

Abstract
Fyn is 59-kDa member of the Src family of kinases that is historically associated with T-cell and neuronal signaling in development and normal cellular physiology. While Src has been heavily studied in cancer, less attention has been traditionally awarded to the other Src kinases such as Fyn. Our group has shown that Fyn is particularly upregulated in prostate cancer in contrast to the alternative members of the Src family. This suggests that it may mediate a number of important processes attributed to Src kinases in prostate cancer or other malignancies. These functions include not only cellular growth and proliferation, but also include morphogenesis and cellular motility. These together suggest a pivotal role for Fyn in both progression and metastasis. As a number of agents in clinical development affect Fyn activation, understanding the role that Fyn may play in prostate cancer and other malignancies may be of great importance in oncology.
1. Introduction
Prostate cancer is the leading cause of cancer in American men accounting for more than 200,000 new cancer diagnoses this year [1]. While the majority of cases are clinically indolent and/or curable with local treatments, a significant number of men will progress to develop often painful and debilitating metastatic disease. Although androgen deprivation therapy and taxane-based chemotherapy are effective, they are not curative and the 27,000 annual deaths from prostate cancer underscore the need for improved therapies. The field of cancer biology has made strides in identifying a number of molecular events and molecules critical to cancer progression.
Tyrosine kinases are an important class of molecules in human biology and particularly relevant to the field of prostate cancer research. Tyrosine kinases (TKs) fall broadly into 2 categories: receptor and non-receptor TKs. Receptor TKs are membrane bound proteins that receive signals from soluble ligands. These include a variety of molecular targets such as the epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), and mesenchymal epithelial transition factor (c-MET). Examples of non-receptor TKs include members of the following families: Abl, Src, focal adhesion kinase, and the Janus kinase. When activated, these tyrosine kinases activate downstream molecular signals that drive processes crucial to growth and motility of cancer cells. Normally, activation of such molecules is tightly regulated [2]. In cancer, receptor and non-receptor kinase activation is often dysregulated leading to altered cellular growth, shape and function - hallmarks of malignancy [3]. Pharmacologic agents that are able to attenuate this uncontrolled signaling have long been pursued as cancer therapies.
Of the Src family kinases (SFKs), Src is the most studied and hence the most commonly discussed in cancer. However, there has been growing interest in the other SFKs in both physiological and pathological states. The role of Src in cancer is thoroughly reviewed in several publications and will not be reviewed here [4]. Instead, we will focus upon developments in understanding the role of Fyn in various biological processes such as cellular motility and morphogenesis. In addition, we will discuss the potential role of Fyn and SFK inhibitors in prostate cancer therapy.
2. The SRC family kinases
2.1. Overview and history
The Src family kinases (SFKs) are among those non-receptor TKs overexpressed in prostate cancer and have long been proposed as molecular targets for therapy [5]. The prototypical member of this family is c-Src (pp60c-src) - the first discovered oncogene. c-Src was originally described by Rous in the early 1900s. Rous originally described a transforming factor present in tissue of sarcoma bearing chickens that drove the formation of tumors in normal chickens. Injection of a tissue homogenate made from tumor-bearing chickens allowed for transmission of this factor. This tissue factor was later known as the Rous Sarcoma Virus (containing v-src). In 1979, J. Michael Bishop and Harold Varmus discovered that normal cellular Src (c-Src) had the potential to be altered in a fashion that allowed it to drive a cancerous phenotype. Their work in elucidating the mechanism of malignant transformation won them the Nobel Prize in medicine in 1989 and opened the field of oncogenesis. Subsequent proteomic studies led to the identification of other members of an entire family of proteins related to Src, collectively known as the SFKs. These members include Fyn, Src, Yes, Fgf, Lyn, Hck, Blk, Lck and Yrk [2]. The features which identify each protein as a member of this family and which define each members’ unique identity are discussed below.
2.2. SFK Structure and activation
These SFK proteins all share a common structure (Fig. 1) and pattern of activation [2]. The domains of these proteins include SH4, SH3, SH2, and SH1 (kinase) domains followed by a short C-terminal regulatory segment. The SH4 domain is the N-terminal domain and is often myristoylated or palmitoylated to allow for association with the cell membrane. A region known as the unique domain located within the N-terminus specifies the identity of each member of the family. This 60 amino-acid region contains the highest degree of variability and is thought to direct protein-protein binding interactions and hence function for each SFK.
Figure 1.
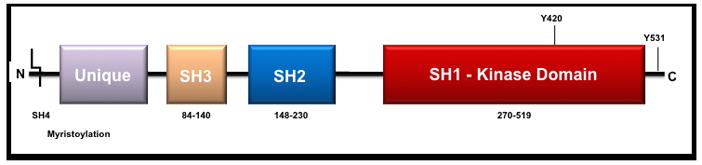
General linear structure of Fyn and the Src-family members.
The SH2 and SH3 domains are highly conserved regions that further mediate protein interactions – the SH2 domain binds phosphotyrosine residues with a general pYEEI sequence while the SH3 domain recognizes PXXP-like sequences. Between the SH2 and SH1 domains is the SH2-kinase linker, which is a loop that functions as a pseudo-SH3-binding site. This domain contains a tyrosine residue (Y416) that is activated by autophosphorylation and is required for optimal activity [6, 7]. The SH1 domain is the site of kinase activity. Following the SH1 domain is a C-terminal regulatory segment. Dephosphorylation of this tyrosine residue (Y527) leads to activation of SFKs via unmasking of an SH1 tyrosine which is also regulated by phophorylation.
The genetic information encoding each member can be quite variable depending on the family member. However, the majority of the genetic and proteomic information is well-preserved with the exception of the unique region. The remainder of this review will focus on the structure, function and role in cancer biology of a specific member of this family, Fyn.
3. Fyn: a brief introduction
3.1. Gene and protein structure
Fyn, (p59-FYN, Slk, Syn, MGC45350, Gene ID 2534) is a 59-kDa protein composed of 537 amino acids whose genetic information is located on chromosome 6q21. Fyn is a member of the Src family originally identified in 1986 as Syn or Slk through probes derived from v-yes and v-fgr [8, 9]. Fyn is primarily localized to the cytoplasmic leaflet of the plasma membrane, where it phosphorylates tyrosine residues on key targets involved in a variety of different signaling pathways.
There are 3 identified transcript isoforms of Fyn. Isoform 1 (isoform a, Fyn(B)) was the first identified and the longest of the 3 genomic sequences. Isoform 2 (isoform b, Fyn(T)) tends to be expressed in T cells and shows a greater ability to mobilize cytoplasmic calcium than isoform 1. [10] These 2 forms differ in the linker region between the SH2 and SH1 domains (exon 7A vs. 7B) accounting for some of the differences in regulation between the two forms [8]. Isoform 2 differs from 1 by approximately 50 amino acids in the region near the end of the SH2 domain and the beginning of the kinase domain. While most tissues express a mixture of the two isoforms [10], Fyn(B) is highly expressed in brain and Fyn(T) is highly expressed in T-cells. Isoform 3 (isoform c) lacking exon 7 (FynΔ7), has been reported. This form has been found in blood cells, but no translated protein has been documented [11]. Additional transcript variations have also been cataloged at this point but have not been associated with a diseased state.
The biological functions of Fyn are diverse (Table 1). Much of the initial work on Fyn centered on its role in immune and neurological function. However, Fyn has also been recognized as an important mediator of mitogenic signaling and regulator of cell cycle entry, growth and proliferation, integrin mediated interactions as well as cell-cell adhesion as will be discussed below.
Table 1.
Biological functions of Fyn
| Function |
|---|
| Growth factor and cytokine receptor signaling |
| Integrin-mediated signaling |
| Cell-cell adhesion |
| Ion channel function |
| Platelet activation |
| T and B cell receptor signaling |
| Axon guidance |
| Fertilization |
| Entry into mitosis |
| Differentiation of natural killer cells, oligodendrocytes and keratinocytes |
3.2. Fyn in cancer
Like Src, overexpression of Fyn has been shown to drive a morphologic transformation in normal cells. Overexpression of Fyn in NIH 3T3 fibroblast cells exhibited a cancer-like phenotype with increased anchorage-independent growth and prominent morphologic changes [12] FYN is overexpressed in various cancers including glioblastoma multiformae, squamous cell carcinoma of the head and neck, and melanoma, [13]. The role of FYN overexpression in these systems, however, has not been well defined.
In addition, our group has shown through a combination of datamining, immunobloting, RT-PCR, and immunohistochemistry that FYN expression is upregulated in the progression to cancer from both normal epithelium and prostate intraepithelial neoplasia (PIN) [14]. In the datamining studies, alternative SRC kinases were explored. LYN, FGR and HCK did not show consistent upregulation in cancer versus normal. There was no significant difference in expression of SRC (P=0.056, P=0.064). LCK, YES and BLK showed strong upregulation in cancer compared to normal epithelium (P=0.00018; 0.00016, 0.019; respectively) but to a lesser degree compared to FYN. The studies on human tissue specimens showed a 2.1-fold increase in FYN expression (P<0.001) in cancer relative to normal prostatic epithelium. Our studies also showed an increase in the signaling partners of Fyn - FAK and paxillin were both upregulated nearly twofold.
4. Biological Functions
4.1. Cell growth and apoptosis
Inhibition of Fyn, like other SFKs, has been associated with decreased cell growth. Expression of kinase-dead-Fyn (KD-Fyn), a specific competitor of endogenous Fyn, reduced primary tumor weights in a mouse squamous cancer model [15]. The PI3K/Akt/PKB is often implicated in cancer cell growth. Fyn and other SFKs are known mediators of growth factor-induced anti-apoptotic activity of Akt/PKB. Knockdown of Fyn, in concert with Src and Yes, resulted in inhibition of Akt activation by EGF [16]. Fyn has been shown to phosphorylate and prevent cleavage of phosphatidylinositol 3-kinase enhancer-activating Akt (PIKE-A), an inhibitor of apoptosis, in HeLa cells [17]. The activation of Fyn has also been shown to be important in prolactin-dependent Akt activation and cell growth [18]. Fyn is thought to relay the anti-apoptotic signals of Akt not only from soluble growth factors but also from interactions downstream of cell-extracellular matrix interactions. Baillat reported that integrin engagement with SW480 cancer cells during early contact with ECM triggered a subset of focal adhesion kinase (FAK) molecules to be recruited to lipid raft domains within the cellular membrane where it interacted with Fyn. Within the lipid raft, Fyn phosphorylation of FAK at Y861 and Y925 lead to FAK recruitment back out of the lipid raft and simultaneous activation of the PI3/Akt pathway [19].
These studies have revealed that the overexpression of Fyn results in promotion of the anti-apoptotic activity of Akt. Although activation of Akt has classically been attributed to inactivation of PTEN, it is becoming apparent that SFKs, such as Fyn, also play a pivotal role. Expectedly, Akt activation is common in many cancers including prostate cancer, and particularly in castration-resistant disease. Higher levels of Akt immunoreactivity in prostate tissue samples have been shown to correlate with higher Gleason scores in prostate cancer [20]. Further studies need to be conducted to elucidate the precise mechanism of how aberrant Fyn function leads to dysregulated Akt activity. Such studies may reveal further novel molecular targets in the treatment of prostate cancer.
4.2. Cell migration
4.2.1. Overview
Understanding how tumor cells interact with and navigate through the extracellular milieu is an important aspect in elucidating how carcinoma in situ progresses to invasive cancers then to metastatic disease. Metastasis is dependent on the ability of cancer cells to migrate and adhere to its local microenvironment. Malignancies of different origins have been shown to use various mechanisms to accomplish this and SFKs play an integral role in the mechanism. In prostate cancer, Fyn and other SFKs have been shown to mediate extracellular interactions driven by various molecules including IL-8, c-Met, EGFR and integrins [5]. Many of these pathways have been shown to be highly dependent on kinase function and constitutive kinase activity contributes to metastatic transformation of prostate cancer. Less is known about the role of Fyn in cell migration and adhesion in cancer, although the growing body of literature as discussed below suggests it may play a prominent role.
4.2.2. Integrins and focal adhesion kinase (FAK)
Integrins are cell surface receptors that interact with the extracellular matrix (ECM) and mediate various intracellular signals that control cellular shape and motility. FAK is a tyrosine kinase recruited to focal adhesion sites and plays a central role in directed cell movement. FAK-mediated cellular motility requires the participation of SFKs [21]. Fyn and Src have been shown to co-immunoprecipitate with FAK [22]. Typically, FAK is recruited to the β-subunit of integrins and following its association with SFK, the SFK-FAK complex formation leads to autophosphorylation at Y397. This complex formation is further activated by various phosphorylation events and such assembly acts as the centerpiece of the cellular machinery coordinating actin fiber formation, focal adhesion formation and ultimately cell shape and motility.
Integrins contribute to cellular motility through the recruitment and activation of several SFK complexes including SFK-FAK and SFK-Shc [19, 23]. The SFK-FAK pathway has been associated with directed chemotaxis while the SFK-Shc pathway has been associated with random haptotaxis [24]. This difference may be related to the means through which each respective pathway impacts rearrangements of actin and cytoskeletal machinery. While FAK activation leads to highly organized actin filaments and focal contacts, Shc activation leads to short actin filaments with fewer focal contacts. It is thought that fine regulation between these two pathways by SFKs as well as other regulator molecules result in normal physiological cellular movement. Although the exact mechanisms behind this observation have not been completely understood, aberration of Fyn/SFK function presumptively leads to dysfunctional cellular movement. Thus, integrins and FAK appear to play key roles in the mediation of Fyn transmitted cellular events impacting shape and motility.
4.2.3. Rac and Rho family GTPases
Downstream of the SFK-FAK activation, a number of molecules affecting cell migration are activated including JUN, nuclear factor κB (NF-κB), B-raf, GEF and Akt/PKB. Therefore, dysfunctional Fyn has the potential to interact with multiple motility effectors. A family of major pathways of interest is the Rho family of GTPases, a subfamily of the of the Ras superfamily. These proteins have been shown to regulate many aspects of intracellular actin dynamics and include Rac1, RhoA and Cdc42. Interactions between Fyn and the Rho family GTPases have been shown to control morphologic differentiation of cells such as oligodendrocytes [25].
RhoA affects stress fiber formation and Cdc42 has been associated with filopodia formation. Rac1 has been shown to control cell motility, affecting actin reorganization at the leading edges of cells. Fyn-deficient (Fyn−/−) mast cells showed a significant defect in cell spreading and lamellipodia formation on fibronectin. In addition, Rac-activation assays showed that Fyn promotes activation of Rac GTPase under stem cell factor (SCF) stimulation [26]. Following αv-integrin stimulation, PTEN has been shown to directly deactivate Fyn leading to downstream regulation of Rac-GTPase activity as described above [27]. Strong interplay between Fyn and the Rho family GTPases such as Rac1 suggests that this may represent another important pathway through which Fyn exerts its effects on cellular shape and motility.
4.2.4. Ras, Erk, MAPKs
While many integrins couple to FAK through their β-subunits as described above, certain integrins, including α5β1, α1β1, α6β4 and αvβ3 are known to couple through their α subunits to the Ras-extracellular signal-regulated kinase (ERK) signaling pathway via Shc and palmitoylated SFKs, such as Fyn and Yes [29, 30]. In this pathway, caveolin-1 functions as a transmembrane adaptor to facilitate the recruitment of SFKs [31]. A palmitoylated SFK then binds, via its SH3 domain, to Shc leading to phosphorylation of Shc at Tyr317 forming an activated complex. The activated complex then combines with GRB2–SOS to activate ERK/MAPK signaling via Ras. Activation of this pathway results in increased cell motility and progression through the G1 stage of the cell cycle in response to mitogens driving cellular growth. This process ties cellular adhesion to cell cycle progression in a process known as anchorage-dependent cell growth. Normal cells need to adhere to serum-derived extracellular matrix components for cell growth in vitro whereas in malignant cells, this requirement is bypassed. Overexpression of Fyn therefore can contribute to dysregulated anchorage-dependent cell growth [31]. Evidence for Fyn involvement in this pathway is supported by the fact that PP1, an SFK inhibitor, will inhibit Fyn over Src at lower concentrations thus preventing the malignant transformation of oncogenic Ras mutants such as v-Ha-Ras [32]. The proposed mechanism for this is inhibition of PAK, a serine/threonine kinase required for malignant transformation of v-Ha-Ras and a key regulator of anchorage-dependent cell growth.
4.3. Cell adhesion, invasion and EMT
Fyn has also been shown to play a role in sensing and responding to the rigidity of extracellular matrix surfaces. The generation of sheer force on rigid cell-matrix interfaces results in recruitment of various focal adhesion proteins leading to increased cell adhesion and cell spreading. Receptor-like protein tyrosine phosphatase-α (RPTP-α) and αvβ3 integrin form a complex at the leading edge of a migrating cell in an ECM rigidity-dependent manner that results in recruitment and activation of Fyn [33, 34]. This recruitment is dependent on the proper functioning of the palmitoylation site on Fyn and the level of Fyn activation is thought to be force-dependent in which greater forces result in greater reinforcement of integrin-cytoskeleton linkages. Malignancies may, in part, spread aggressively due to overexpression of Fyn causing an exaggerated sensing response to the rigidity the extracellular matrix.
Epithelial-mesenchymal transition (EMT) is the process in which cells convert from an epithelial to a mesenchymal phenotype. Key features of EMT include loss of cell adhesion, a switch from E-cadherin to N-cadherin expression, and an increase in cell motility [35]. Furthermore, a characteristic upregulation of the neural cell adhesion molecule (NCAM) expression is also commonly known. EMT is necessary for a number of physiologic processes during development but is also seen in progression from localized cancer to metastatic disease.
Fyn has been reported to play a role in EMT. Recently, Lehembre suggested that at low concentrations of NCAM, a series of events occur including a complex formation outside of lipid rafts between NCAM and FGFR, the downstream activation of the MAPK pathway as well as sustained levels of cellular adhesion [36]. However, in EMT, the loss of E-cadherin results in the overexpression of NCAM leading to its relocalization into lipid rafts. This event, in turn, results in increased motility due to association and activation of Fyn together with the downstream activation of FAK.
Matrix metalloproteinases (MMPs) are regulators of the interface between epithelial cells and their underlying ECM. Dysregulation of MMP function is commonly observed during metastatic progression as they facilitate invasion into metastatic sites by degrading the ECM in pathologic states [37]. β6 integrin has also been demonstrated to directly phosphorylate and activate Fyn. This results in downstream upregulation of matrix metalloproteinase-3 (MMP-3) leading to increased cell proliferation and progression to metastatic disease in vivo [15]. Taken together, these findings suggest a role for Fyn as a mediator of metastatic progression of disease apart from local tumor growth.
5. SFK inhibitors in preclinical and clinical models
Tyrosine kinase inhibitors PP1 and PP2 are the earliest reported SFK-selective tyrosine kinase inhibitors. They have been extremely important in elucidating the role of SFK in signal transduction [38]. Since then, a number of signal transduction inhibitors have been synthesized and are now being brought forward into clinical studies (table 2).
Table 2.
SRC kinase inhibitors
| Name | Reference | Manufacturer | Comments |
|---|---|---|---|
| Clinically studied | |||
| Dasatinib | [46] | Bristol-Myers-Squibb | FDA approved for imitinib-resistant CML |
| AZD0530 | [47] | Astra Zeneca | In phase I/II clinical studies |
| Bosutinib (SKI-606) | [48] | Wyeth | Phase I |
| KX2-391 | [43] | Kinex | Phase I/II clinical studies |
| Preclinical only | |||
| PP1 | [38] | Not usable clinically | |
| PP2 | [38] | Not usable clinically | |
| AP23846 | [49] | ARIAD | |
| Herbimycin A | benzochinoid antibiotic related to geldanamycin | ||
| CGP76030 | [50] | Novartis | |
| 1l (Nbenzyl- 1-(2-chloro-2-phenylethyl)-1H-pyrazolo[3,4-d]pyrimidin- 4-amine | [51] | ||
| 7-(2,6-dichlorophenyl)-5-methylbenzo [1,2,4]triazin-3-yl]- [4-(2-pyrrolidin-1-ylethoxy)phenyl]-amine | [17] | TargeGen, WuXi PharmaTech | |
Agents targeted specifically against Fyn have not been developed clinically at this time. However, SFK inhibitors known to inhibit Fyn activation have been tested in pre-clinical and clinical models. Dasatinib is a FDA approved and commercially available SRC/ABL inhibitor [39] that impairs cell migration [40] and inhibits FAK and p130CAS phosphorylation in DU145 and LNCaP prostate cancer cell lines. This may be attributed to the effects of FAK and p130CAS on integrin interaction. Another SFK inhibitor, AZD0530, inhibits growth by inducing G1-arrest in 22Rv1, DU145, LAPC-4, LNCaP, and PC3 prostate cancer cell lines [41]. DU145 cells treated with AZD0530 showed decreased invasion in a Boyden chamber assay and decreased FAK and p130CAS phosphorylation. The investigators in both studies did not specifically determine which SFKs were responsible for the observed phenomenon. In fact, this is a particularly difficult distinction to establish as the typically utilized pSRC (Y419) antibody crossreacts with all active SFKs. Both studies suggested a strong correlation with activation of FAK, a known binding partner of Fyn. Given this and the high relative expression of Fyn in prostate cancer models, it is reasonable to hypothesize that Fyn is the major regulator of these processes.
Further work targeted at understanding the role of dasatinib in prostate cancer has been pursued. Park showed that in a murine orthotopic metastasis model, the use of dasatinib was associated with both decreased activation of Src and Lyn and resulted in decreased lymph node metastases from PC3-M cells [42]. This same group showed similar findings with the novel SFK inhibitor, KX2-391 [43]. Interestingly, Park’s work shows that Lyn and Src function differently in that Lyn regulated metastasis apart from growth whereas Src regulated growth apart from metastasis. The investigators, however, did not query other SFKs such as Fyn to determine what role Fyn may have played in this behavior.
Both dasatinib and AZD0530 have been studied as single agent therapies for castrate resistant prostate cancer (CRPC). Our group participated in a study reported by Lara [44] which was a single agent phase II clinical study of AZD0530 based on the preclinical data showing the inhibition of growth and migration described above. The study was powered to detect a PSA response rate of 15% or greater. None of the patients treated exhibited such a response by PSA. However, it is important to note that the trial was not designed to look at alternative outcomes such as new metastasis which would be may be more relevant to SFK inhibition.
In addition to inhibiting Fyn, dasatinib also inhibits other SFKs such as LCK and SRC. Our group also participated in a single agent phase II study in chemotherapy naïve patients with CRPC reported by Yu [45]. This study showed that the disease control rate for 15 RECIST- evaluable patients was 67% (10 had stable disease). Of 27 patients with bone scans at 12 weeks, 16 were stable and 1 was improved. Two of 5 patients with greater than 2 bone scans at 24 weeks had stable disease. An improved PSA doubling time was seen in 29 of 36 patients (80.1%). The mode of action of dasatinib in this population is not as clear, but the effects reported are more consistent with what is recognized in the role that SFKs such as Fyn may play in prostate cancer. Collectively, these data support ongoing evaluation of SFK inhibitors in prostate cancer.
6. Conclusion
While Src has long been recognized as an important oncogene, little attention has been given to its family members such as Fyn, which is likely to be more relevant than cSrc in prostate cancer. Our initial work shows a particular upregulation of Fyn in prostate cancer. Given the above data showing expression and putative role for Fyn in prostate cancer progression and with the availability of pharmacologic agents to manipulate this target, it is reasonable and timely to test the utility of this molecular target. The additional information, from ongoing studies of Fyn in prostate cancer clarify the role it plays in the disease process to optimize Fyn-directed therapeutics. Regretfully, pharmacologic developments of SFK inhibitors have focused upon inhibition of cSrc rather than other SFKs that may be more relevant to human disease such as Fyn. As more is learned of the specific role that Fyn plays in human disease, we hope that agents specifically targeted at Fyn may be advanced in preclinical and clinical development.
Figure 2.
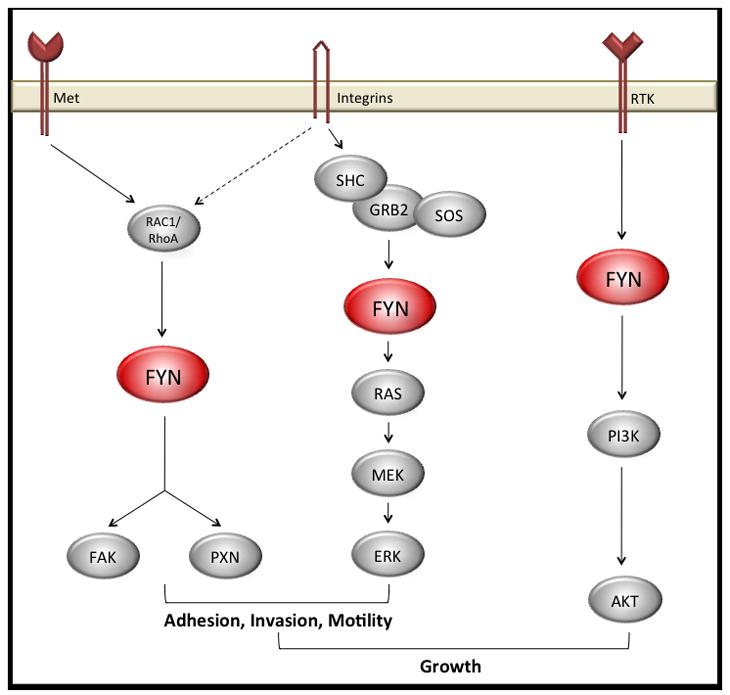
FYN is mediates signals from cell surface receptors to a number of critical growth and motility pathways.
References
- 1.Jemal A, et al. Cancer statistics, 2007. CA Cancer J Clin. 2007;57(1):43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23(48):7918–27. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 3.Vlahovic G, Crawford J. Activation of tyrosine kinases in cancer. Oncologist. 2003;8(6):531–8. doi: 10.1634/theoncologist.8-6-531. [DOI] [PubMed] [Google Scholar]
- 4.Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4(6):470–80. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 5.Chang YM, Kung HJ, Evans CP. Nonreceptor tyrosine kinases in prostate cancer. Neoplasia. 2007;9(2):90–100. doi: 10.1593/neo.06694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson LN, Noble ME, Owen DJ. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85(2):149–58. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 7.Cowan-Jacob SW, et al. The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure. 2005;13(6):861–71. doi: 10.1016/j.str.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 8.Davidson D, Fournel M, Veillette A. Oncogenic activation of p59fyn tyrosine protein kinase by mutation of its carboxyl-terminal site of tyrosine phosphorylation, tyrosine 528. J Biol Chem. 1994;269(14):10956–63. [PubMed] [Google Scholar]
- 9.Semba K, et al. yes-related protooncogene, syn, belongs to the protein-tyrosine kinase family. Proc Natl Acad Sci U S A. 1986;83(15):5459–63. doi: 10.1073/pnas.83.15.5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 11.Goldsmith JF, Hall CG, Atkinson TP. Identification of an alternatively spliced isoform of the fyn tyrosine kinase. Biochem Biophys Res Commun. 2002;298(4):501–4. doi: 10.1016/s0006-291x(02)02510-x. [DOI] [PubMed] [Google Scholar]
- 12.Kawakami T, et al. Acquisition of transforming properties by FYN, a normal SRC-related human gene. Proc Natl Acad Sci U S A. 1988;85(11):3870–4. doi: 10.1073/pnas.85.11.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ONCOMINE database
- 14.Posadas EM, et al. BJ Urol Intl. 2008. FYN is overexpressed in human prostate cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, et al. Alphavbeta6-Fyn signaling promotes oral cancer progression. J Biol Chem. 2003;278(43):41646–53. doi: 10.1074/jbc.M306274200. [DOI] [PubMed] [Google Scholar]
- 16.Chen R, et al. Regulation of Akt/PKB activation by tyrosine phosphorylation. J Biol Chem. 2001;276(34):31858–62. doi: 10.1074/jbc.C100271200. [DOI] [PubMed] [Google Scholar]
- 17.Noronha G, et al. Discovery of [7-(2,6-dichlorophenyl)-5-methylbenzo [1,2,4]triazin-3-yl]-[4-(2-pyrrolidin-1-ylethoxy)phenyl]amine--a potent, orally active Src kinase inhibitor with anti-tumor activity in preclinical assays. Bioorg Med Chem Lett. 2007;17(3):602–8. doi: 10.1016/j.bmcl.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 18.Fresno Vara JA, et al. Src family kinases are required for prolactin induction of cell proliferation. Mol Biol Cell. 2001;12(7):2171–83. doi: 10.1091/mbc.12.7.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baillat G, et al. Early adhesion induces interaction of FAK and Fyn in lipid domains and activates raft-dependent Akt signaling in SW480 colon cancer cells. Biochim Biophys Acta. 2008;1783(12):2323–31. doi: 10.1016/j.bbamcr.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 20.Liao Y, et al. Increase of AKT/PKB expression correlates with gleason pattern in human prostate cancer. Int J Cancer. 2003;107(4):676–80. doi: 10.1002/ijc.11471. [DOI] [PubMed] [Google Scholar]
- 21.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6(1):56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 22.Cary LA, Chang JF, Guan JL. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J Cell Sci. 1996;109( Pt 7):1787–94. doi: 10.1242/jcs.109.7.1787. [DOI] [PubMed] [Google Scholar]
- 23.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5(10):816–26. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 24.Gu J, et al. Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J Cell Biol. 1999;146(2):389–403. doi: 10.1083/jcb.146.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang X, Draghi NA, Resh MD. Signaling from integrins to Fyn to Rho family GTPases regulates morphologic differentiation of oligodendrocytes. J Neurosci. 2004;24(32):7140–9. doi: 10.1523/JNEUROSCI.5319-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samayawardhena LA, Kapur R, Craig AW. Involvement of Fyn kinase in Kit and integrin-mediated Rac activation, cytoskeletal reorganization, and chemotaxis of mast cells. Blood. 2007;109(9):3679–86. doi: 10.1182/blood-2006-11-057315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dey N, et al. The protein phosphatase activity of PTEN regulates SRC family kinases and controls glioma migration. Cancer Res. 2008;68(6):1862–71. doi: 10.1158/0008-5472.CAN-07-1182. [DOI] [PubMed] [Google Scholar]
- 28.Vacaresse N, et al. Activation of c-Src and Fyn kinases by protein-tyrosine phosphatase RPTPalpha is substrate-specific and compatible with lipid raft localization. J Biol Chem. 2008;283(51):35815–24. doi: 10.1074/jbc.M807964200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mettouchi A, et al. Integrin-specific activation of Rac controls progression through the G(1) phase of the cell cycle. Mol Cell. 2001;8(1):115–27. doi: 10.1016/s1097-2765(01)00285-4. [DOI] [PubMed] [Google Scholar]
- 30.Mariotti A, et al. EGF-R signaling through Fyn kinase disrupts the function of integrin alpha6beta4 at hemidesmosomes: role in epithelial cell migration and carcinoma invasion. J Cell Biol. 2001;155(3):447–58. doi: 10.1083/jcb.200105017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wary KK, et al. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell. 1998;94(5):625–34. doi: 10.1016/s0092-8674(00)81604-9. [DOI] [PubMed] [Google Scholar]
- 32.He H, et al. An anti-Ras cancer potential of PP1, an inhibitor specific for Src family kinases: in vitro and in vivo studies. Cancer J. 2000;6(4):243–8. [PubMed] [Google Scholar]
- 33.Jiang G, et al. Rigidity sensing at the leading edge through alphavbeta3 integrins and RPTPalpha. Biophys J. 2006;90(5):1804–9. doi: 10.1529/biophysj.105.072462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kostic A, Sheetz MP. Fibronectin rigidity response through Fyn and p130Cas recruitment to the leading edge. Mol Biol Cell. 2006;17(6):2684–95. doi: 10.1091/mbc.E05-12-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vernon AE, LaBonne C. Tumor metastasis: a new twist on epithelial-mesenchymal transitions. Curr Biol. 2004;14(17):R719–21. doi: 10.1016/j.cub.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 36.Lehembre F, et al. NCAM-induced focal adhesion assembly: a functional switch upon loss of E-cadherin. EMBO J. 2008;27(19):2603–15. doi: 10.1038/emboj.2008.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25(1):9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- 38.Hanke JH, et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271(2):695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 39.Lombardo LJ, et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2- hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47(27):6658–61. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 40.Nam S, et al. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 2005;65(20):9185–9. doi: 10.1158/0008-5472.CAN-05-1731. [DOI] [PubMed] [Google Scholar]
- 41.Chang YM, et al. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene. 2008;27(49):6365–75. doi: 10.1038/onc.2008.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park SI, et al. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 2008;68(9):3323–33. doi: 10.1158/0008-5472.CAN-07-2997. [DOI] [PubMed] [Google Scholar]
- 43.Park SI, Kopetz S, Gallick GE. A novel Src family kinase inhibitor (KX2-391) targeting the peptide substrate binding domain inhibits prostate cancer growth and lymph node metastasis in vivo. Proceedings of the 100th Annual Meetings of the American Assocaition for Cancer Research; 2009; Denver, CO. [Google Scholar]
- 44.Lara PN, Jr, et al. A phase II trial of the Src-kinase inhibitor AZD0530 in patients with advanced castration-resistant prostate cancer: a California Cancer Consortium study. Anticancer Drugs. 2009;20(3):179–84. doi: 10.1097/CAD.0b013e328325a867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu EY, Wilding EPG, Gross M, Culine S, Massard C, Hudes GR, Cheng S, Paliwal P, Sternberg CN. J Clin Oncol. suppl. Vol. 26. 2008. May 20, Dasatinib in patients with hormone-refractory progressive prostate cancer: A phase II study; p. 2008. abstr 5156. [Google Scholar]
- 46.Talpaz M, et al. Dasatinib in imatinib-resistant Philadelphia chromosome- positive leukemias. N Engl J Med. 2006;354(24):2531–41. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 47.Tabernero J, et al. Phase I study of AZD0530, an oral potent inhibitor of Src kinase: First demonstration of inhibition of Src activity in human cancers. Journal of Clinical Oncology; 2007 ASCO Annual Meeting Proceedings Part I; 2007. p. 3520. [Google Scholar]
- 48.Messersmith WA, et al. Bosutinib (SKI-606), a dual Src/Abl tyrosine kinase inhibitor: Preliminary results from a phase 1 study in patients with advanced malignant solid tumors. 2007 ASCO Annual Meeting Procedings part I; 2007. p. 3552. [Google Scholar]
- 49.Summy JM, et al. AP23846, a novel and highly potent Src family kinase inhibitor, reduces vascular endothelial growth factor and interleukin-8 expression in human solid tumor cell lines and abrogates downstream angiogenic processes. Mol Cancer Ther. 2005;4(12):1900–11. doi: 10.1158/1535-7163.MCT-05-0171. [DOI] [PubMed] [Google Scholar]
- 50.Morinaga K, et al. Overcoming imatinib resistance using Src inhibitor CGP76030, Abl inhibitor nilotinib and Abl/Lyn inhibitor INNO-406 in newly established K562 variants with BCR-ABL gene amplification. Int J Cancer. 2008;122(11):2621–7. doi: 10.1002/ijc.23435. [DOI] [PubMed] [Google Scholar]
- 51.Donnini S, et al. Pyrazolo-pyrimidine-derived c-Src inhibitor reduces angiogenesis and survival of squamous carcinoma cells by suppressing vascular endothelial growth factor production and signaling. Int J Cancer. 2007;120(5):995–1004. doi: 10.1002/ijc.22410. [DOI] [PubMed] [Google Scholar]