences in DDC-induced oxidative stress. (A) Changes in hepatic protein expression between male and female control and DDC-fed mice as determined by Coomassie stain of liver lysates. The expression of a protein, identified by mass spectrometry as GSTM1 and denoted by the arrow, is markedly induced after DDC treatment. (B) Confirmation of GSTM1 induction (male > female) by immunoblotting. (C) Measurement of total GST activity in liver lysates of control and DDC-fed mice (3 mice/gender/condition). (D) Quantitative measurement of lipid peroxidation in the livers of control and DDC-fed mice (4 mice/gender/condition). *P<0.05, **P<0.01 compared to female group, one way ANOVA.
Gender differences in liver microsomal cytochrome P450-mediated DDC metabolism
Given the well-documented gender difference in expression and activity of the hepatic cytochrome P450 (CYP) monooxygenases18, which are known to form reactive oxygen species19, we examined the CYP-mediated transformation of DDC. A single mono-oxygenated metabolite was detected by LC/MS analysis upon DDC incubation with liver microsomes from untreated mice in the presence of NADPH (Fig. 4A-C). Formation of the metabolite was significantly greater with the female microsomes (Fig. 4A), which could result from either a differential expression of a single CYP responsible for the metabolism, or the participation of different CYPs in the male and female mice in product formation. The latter was excluded, since microsomal pre-incubation with the CYP3A-selective inhibitor ketoconazole20 abolished metabolite formation in a dose-dependent manner (IC50 = 14–22 nM), regardless of gender (Fig. 4D). The in vivo relevance of this metabolic pathway was confirmed by quantification of DDC and the metabolite in livers of mice that were fed DDC for either 10 or 90 days (Fig. 4E,F). After 10 days of feeding, DDC levels were similar between genders, but there was approximately 5 times more metabolite in the female livers. After 90 days, the DDC concentration was significantly lower relative to 10 days, but there was no statistically significant difference between genders. Relative to 10 days, metabolite levels decreased in the female, and increased in the male mice after 90 days of DDC feeding.
Fig. 4.

Gender differences in hepatic DDC metabolism. (A) NADPH-dependent in vitro metabolism of DDC by pooled liver microsomes from untreated FVB/N mice (3 mice/gender/condition) as determined by ESI-LC/MS analysis. The ion chromatogram shows the peaks from DDC and the DDC metabolite, which had mass to charge ratios (m/z) of 267.9 and 284.8, respectively. (B) Tandem MS of DDC, including the structure of DDC and potential oxidation sites as denoted by asterisks. The proposed oxidation sites are based on the major ms/ms fragments of DDC and the metabolite (C), which signify a mass difference of 46 (loss of CH3CH2OH) from the corresponding precursor ions. (D) Dose-dependent inhibition of microsomal DDC metabolism by the CYP3A-selective inhibitor ketoconazole. Inhibition is expressed as a percent of metabolite formed relative to vehicle control. (E,F) Measurement of the levels of DDC and the metabolite in the livers of mice fed DDC for 10 or 90 days (n=3). **P<0.01 compared to female group, one way ANOVA.
Gender differences in the basal and DDC-altered CYP expression and activity
The observed time- and gender-dependent differences in DDC metabolism prompted further investigation into the fate of the CYPs in these mice. Total microsomal CYP content was two-fold greater in untreated female mice (Fig. 5A,D). DDC feeding for 10 and 90 days diminished the CYP levels in the female mice to 42% and 57% of control, respectively (Fig. 5B-D). In contrast, the total male liver CYP content decreased to 76% of control after 10 days, but increased to 116% of control after 90 days of DDC feeding (Fig. 5B-D). The drug-metabolizing CYP activity of untreated female mice was approximately two-fold greater relative to the male mice and DDC produced a decrease of approximately 10-fold and 20-fold in the male and female mice, respectively (Fig. 5E). Immunoblot analysis showed that, with the exception of CYP2E1, which was similar in untreated mice regardless of gender, the expression of all other CYPs tested was significantly lower in the microsomes of untreated male mice. The striking difference in the expression of CYP3A (Fig. 5F), the enzyme responsible for DDC metabolism (Fig. 4D), supports the data demonstrating increased DDC metabolism in the female mice under basal conditions (Fig. 4A). Upon DDC treatment, all CYP isoforms tested were largely decreased in the female mice, whereas an isoform-dependent effect on the expression of the male mouse CYPs was observed, such that 2A/3A were upregulated, 2E1 was unchanged, and 2C/7A1 were downregulated (Fig. 5F). The increased amount of DDC metabolite in male mice after 90 days of DDC feeding (Fig. 4F) is in agreement with the observed induction of CYP3A and correlates with the elevated oxidative stress (Fig. 3). Additionally, sustained expression of CYP2E1 in male DDC-treated mice, in contrast to its attenuated expression in female mice, likely contributes to the generation of more LOOHs in male mice (Fig. 3D), given the well-known involvement of this enzyme in ALD-associated oxidative stress21. The downregulation of cholesterol 7α-hydroxylase (CYP7A1) may also be related to the formation of LOOHs and MDBs, since this enzyme is also downregulated in a rat model of Wilson disease22, another liver disorder characterized by the presence of MDBs and LOOHs.
Fig. 5.

Gender differences in the basal and DDC-altered microsomal CYP expression and activity. (A-C) Reduced CO spectra of pooled microsomes prepared from mice (n=3) that were untreated (A), or fed DDC for 10 days (B) or 90 days (C). (D) Quantification of the total microsomal CYP content based on the reduced CO spectra. ***P<0.001 compared to female group in the same treatment, one way ANOVA. (E) Measurement of the drug metabolizing microsomal CYP activity using the fluorogenic probe substrates 7-BFC, 7-EFC and 7-MFC, as described in Materials and Methods. The values reflect the average of the activity for the 3 substrates. ***P<0.001 compared to female group in the same treatment, one way ANOVA. (F) Immunoblot detection of CYP proteins in liver microsomes of control (0) and DDC-fed mice (10, 90). Lanes are marked with the name of the CYP isoforms against which the antibody was targeted.
DDC-mediated CYP inactivation potentially leading to time- and gender-dependent liver protoporphyrin IX accumulation
A plausible mechanism for the DDC-induced diminishment of CYPs is enzymatic inactivation, followed by proteasomal degradation23, 24. Therefore, we tested the ability of DDC to inactivate human CYP2C9 and CYP3A4, since these isoforms make up the majority of liver CYP protein25. Incubation of CYP2C9 with DDC (10–100 μM) inhibited the CYP2C9 activity in a dose-dependent manner but had no effect on the CYP3A4 activity at these concentrations (Fig. 6A). However, given the role of the female-predominant mouse CYP3A in metabolizing DDC, and the significant degradation of female CYPs, we considered whether DDC metabolism contributes to CYP inactivation. Pre-incubating DDC with CYP3A4, which is able to metabolize DDC (Supplementary Fig. 3), followed by addition of the reaction mixture to CYP2C9, resulted in greater CYP2C9 inactivation when NADPH was included in the CYP3A4 reaction (i.e., including metabolized DDC) compared to NADPH absence (with only unmetabolized DDC) (Fig. 6B). These data demonstrate the importance of DDC metabolism by CYP3A in the inactivation of other CYP isoforms and help to explain the significant decrease in the female microsomal CYPs.
Fig. 6.

DDC-mediated CYP inactivation and liver protoporphyrin IX (PPIX) accumulation. (A) The effect of DDC on CYP2C9 and CYP3A4 activity, as measured by the ability of the enzymes to metabolize 7-MFC and 7-BFC, respectively. **P<0.01, ***P<0.001 compared to vehicle (0 μM), two-way ANOVA. (B) The effect of pre-incubating DDC with CYP3A4 in the absence (unmetabolized) or presence (metabolized) of NADPH on CYP2C9 inactivation. **P<0.01, unpaired t-test. (C) Quantitative measurement of liver PPIX accumulation in mice fed DDC for 10 or 90 days (n=3). PPIX was not detected in control livers (not shown). **P<0.01 compared to female group in the same treatment, one way ANOVA.
Another hallmark consequence of DDC treatment is protoporphyrin IX (PPIX) accumulation due to inhibition of ferrochelatase, which catalyzes the terminal step in the heme biosynthetic pathway by converting PPIX to heme26. We investigated potential gender differences in PPIX accumulation. After the 10-day DDC treatment, PPIX levels were similar between genders, but the female mice had more than 2-fold greater levels of liver PPIX after the 90-day treatment relative to the male mice (Fig. 6C). This time-dependent increase in PPIX accumulation parallels the sustained DDC-mediated CYP degradation observed in the female mice (Fig. 5).
Discussion
Mechanisms that contribute to gender dimorphism in MDB formation
Gaining a better understanding of the cell biological and disease-related significance of MDBs has the potential to lead to new therapies for the treatment of MDB-associated liver disease as well as protein aggregation disorders affecting other organs. For example, rapamycin-mediated activation of autophagy prevents DDC-induced mouse MDB formation and leads to resorption of existing MDBs, while proteasomal inhibitors, such as bortezomib, induce MDB formation27. However, the upstream and genetic factors that are involved in MDB formation are currently in the early stages of becoming better understood.
Although DDC is not used clinically, it likely shares common functional mechanisms with protein aggregate-inducing xenobiotics to which humans are exposed28. The findings herein using K8-overexpressing and non-transgenic DDC models support the contribution of CYP-dependent metabolism, oxidative stress, and differences in K18 and p62 induction in the gender dimorphic mouse MDB formation (Fig. 7). Our findings are consistent with the conclusion that MDB formation results, at least in part, from oxidative stress induced by DDC via CYP-mediated and gender-dependent metabolism, and is indirectly affected by the drug’s porphyrinogenicity. Furthermore, the relative DDC-induced increase in K18 in female livers, in comparison to male livers, is another likely contributor to the gender differences in MDB formation. The increase in p62 mRNA and protein in the livers of male mice, which is MDB-promoting, represents a response to elevated oxidative stress based on prior reports regarding the regulation of p62 and its presence in MDBs and similar inclusions8, 9, 29–31.
Fig. 7.

The contribution of hepatic metabolism, K18 and p62 induction to the gender difference in DDC-induced porphyria and MDB formation. When administered as a dietary component, DDC undergoes CYP3A-dependent liver metabolism, which occurs more extensively in female mice at feeding onset due to greater CYP3A expression. Prolonged DDC feeding causes CYP3A downregulation in female, and upregulation in male livers. The upregulation of male CYP3A, in combination with sustained expression of other CYPs (such as 2E1) leads to the generation of more reactive oxygen species, ultimately resulting in more MDB formation. Conversely, the sustained degradation of female CYPs during DDC feeding results in reduced oxidative stress and less MDBs. Also as a result of CYP degradation, there is an elevated accumulation of PPIX in female mice due to inhibition of ferrochelatase by N-methyl protoporphyrin (N-methyl PP), an intermediate generated during CYP inactivation and degradation by DDC. Increased PPIX accumulation attenuates oxidative stress in the female mouse livers. Furthermore, the relative DDC-induced increases in K18 in female livers and p62 in male livers are likely additional contributors to the gender differences in MDB formation.
Oxidative stress is a common pathologic finding of virtually all protein aggregation disorders, including neurodegeneration32, inclusion-body myopathies33 and MDB-related liver diseases5. The current findings demonstrate increased oxidative damage in male livers in association with MDB formation, based upon assessment of several parameters related to oxidative injury (GST expression and activity and lipid hydroperoxides). In light of the important role of oxidative stress in protein aggregation pathologies32, the inter-individual and gender-dependent variability in CYP activity18 and extrahepatic CYP expression34, these findings may extend to human liver disease where gender and genetic background differences in MDB formation have not been investigated, and to other inclusion-associated diseases such as myopathies and neurodegeneration.
Porphyria and the ductular reaction in MDB formation
Protoporphyrin can act as either a pro- or an anti-oxidant, in the presence and absence of photosensitization, respectively35. In the liver (i.e. dark environment), PPIX accumulation protects against lipid peroxidation by scavenging peroxyl radicals, thus exerting an anti-oxidant effect similar to bilirubin35. This is reflected in our observation that the two-fold higher levels of liver PPIX in the female DDC-treated mice (90 days) coincide with approximately two-fold less LOOHs compared to male mice. The indirect, oxidative injury-modifying, effect of the porphyria on MDB formation in the DDC model supports the relevance of this animal model to alcoholic liver disease, where oxidative stress and MDBs are observed in the absence of porphyria2, 21.
Another noteworthy histological feature that was more prominent in female livers after DDC feeding, in association with decreased formation of MDBs, is the presence of ductular reaction (Table 1). Ductular reaction reflects an accumulation of increased matrix, inflammatory cells, and K19 positive epithelial cells in the periportal regions of the liver as a proliferative response to many forms of liver injury36. The association of ductular reaction with fibrosis progression may provide a parallel if extrapolated to human female predisposition to alcoholic liver disease progression.
Comparison of gender association of MDB formation with other inclusion diseases
Aside from keratins, several other IF proteins, including glial fibrillary acidic protein and desmin appear as components of inclusions found in Alexander disease (Rosenthal fibers) and desminopathies (Desmin bodies)36, 37. However, it is not known whether gender differences contribute to the formation of these inclusions. In addition, MDBs share some similarities with cytoplasmic tubular aggregates (TAs) associated with inherited and alcohol- and drug-induced myopathies38, and sporadic inclusion body myositis (s-IBM) inclusions33.
The TAs of skeletal muscle are sarcoplasmic reticulum-derived inclusions that contain heat shock proteins, tau and ubiquitin, although there is no evidence for the presence of intermediate filaments38. Male-exclusive presence of TAs has been demonstrated in at least three mouse models, including the senescence-accelerated (SAM), the caveolin-deficient, and the congenic mice of the MRL+/+ strain39–41. Also, TAs were present in human muscle biopsies obtained from only males in a large cohort of patients who had muscle aches and cramps as their chief complaint42. Another muscle-related disorder with inclusions similar to MDBs is s-IBM, the most common debilitating muscle disease among elderly patients33. Presence of the intermediate filament proteins desmin and synemin within vacuolated fibers of s-IBM patients has also been demonstrated, along with increased expression and cross-linking by transglutaminases43, 44. Notably, there is a strong genetic component in the epidemiology of s-IBM45, and the disease has a male-to-female preponderance estimated to be 2:146.
Collectively, our findings tie together observations related to DDC-induced MDB formation, porphyria and oxidative injury that have been reported in numerous publications since the initial description of the DDC and griseofulvin models in the mid 1970’s1. The findings herein warrant the assessment of potential gender differences in human MDB formation and may extend to other cytoplasmic inclusions that are found in association with several neurological and muscle disorders.
Supplementary Material
Acknowledgments
Financial Support: NIH grant DK52951 and the Department of Veterans Affairs (M.B.O.) and a University of Michigan Postdoctoral Translational Scholars Program Award UL1RR024986 (N.T.S.).
Nonstandard abbreviations used
- Ab
antibody
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- ASH
alcoholic steatohepatitis
- AST
aspartate aminotransferase
- CYP
cytochrome P450
- DDC
3,5-diethoxycarbonyl-1,4-dihydrocollidine
- E2
estradiol
- EPP
erythropoietic porphyria
- FL
immunofluorescence
- GST
glutathione S-transferase
- H&E
hematoxylin and eosin
- Hsp
heat shock protein
- IF
intermediate filament
- K
keratin
- LOOH
lipid hydroperoxide
- MDB
Mallory-Denk body
- MS
mass spectrometry
- PPIX
protoporphyrin IX
- qPCR
real-time quantitative PCR
- s-IBM
sporadic inclusion body myositis
- TA
tubular aggregate
- TG2
transglutaminase-2
- Ub
ubiquitin
Footnotes
Disclosures: The authors report no conflicts of interest.
Author Involvement: Study concept and design: SH, MBO
Acquisition of data: SH, NTS, EMB
Analysis and interpretation of data: SH, NTS, EMB, PFH, MBO
Drafting of the manuscript: SH, NTS
Critical revision of the manuscript for intellectual content: SH, NTS, EMB, PFH, MBO
Statistical analysis: SH, NTS
Obtained funding: MBO, NTS
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zatloukal K, French SW, Stumptner C, et al. From Mallory to Mallory-Denk bodies: what, how and why? Exp Cell Res. 2007;313:2033–49. doi: 10.1016/j.yexcr.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 2.Brunt EM. Alcoholic and nonalcoholic steatohepatitis. Clin Liver Dis. 2002;6:399–420. vii. doi: 10.1016/s1089-3261(02)00002-8. [DOI] [PubMed] [Google Scholar]
- 3.Denk H, Gschnait F, Wolff K. Hepatocellar hyalin (Mallory bodies) in long term griseofulvin-treated mice: a new experimental model for the study of hyalin formation. Lab Invest. 1975;32:773–6. [PubMed] [Google Scholar]
- 4.Yokoo H, Harwood TR, Racker D, et al. Experimental production of Mallory bodies in mice by diet containing 3,5-diethoxycarbonyl-1,4-dihydrocollidine. Gastroenterology. 1982;83:109–13. [PubMed] [Google Scholar]
- 5.Ku NO, Strnad P, Zhong BH, et al. Keratins let liver live: Mutations predispose to liver disease and crosslinking generates Mallory-Denk bodies. Hepatology. 2007;46:1639–49. doi: 10.1002/hep.21976. [DOI] [PubMed] [Google Scholar]
- 6.Nakamichi I, Toivola DM, Strnad P, et al. Keratin 8 overexpression promotes mouse Mallory body formation. J Cell Biol. 2005;171:931–7. doi: 10.1083/jcb.200507093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magin TM, Schroder R, Leitgeb S, et al. Lessons from keratin 18 knockout mice: formation of novel keratin filaments, secondary loss of keratin 7 and accumulation of liver-specific keratin 8-positive aggregates. J Cell Biol. 1998;140:1441–51. doi: 10.1083/jcb.140.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stumptner C, Fuchsbichler A, Zatloukal K, et al. In vitro production of Mallory bodies and intracellular hyaline bodies: the central role of sequestosome 1/p62. Hepatology. 2007;46:851–60. doi: 10.1002/hep.21744. [DOI] [PubMed] [Google Scholar]
- 9.Stumptner C, Fuchsbichler A, Heid H, et al. Mallory body--a disease-associated type of sequestosome. Hepatology. 2002;35:1053–62. doi: 10.1053/jhep.2002.32674. [DOI] [PubMed] [Google Scholar]
- 10.Hanada S, Strnad P, Brunt EM, et al. The genetic background modulates susceptibility to mouse liver Mallory-Denk body formation and liver injury. Hepatology. 2008;48:943–52. doi: 10.1002/hep.22436. [DOI] [PubMed] [Google Scholar]
- 11.Mandayam S, Jamal MM, Morgan TR. Epidemiology of alcoholic liver disease. Semin Liver Dis. 2004;24:217–32. doi: 10.1055/s-2004-832936. [DOI] [PubMed] [Google Scholar]
- 12.Ku NO, Toivola DM, Zhou Q, et al. Studying simple epithelial keratins in cells and tissues. Methods Cell Biol. 2004;78:489–517. doi: 10.1016/s0091-679x(04)78017-6. [DOI] [PubMed] [Google Scholar]
- 13.Jia L, Liu X. The conduct of drug metabolism studies considered good practice (II): in vitro experiments. Curr Drug Metab. 2007;8:822–9. doi: 10.2174/138920007782798207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Omura T, Sato R. The Carbon Monoxide-Binding Pigment of Liver Microsomes. I. Evidence for Its Hemoprotein Nature. J Biol Chem. 1964;239:2370–8. [PubMed] [Google Scholar]
- 15.Sridar C, Goosen TC, Kent UM, et al. Silybin inactivates cytochromes P450 3A4 and 2C9 and inhibits major hepatic glucuronosyltransferases. Drug Metab Dispos. 2004;32:587–94. doi: 10.1124/dmd.32.6.587. [DOI] [PubMed] [Google Scholar]
- 16.Omary MB, Ku NO, Strnad P, et al. Toward unraveling the complexity of simple epithelial keratins in human disease. J Clin Invest. 2009;119:1794–805. doi: 10.1172/JCI37762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whalen R, Boyer TD. Human glutathione S-transferases. Semin Liver Dis. 1998;18:345–58. doi: 10.1055/s-2007-1007169. [DOI] [PubMed] [Google Scholar]
- 18.Scandlyn MJ, Stuart EC, Rosengren RJ. Sex-specific differences in CYP450 isoforms in humans. Expert Opin Drug Metab Toxicol. 2008;4:413–24. doi: 10.1517/17425255.4.4.413. [DOI] [PubMed] [Google Scholar]
- 19.Zangar RC, Davydov DR, Verma S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol Appl Pharmacol. 2004;199:316–31. doi: 10.1016/j.taap.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 20.Liu YT, Hao HP, Liu CX, et al. Drugs as CYP3A probes, inducers, and inhibitors. Drug Metab Rev. 2007;39:699–721. doi: 10.1080/03602530701690374. [DOI] [PubMed] [Google Scholar]
- 21.Wu D, Cederbaum AI. Oxidative stress and alcoholic liver disease. Semin Liver Dis. 2009;29:141–54. doi: 10.1055/s-0029-1214370. [DOI] [PubMed] [Google Scholar]
- 22.Levy E, Brunet S, Alvarez F, et al. Abnormal hepatobiliary and circulating lipid metabolism in the Long-Evans Cinnamon rat model of Wilson’s disease. Life Sci. 2007;80:1472–83. doi: 10.1016/j.lfs.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 23.Ortiz de Montellano PR, Beilan HS, Kunze KL. N-Alkylprotoporphyrin IX formation in 3,5-dicarbethoxy-1,4-dihydrocollidine-treated rats. Transfer of the alkyl group from the substrate to the porphyrin. J Biol Chem. 1981;256:6708–13. [PubMed] [Google Scholar]
- 24.Correia MA, Sadeghi S, Mundo-Paredes E. Cytochrome P450 ubiquitination: branding for the proteolytic slaughter? Annu Rev Pharmacol Toxicol. 2005;45:439–64. doi: 10.1146/annurev.pharmtox.45.120403.100127. [DOI] [PubMed] [Google Scholar]
- 25.Wienkers LC, Heath TG. Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discov. 2005;4:825–33. doi: 10.1038/nrd1851. [DOI] [PubMed] [Google Scholar]
- 26.Cole SP, Marks GS. Ferrochelatase and N-alkylated porphyrins. Mol Cell Biochem. 1984;64:127–37. doi: 10.1007/BF00224769. [DOI] [PubMed] [Google Scholar]
- 27.Harada M, Hanada S, Toivola DM, et al. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology. 2008;47:2026–35. doi: 10.1002/hep.22294. [DOI] [PubMed] [Google Scholar]
- 28.Jaeschke H, Gores GJ, Cederbaum AI, et al. Mechanisms of hepatotoxicity. Toxicol Sci. 2002;65:166–76. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- 29.Heo SR, Han AM, Kwon YK, et al. p62 protects SH-SY5Y neuroblastoma cells against H2O2-induced injury through the PDK1/Akt pathway. Neurosci Lett. 2009;450:45–50. doi: 10.1016/j.neulet.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Nogalska A, Terracciano C, D’Agostino C, et al. p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol. 2009;118:407–13. doi: 10.1007/s00401-009-0564-6. [DOI] [PubMed] [Google Scholar]
- 31.Olive M, van Leeuwen FW, Janue A, et al. Expression of mutant ubiquitin (UBB+1) and p62 in myotilinopathies and desminopathies. Neuropathol Appl Neurobiol. 2008;34:76–87. doi: 10.1111/j.1365-2990.2007.00864.x. [DOI] [PubMed] [Google Scholar]
- 32.Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10 (Suppl):S18–25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 33.Askanas V, Engel WK. Inclusion-body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition. Neurology. 2006;66:S39–48. doi: 10.1212/01.wnl.0000192128.13875.1e. [DOI] [PubMed] [Google Scholar]
- 34.Pavek P, Dvorak Z. Xenobiotic-induced transcriptional regulation of xenobiotic metabolizing enzymes of the cytochrome P450 superfamily in human extrahepatic tissues. Curr Drug Metab. 2008;9:129–43. doi: 10.2174/138920008783571774. [DOI] [PubMed] [Google Scholar]
- 35.Williams M, Krootjes BB, van Steveninck J, et al. The pro- and antioxidant properties of protoporphyrin IX. Biochim Biophys Acta. 1994;1211:310–6. doi: 10.1016/0005-2760(94)90155-4. [DOI] [PubMed] [Google Scholar]
- 36.Desmet VJ. The amazing universe of hepatic microstructure. Hepatology. 2009;50:333–44. doi: 10.1002/hep.23152. [DOI] [PubMed] [Google Scholar]
- 37.Liem RK, Messing A. Dysfunctions of neuronal and glial intermediate filaments in disease. J Clin Invest. 2009;119:1814–24. doi: 10.1172/JCI38003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest. 2009;119:1806–13. doi: 10.1172/JCI38027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pavlovicova M, Novotova M, Zahradnik I. Structure and composition of tubular aggregates of skeletal muscle fibres. Gen Physiol Biophys. 2003;22:425–40. [PubMed] [Google Scholar]
- 40.Nishikawa T, Takahashi JA, Matsushita T, et al. Tubular aggregates in the skeletal muscle of the senescence-accelerated mouse; SAM. Mech Ageing Dev. 2000;114:89–99. doi: 10.1016/s0047-6374(00)00088-9. [DOI] [PubMed] [Google Scholar]
- 41.Schubert W, Sotgia F, Cohen AW, et al. Caveolin-1(-/-)- and caveolin-2(-/-)deficient mice both display numerous skeletal muscle abnormalities, with tubular aggregate formation. Am J Pathol. 2007;170:316–33. doi: 10.2353/ajpath.2007.060687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuncl RW, Pestronk A, Lane J, et al. The MRL +/+ mouse: a new model of tubular aggregates which are gender- and age-related. Acta Neuropathol. 1989;78:615–20. doi: 10.1007/BF00691288. [DOI] [PubMed] [Google Scholar]
- 43.Rosenberg NL, Neville HE, Ringel SP. Tubular aggregates. Their association with neuromuscular diseases, including the syndrome of myalgias/cramps. Arch Neurol. 1985;42:973–6. doi: 10.1001/archneur.1985.04060090055014. [DOI] [PubMed] [Google Scholar]
- 44.Olive M, Goldfarb L, Dagvadorj A, et al. Expression of the intermediate filament protein synemin in myofibrillar myopathies and other muscle diseases. Acta Neuropathol. 2003;106:1–7. doi: 10.1007/s00401-003-0695-0. [DOI] [PubMed] [Google Scholar]
- 45.Choi YC, Park GT, Kim TS, et al. Sporadic inclusion body myositis correlates with increased expression and cross-linking by transglutaminases 1 and 2. J Biol Chem. 2000;275:8703–10. doi: 10.1074/jbc.275.12.8703. [DOI] [PubMed] [Google Scholar]
- 46.Needham M, Mastaglia FL. Inclusion body myositis: current pathogenetic concepts and diagnostic and therapeutic approaches. Lancet Neurol. 2007;6:620–31. doi: 10.1016/S1474-4422(07)70171-0. [DOI] [PubMed] [Google Scholar]
- 47.Badrising UA, Maat-Schieman M, van Duinen SG, et al. Epidemiology of inclusion body myositis in the Netherlands: a nationwide study. Neurology. 2000;55:1385–7. doi: 10.1212/wnl.55.9.1385. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.