

Abstract
The application of gas chromatography–mass spectrometry (GC–MS) to the ‘global’ analysis of metabolites in complex samples (i.e. metabolomics) has now become routine. The generation of these data-rich profiles demands new strategies in data mining and standardisation of experimental and reporting aspects across laboratories. As part of the META-PHOR project’s (METAbolomics for Plants Health and OutReach: http://www.meta-phor.eu/) priorities towards robust technology development, a GC–MS ring experiment based upon three complex matrices (melon, broccoli and rice) was launched. All sample preparation, data processing, multivariate analyses and comparisons of major metabolite features followed standardised protocols, identical models of GC (Agilent 6890N) and TOF/MS (Leco Pegasus III) were also employed. In addition comprehensive GC×GC–TOF/MS was compared with 1 dimensional GC–TOF/MS. Comparisons of the paired data from the various laboratories were made with a single data processing and analysis method providing an unbiased assessment of analytical method variants and inter-laboratory reproducibility. A range of processing and statistical methods were also assessed with a single exemplary dataset revealing near equal performance between them. Further investigations of long-term reproducibility are required, though the future generation of global and valid metabolomics databases offers much promise.
Keywords: Metabolomics, GC–MS, GC×GC–MS, Inter-laboratory reproducibility, Rice, Melon, Broccoli
Introduction
Gas chromatography–electron impact–mass spectrometry (GC–EI–MS) has been an established technique for several decades. However, its application to ‘global’ metabolite analysis in complex samples has only become routine in the past 10 years of plant science (Fiehn et al. 2000a), and perhaps more recently for animal studies (Dunn 2008), although biofluid analysis first occurred in the 1960s (Horning 1968; Pauling et al. 1971). GC–EI–MS profiling has been greatly facilitated by high data acquisition rate GC–EI–time of flight (TOF)/MS and reproducible derivatisation procedures suited to polar metabolites (Roessner et al. 2000). Since the recognition of Sauter et al. (1988) groundbreaking work on herbicide mode of action, Max Plank Institute of Molecular Plant Physiology have strived to update the method establishing robust SOP’s using first quadrupole and later TOF based GC–EI–MS (Fiehn et al. 2000a, b; Fernie et al. 2004; Lisec et al. 2006; Erban et al. 2007). TOF mass analysers give increased sensitivity and very data-rich metabolite profiles, which subsequently demands new strategies in data mining. Standard operating procedures (SOP) are well established for targeted methods however there is a need for standardisation across laboratories for all aspects of metabolomics work. Suggestions have recently been made by the metabolomics standards initiative (MSI; Fiehn et al. 2007a) which is developing minimal reporting standards in data generation (Fiehn et al. 2008), exchange (Jenkins et al. 2004; Hardy and Taylor 2007), analysis (Goodacre et al. 2007) and reporting (Fiehn et al. 2007b; Sumner et al. 2007).
Food quality traits such as fragrance, taste, appearance, shelf-life and nutritional content are determined by their biochemical composition and thus reflected in their metabolite profiles (Hall 2006, 2007). Metabolomics has proven to be an appropriate tool for the extensive analysis of plant and food composition (Dixon et al. 2006; Schauer and Fernie 2006). META-PHOR (http://www.meta-phor.eu/) (Hall 2007; Hall et al. 2008) aims at developing technological platforms and associated methods to provide a tool to monitor food nutritional quality and safety, whilst adhering to all work guidelines of the MSI. Three target species were selected; melon for its matrix complexity and dominance of sugars and the analytical challenges which result, broccoli for its extreme complexity and metabolite richness (especially ‘nutraceuticals’), and the rice grain due to its position as the major staple food.
As part of the META-PHOR project priority towards technology development (Hall 2007) a series of ring experiments comparing, proton-nuclear magnetic resonance (1H-NMR), liquid chromatography (LC)–MS, and GC–EI–TOF/MS have been initiated. The GC–EI–TOF/MS ring experiment was undertaken by the University of Manchester UK (UMAN), Max Plank Institute of Molecular Plant Physiology, Golm DE (MPIMP), and LECO Instruments Mönchengladbach DE (LECO). Each of these groups had SOP’s established for variants of an initial analytical methodology (Fiehn et al. 2000a) largely resulting from the different research activities each focuses upon. The UMAN method was optimised for primary metabolite detection in yeast media whilst maintaining analysis times of less than 20 min (O’Hagan et al. 2005). The MPIMP method was optimised towards maintaining maximum metabolite coverage with polar extracts from plants (Lisec et al. 2006; Erban et al. 2007). The LECO GC–EI–TOF/MS method was optimised for maximal metabolite coverage regardless of the sample matrix.
The ring experiment study design included a standardised protocol (Erban et al. 2007) for sample preparation (Fiehn et al. 2000a) and multivariate analyses, i.e. principal components analysis (PCA) and independent component analysis (ICA) and comparisons of major metabolite features. PCA is a statistical technique for sample classification which reduces multivariate data sets to a small number of variables (PCs) which comprise the major variances in the data set (Jolliffe 1986). ICA is a variant of PCA which additionally allows the unsupervised search for best bimodal sample partitions. ICA is well suited to the confirmation of known experimental sample classes but allows also the discovery of unexpected classes or trends (Stone 2002; Scholz et al. 2004; Scholz et al. 2005; Trygg et al. 2006). Each independent component (IC) encodes a single partition among samples from which a loadings analysis unravels which signals are most relevant for the distinction of the embedded sample partitions. Since PCA and ICA do not use sample class information they are so-called unsupervised methods and thereby are ideal for non-biased reproducibility analysis.
To the best of the authors’ knowledge, this is one of the first ring experiments in the metabolomics field to concentrate upon reproducibility of major differential metabolite features suitable for food sample classifications from a common set of extracts by GC–EI–TOF/MS. By making comparisons of the different laboratories data with ICA, reproducibility can be demonstrated for the unambiguous discrimination of the three plant matrices, indicating that the short-term inter-laboratory reproducibility of GC–EI–TOF/MS based metabolomics is high and thus has great promise for the current efforts being made towards the generation of global metabolomics databases.
Methods
Plant materials
The French melon varieties, Cucumis melo cv. Cézanne and Escrito, were commercial F1 hybrids. Seeds were obtained from Clause-Tézier (FR). Plants were grown by the French National Institute for Agricultural Research (INRA) in an open field in the South-West of France (Moissac, Bordeaux, 44° N × 1° E) between April and August 2006. The soil type was clay and limestone, the plant density was 9,200 plants/ha. The Cézanne cultivar, but not Escrito, was protected with a polyethylene sheet. The Israeli melon varieties, C. melo cv. Noy Yize’el and Tam Dew, were obtained from the germplasm collection at the Agricultural Research Organisation (ARO), Volcani Centre (IL). Plants were grown in a standardised green house (32° N × 35° E) between June and September 2006. The soil type was volcanic tuff and peat (1:1), the plant density was 20,000 plants/ha. French broccoli cultivars, Brassica oleracea cv. Monaco and Chevalier (seed obtained from Syngenta and Seminis (FR) respectively) were grown by INRA in an open field (Toull lan, Bordeaux, 48° N × 3° E) between June and September 2006. The soil type was Eolian silt (12% clay, 16% fine silt, 44% coarse silt, 24% fine sand), the plant density was 2,500 plants/ha. Rice cultivars, Oryza sativa cv. Hom Nang Nouane (HNN), Kay Noy (KNL) and TSN1 seed stocks were obtained from the International Rice Research Institute (IRRI) the Philippines and grown by the Laos National Agricultural Research Centre (NARC) in open paddy fields in the Saythany District of Vientiane (17° N × 102° E) from 1st September until 1st December 2006, the soil type was clay. Fertilisation involved nitrogen supplementation at three time points (0, 4, and 8 weeks) throughout the three month growth period. Four different nitrogen fertilisation regimes (0–30–30 kg/ha; 30–30–30 kg/ha; 60–30–30 kg/ha; 90–30–30 kg/ha) were applied to separated plots for each rice cultivar. For all species (unless otherwise detailed), irrigation, watering, fertilisation and pathogen–pest control were performed according to commercial practices.
For each cultivar, 50 melons were harvested at commercial maturity between July and August 2006 (French varieties) and August and September 2006 (Israeli varieties). Broccoli florets were harvested in mid September 2006. Both melons and broccoli were transported in insulated boxes and upon arrival processed within 2 h. For each cultivar, 36 fruits or 1.5 kg of floret were selected depending on the size, weight and colour in order to make three homogeneous lots (biological replicates) of 11 fruits or 1.5 kg of pooled floret each. For every biological replicate, fruits and florets were rapidly washed for 1–2 min with tap water (~10°C) and air dried. One quarter of each melon was taken, the skin was removed and the flesh cut in 2 cm × 2 cm cubes, the broccoli floret was also cut into small pieces, the samples were then flash frozen in liquid nitrogen. All samples were next ground (UMC5 grinder, STEPHANTM, Lognes, FR) to a homogeneous fine powder. When the rice grain had reached 22% moisture (December 2006), the panicles were harvested and threshed, 1 kg of grain was collected per cultivar and per nitrogen treatment. The grain was equilibrated at room temperature for six weeks to reduce variability in moisture content. For each of the 12 biological samples, the rice grain was ground for ~30 s in an IKA grinder A11 basic (Staufen, DE) fitted with a metallic cup to which liquid nitrogen was added, ensuring the material remained frozen. The fine rice flour was further flash frozen in liquid nitrogen. Ground samples for all species were immediately shipped on dry ice and stored at −80°C on receipt. Sample extraction was undertaken within three months of sample receipt. A full list of the samples analysed is provided (Table 1).
Table 1.
Sample details
| Latin name | Species | Nitrogen supplementation | Bilogical replicate |
|---|---|---|---|
| Cucumis melo var. cantaloupensis Noy Yizre’el | Melon (IL) | NA | 1 |
| Cucumis melo var. cantaloupensis Noy Yizre’el | Melon (IL) | NA | 2 |
| Cucumis melo var. cantaloupensis Noy Yizre’el | Melon (IL) | NA | 3 |
| Cucumis melo var. inodorous Tam Dew | Melon (IL) | NA | 1 |
| Cucumis melo var. inodorous Tam Dew | Melon (IL) | NA | 2 |
| Cucumis melo var. inodorous Tam Dew | Melon (IL) | NA | 3 |
| Cucumis melo var. cantaloupensis Cezanne | Melon (FR) | NA | 1 |
| Cucumis melo var. cantaloupensis Cezanne | Melon (FR) | NA | 2 |
| Cucumis melo var. cantaloupensis Cezanne | Melon (FR) | NA | 3 |
| Cucumis melo var. cantaloupensis Escrito | Melon (FR) | NA | 1 |
| Cucumis melo var. cantaloupensis Escrito | Melon (FR) | NA | 2 |
| Cucumis melo var. cantaloupensis Escrito | Melon (FR) | NA | 3 |
| Brassica oleracea Botrytis cymosa var. Chevalier | Broccoli (FR) | NA | 1 |
| Brassica oleracea Botrytis cymosa var. Chevalier | Broccoli (FR) | NA | 2 |
| Brassica oleracea Botrytis cymosa var. Chevalier | Broccoli (FR) | NA | 3 |
| Brassica oleracea Botrytis cymosa var. Monaco | Broccoli (FR) | NA | 1 |
| Brassica oleracea Botrytis cymosa var. Monaco | Broccoli (FR) | NA | 2 |
| Brassica oleracea Botrytis cymosa var. Monaco | Broccoli (FR) | NA | 3 |
| Oryza sativa cv. Hom Nang Nouane (HNN) | Rice | Nitrogen 00–30–30 kg/ha (3 treatments) | 1 |
| Oryza sativa cv. Hom Nang Nouane (HNN) | Rice | Nitrogen 30–30–30 kg/ha (3 treatments) | 1 |
| Oryza sativa cv. Hom Nang Nouane (HNN) | Rice | Nitrogen 60–30–30 kg/ha (3 treatments) | 1 |
| Oryza sativa cv. Hom Nang Nouane (HNN) | Rice | Nitrogen 90–30–30 kg/ha (3 treatments) | 1 |
| Oryza sativa cv. Kay Noy (KNL) | Rice | Nitrogen 00–30–30 kg/ha (3 treatments) | 1 |
| Oryza sativa cv. Kay Noy (KNL) | Rice | Nitrogen 30–30–30 kg/ha (3 treatments) | 1 |
| Oryza sativa cv. Kay Noy (KNL) | Rice | Nitrogen 60–30–30 kg/ha (3 treatments) | 1 |
| Oryza sativa cv. Kay Noy (KNL) | Rice | Nitrogen 90–30–30 kg/ha (3 treatments) | 1 |
| Oryza sativa cv. TSN1 | Rice | Nitrogen 00–30–30 kg/ha (3 treatments) | 1 |
| Oryza sativa cv. TSN1 | Rice | Nitrogen 30–30–30 kg/ha (3 treatments) | 1 |
| Oryza sativa cv. TSN1 | Rice | Nitrogen 60–30–30 kg/ha (3 treatments) | 1 |
| Oryza sativa cv. TSN1 | Rice | Nitrogen 90–30–30 kg/ha (3 treatments) | 1 |
Chemicals
UMAN obtained succinic-d4 acid, glycine-d5 and malonic-d2 acid standard metabolites (all of 99% purity or greater: 1:1:1 working stock of each standard of a final concentration of 0.5 mg/ml), along with all solvents (HPLC grade), O-methylhydroxylamine chloride, N-acetyl-N(trimethylsilyl)-trifluoroacetamide, pyridine and n-alkane time series from Sigma-Aldrich (Gillingham, UK). LECO and MPIMP obtained O-methylhydroxylamine chloride and n-alkane series from Sigma-Aldrich (Deisenhofen, DE), N-acetyl-N-(trimethylsilyl)-trifluoroacetamide from Macherey-Nagel (Düren, DE), and pyridine from Merck (Darmstadt, DE). The use of solvents and reagents from different manufacturers and locations represents a realistic evaluation of laboratory-to-laboratory robustness.
Sample extraction
Since the ring experiment was focused on an evaluation of data-acquisition and processing methods all extractions were conducted by a single laboratory and technician. The extraction procedure precisely followed that of Lisec et al. (2006), which was developed from the protocol of Fiehn et al. (2000a). Briefly, metabolites were extracted from 100 mg fresh weight (FW) for all plant tissue types with methanol and water. Polar metabolites were separated using chloroform purification. Three technical repeat samples each were combined and mixed well giving ~7 ml of polar phase ‘super’-extract, 1 ml was then transferred to clean 2 ml microcentrifuge tubes (Greiner Bio-One Ltd., Stonehouse, Glos., UK) to which 100 μl of the fore mentioned deuterated internal standard solution (cf. Sect. 2.2) was added. Samples were dried by vacuum centrifugation, Eppendorf Concentrator 5301, set on function 1 at 30°C for 8 h and stored at −80°C. The only alteration from the protocol of Lisec et al. (2006) was that ribitol was not used as an internal standard. Samples were shipped on dry ice from UMAN to LECO (Mönchengladbach, DE) and MPIMP (Potsdam-Golm, DE) where they were stored dry at −80°C until analysis. Sample analysis was completed by each lab within one month of receiving the extracts.
Analytical methods
Analytical methods are numbered and abbreviated by a capital L prefix in square brackets and detailed in Table 2. Common procedures of all method variations were as follows: Samples were removed from −80°C storage and placed in a speed vacuum concentrator for 1 h to remove residual condensation and water. The dried samples were derivatised with O-methylhydroxylamine and N-acetyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA). Further details are presented in Table 2. All samples were run on a GC–EI–TOF/MS instrument with an Agilent 6890N gas chromatograph and a LECO Pegasus III TOF mass spectrometer using the manufacturer’s ChromaTOF software (versions 2.12, 2.22, 3.34; LECO, St. Joseph, MI, USA).
Table 2.
Method parameters highlighting variations in GC–TOF/MS data-acquisition
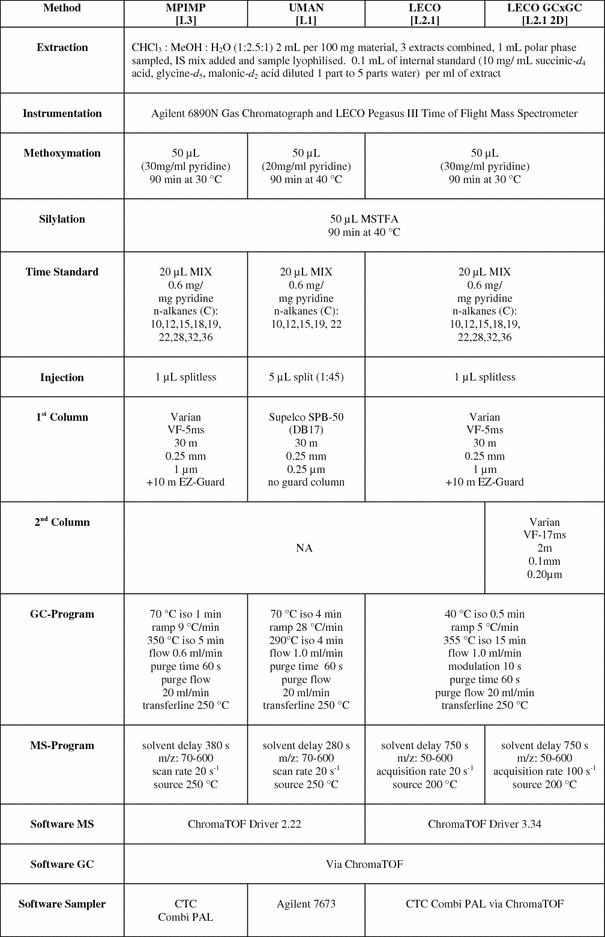
The UMAN (laboratory 1 [L1]) GC–EI–TOF/MS instrument conditions and parameters (Table 2) were as previously described for the optimised method of O’Hagan et al. (2005). This applies a higher polarity column and different injection system when compared to the other methods. MPIMP (laboratory 3 [L3]) GC–EI–TOF/MS instrument conditions and parameters (Table 2) were the same as previously described by Erban et al. (2007). LECO (laboratory 2) GC–EI–TOF/MS [L2.1] instrument conditions and parameters were essentially the same as MPIMP’s with a slightly reduced oven temperature ramp rate and thus longer chromatographic separation time (Table 2). All of the instrument conditions and parameters for the Pegasus 4D GC×GC–TOF/MS [L2.1 2D] analysis undertaken by LECO were standard (Table 2).
Data processing and statistical analysis
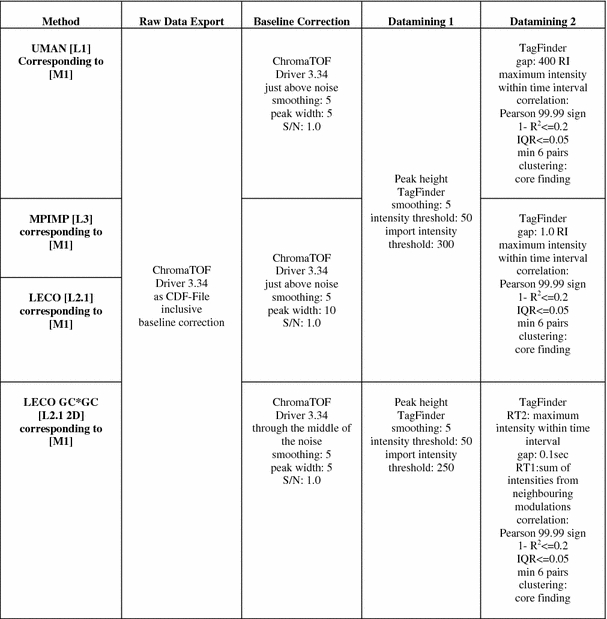
Processing methods are numbered and abbreviated by a capital M prefix in square brackets and details are given in Table 3. Peak heights of mass (m/z) fragments were normalised using the succinic-d4 acid stable isotope labelled standard (cf. Sect. 2.2). Annotation of peak identity was manually supervised using the TagFinder visualisations for mass spectral matching of so-called time groups and clusters (Lüdemann et al. 2008). Identification afforded a minimum of three correlating fragments in a cluster or time group and less than 5% of time deviation between the expected retention index (RI) of a spectral library of reference compounds of the Golm Metabolome Database (http://csbdb.mpimp-golm.mpg.de/csbdb/gmd/gmd.html) (Kopka et al. 2005). Initial visual and statistical analyses of the data were performed with the Multi Experiment Viewer software (Saeed et al. 2003, 2006) and MetAlign (de Vos et al. 2007; Lommen et al. 2007; Lommen 2009). The pre-processing software tool MetAlign (http://www.metalign.nl/UK/) offers two possibilities for interaction with other software: A, de-noising and baseline correction, which maintains the peak shape information (compatible with deconvolution software and Tagfinder); B, de-noising, baseline correction, peak-picking, alignment and export to an Excel format (compatible with Tagfinder and multivariate analysis software) (Lommen 2009). PCA and ICA were performed according to Scholz et al. (2004) using the MetaGenalyse web-service (Daub et al. 2003). The detailed data processing and statistical analysis methods, [M1] to [M7], are summarised in Tables 3 and 4.
Table 3.
Method variations of data pre-processing
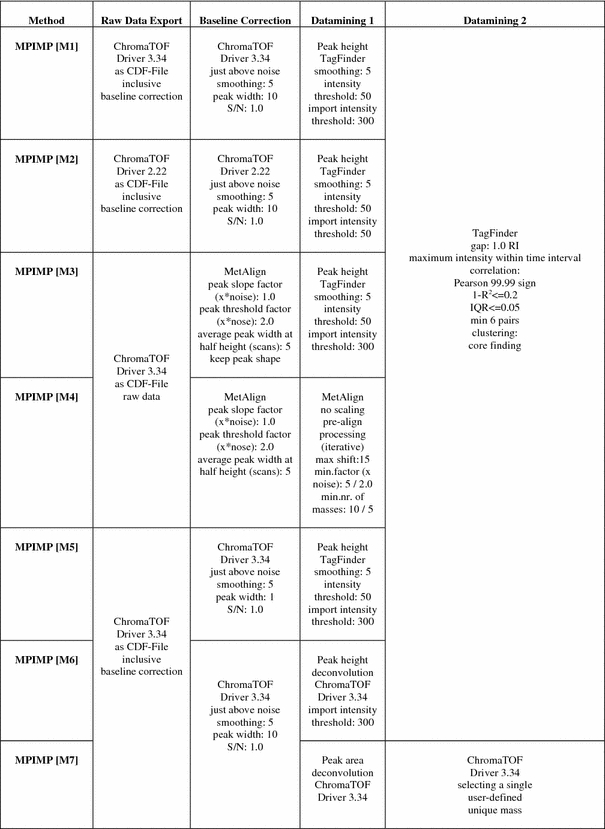
Table 4.
Method variations of data mining relevant for laboratory comparisons
Results and discussion
GC–TOF/MS is a routine technology in analytics with well established standard procedures, nevertheless the use of these data in metabolomics, especially with regard to data exchange between laboratories, demands new strategies in data mining. The aim of our work, based on GC–EI–TOF/MS analysis of identical sample sets, is to demonstrate the reproducibility of sample classification results acquired in different laboratories. Thus data were generated in the three laboratories with different data mining strategies including non-targeted approaches without deconvolution. This was since previous reports (e.g., Lisec et al. 2006; Lu et al. 2008) discovered outlying deconvolutions and cautioned against the non-critical use of deconvoluted mass spectral intensities for relative quantification. Therefore our method of using sample classification of all detected mass features for laboratory-to-laboratory comparison differs from the approach taken in classical ring experiments where deconvoluted quantified data for specific target analytes are compared.
Analytes detected by GC–EI–TOF/MS and its potential for application to food quality assurance
Across the META-PHOR target species of rice grain, melon fruit and broccoli floret, when analysed with GC–EI–TOF/MS typical MSTFA derived GC amenable analytes (being non thermo-labile and within the instruments upper mass range of ~700 m/z) are observed. The typical metabolite groups that are detected include amino, organic, nucleic and fatty acids, as well as monosaccharides, disaccharides, sugar phosphates, sugar alcohols, and polyols. In the case of melon fruit a large number of these metabolite groups are related significantly to the fruit flavour and quality. For example monosacharides, disaccharides, and sugar alcohols all contribute to the sweet flavoured flesh of melon fruit a key quality trait to the consumer (Gao et al. 1999; Stepansky et al. 1999), and are indeed detected as being significantly more concentrated in the fruit inner mesocarp than the outer mesocarp and epicarp (Biais et al. 2009). Secondly the amino acid profile of the fruit is indicative of its fragrant qualities with many VOC’s such as esters and aldehydes being derived from amino acids such as alanine and valine.
Amino acids, organic acids, mono and disaccharides are also significant indicators of broccoli floret flavour and quality. Unfortunately many nutraceuticals within broccoli such as the flavones, flavanoids and glucosinalates, are large compounds and outside of the mass range of typical GC–EI–MS instrumentation, such nutraceutical compounds are much more amenable to detection via LC–MS (deVos et al. 2007; Jansen et al. 2008). The quality of rice grain is largely reflected in its starch and vitamin content, thus techniques such as LC–inductively coupled plasma (ICP)–MS which is capable of elemental profiling is required for its quality assessment. Since the market value of rice is largely determined by the fragrant nature of the rice variety, again VOC analysis is essential for determining phenotypic measures of market price and quality.
For a metabolomics screen to assess food quality and safety GC–EI–TOF/MS alone will not provide enough information across a large enough range of metabolite groups. Therefore, META-PHOR recommends multi-platform based analysis with: 1H-NMR, GC–EI–TOF/MS, LC–TOF/MS, VOC analysis via thermal desorption (TD) or solid phase micro extraction (SPME) analyte trapping followed by GC–EI–MS, various high resolution MS trap based techniques for the proceeding analyte identification, and where elemental composition analysis is required LC–ICP–MS is also applied.
Demonstration of global repeatability of GC–EI–TOF/MS based plant metabolomics using independent component analysis (ICA)
Through the use of ICA it was demonstrated that the global repeatability of the sample sets analysed by GC–EI–TOF/MS between the laboratories was high (Fig. 1). The data employed in the generation of Fig. 1a and b from UMAN (laboratory 1 [L1]) (Table 2) differ in data mining strategy. Figure 1a is based on a targeted method using deconvolved data as described by Lisec et al., (2006), corresponding to data analysis method [M7] (Tables 3, 4). Figure 1b is based on the same acquired raw data but processed with a non-targeted fingerprinting approach, thus enabling the analysis of all acquired mass spectral features from the data set and subsequent application to comprehensive statistical analysis (data analysis method [M1]) (Tables 3, 4) (Scholz et al. 2004; Pongsuwan et al. 2007).
Fig. 1.
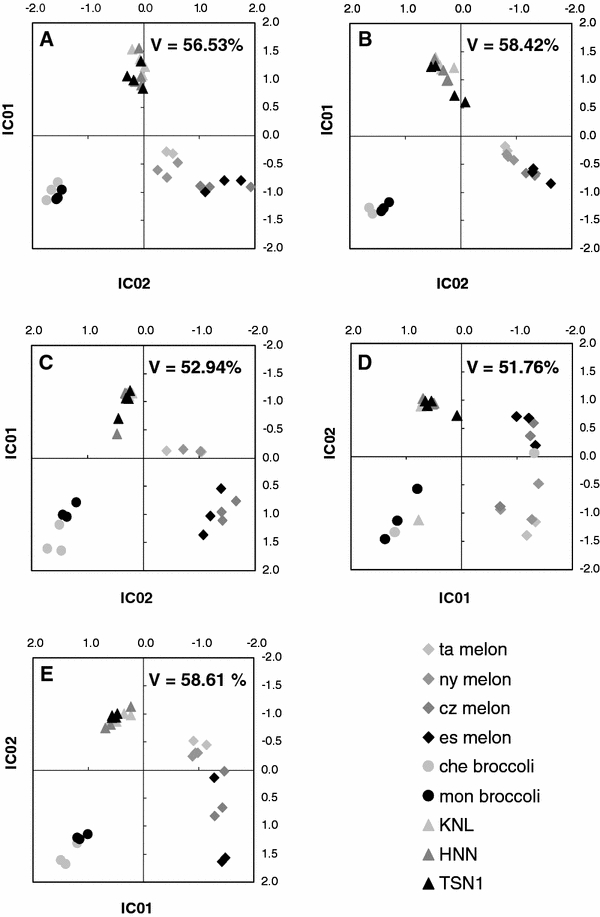
Comparative independent component analysis demonstrates the reproducibility of sample discrimination between laboratories and method variations. a–e shows independent component analyses based on the first two principal components of a PCA preprocessing. The visualised percentage of total variance (V) is indicated. a shows data of UMAN after metabolite targeted data processing, method combination [L1] and [M7]. b is based on fingerprinting the data set of UMAN with methods [L1] and [M1]. c compares fingerprinting data of LECO with method [L2.1] and [M1] to GC×GC-fingerprinting data (d) of the same laboratory using method [L2.1 2D] and [M1]. e demonstrates the fingerprinting results of MPIMP using the method combination [L3] and [M2]
Figure 1b–d based on data analysis method [M1], and Fig. 1e based on data analysis method [M2] (Tables 3, 4), are all generated by the non-targeted approach with data from all three laboratories [L1] to [L3] (Table 2). Noise reduction was performed by applying a criterion to find at least three unique and mutually correlating mass fragments per analyte for peak height based quantification. By contrast, Fig. 1a generated via data processing method [M7] (Tables 3, 4) is based upon a defined, pre-selected single unique mass for peak area based quantification. A maximum normalised response value was calculated from the available unique masses found via the underlying correlation and cluster analyses performed within TagFinder (Lüdemann et al. 2008). Annotation was manually supervised testing mass spectral similarity between the reference library (Kopka et al. 2005) and the measured feature and retention index behaviour.
Figure 1c and d compare the LECO (laboratory 2 [L2]) methods [L2.1] and [L2.1 2D] (Table 2) respectively, using data processing method [M1] (Tables 3, 4). Chromatography is longer, with a less polar column, and splitless injection, in contrast to the UMAN method [L1]. Figure 1c and e are based essentially on the same technical settings but generated by different laboratories (LECO [L2.1] and data processing method [M1], and MPIMP [L3] and data processing method [M2]; Tables 3, 4). All of the four data sets (3 × GC–EI–TOF/MS and 1 × GC×GC–EI–TOF/MS) were aligned according to retention index, normalised to the succinic-d4 acid as this standard was ideal under all chromatography regimes, mean centred by each mass feature and finally log10 transformed. Missing data were replaced with “0” before uploading into MetaGeneAlyse for PCA and ICA (Daub et al. 2003). Note that the plots axes are scaled to the same scores range allowing comparative visualisation. The comparison between laboratories as well as between one (GC) and two (GC×GC) dimensional chromatography show good reproducibility and using unsupervised ICA clear and highly similar sample classifications were achieved for all data sets.
Assessments of technical reproducibility
For further and more detailed analysis, the subset of rice data was evaluated alone since the direct simultaneous analysis of the highly different matrices of broccoli, melon and rice, with respect to the high qualitative and quantitative differences in composition, reduces the availability of unique masses which can be employed for quantification. Therefore analysing a sub-set of the data according to biological matrix is advised. First a detailed non targeted evaluation of technical reproducibility is shown in Fig. 2. All mass spectral features with pair-wise availability after respective processing are plotted in Fig. 2a and b. In Fig. 2a, using the MPIMP instrument method [L3] (Table 2) and data processing method [M2] (Table 3), two similar biological rice samples with a minimum fragment intensity of 50 are compared. Figure 2b demonstrates the technical reproducibility of two identical analytical replicates, i.e. based on one biological extract redundant from derivate variation, taken from a MPIMP reproducibility experiment with a total of 29 analysed replicates.
Fig. 2.
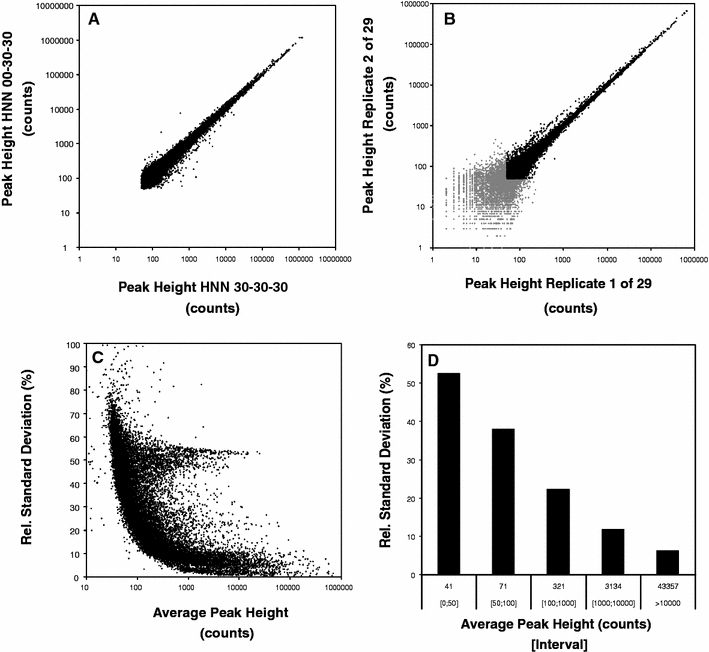
Analyses of technical replicate profiles. a, b compares the reproducibility of all mass spectral features from technical replicates (b) to biological replicates of highly similar rice samples (a, cf. to samples of Figs. 4, 5). The peak-heights (counts) of all aligned acquired mass fragments are plotted. a is limited to 50 counts minimum using the baseline correction integrated in method [M2]. b also processed by [M2] demonstrates the validity of the 50 count cut-off (grey format). c summarises the relative standard deviations (RSDs) of all aligned mass spectral features from an MPIMP experiment comprising 29 technological replicate chromatograms. Note that the population of intense features at 50–60% RSD is caused by reagent contaminations. d demonstrates the expected technological RSDs with regard to choice of peak intensity (count) range as a histogram
A strong impact of the signal-to-noise threshold can be observed in Fig. 2b–d. With increasing fragment intensity from 1 to 106 the technical variability decreases dramatically from approximately 50% down to 5% (based on a minimum of six data points out of 29 replicates). The bi-modal behaviour of quantitative variability observed in Fig. 2c where some of the high intensity fragments show increased relative standard deviation (RSD) can be traced back to the replicate specific concentration of artefact polysiloxanes generated commonly by column bleed or silylation reagents independently of sample composition. In typical metabolite profiling experiments high RSD mass fragments are ignored as these can be identified and removed from further analysis using characteristic mass spectra. Since high RSD artefact mass fragments may impact upon the PCA and ICA of non-targeted fingerprinting studies, routine exclusion prior to statistical analyses is recommended. However, artefact exclusion may not always be necessary, the comparative ICA for this study (Fig. 1) were performed including mass fragments of both artefacts and internal standards and yet reproducible sample classification was obtained.
Chemical stability of derivatives based on different amounts of silylated groups or isomerism from methoxymation is represented in Fig. 3. As the relative quantification of amino acids using GC–MS based profiling has been controversially discussed (Noctor et al. 2007), we used glutamic acid as one example to compare between laboratories. For glutamic acid two major detectable derivatives with two and three silylated groups are plotted in Fig. 3a. Figure 3a shows adequate reproducibility between the different participating laboratories of the META-PHOR ring experiment and their datasets based upon instrument methods [L1] to [L3] (Table 2), and data processing method [M1] (Table 3). Although not easily achievable a stable isotope labelled standard for each metabolite class detected is ultimately advisable to improve precision. It should be noted that glutamic acid can also form not only a four times silylated derivative but may also generate varying amounts of the cyclic pyroglutamic acid during derivatisation and analysis under high temperatures. Much less chemically affected and therefore not a matter of discussion is the stability of glucose derivatives, as shown in Fig. 3b based upon instrument methods [L1] to [L3] (Table 2) and data processing method [M1] (Table 3). Here the derivatives are based on the geometric cis/trans-isomerism of the methoxymated carboxyl-group.
Fig. 3.
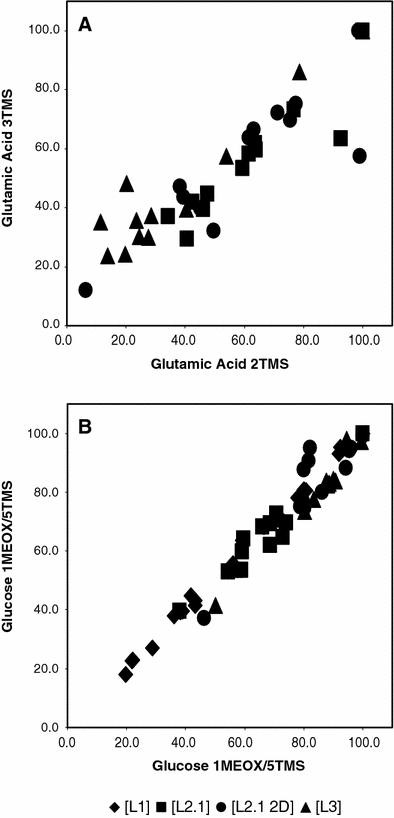
Stability of alternative chemical derivatives. The normalised responses after internal standardisation of alternative glutamate (a) and glucose (b) derivatives are shown. The high agreement of the METAPHOR data [L1], [L2.1], [L2.1 2D], [L3], processed by [M1] is demonstrated. For analysis of the resilient biological matrices or unstable metabolite derivatives, specific stable isotope labelled standards will enhance accuracy. Note that glutamic acid 2TMS was not detectable in [L1]
When comparing the analytical methodologies employed across the ring experiment, unsurprisingly the medium throughput methods (Lisec et al. 2006; Erban et al. 2007) of MPIMP [L3] and LECO [L2.1] were more appropriate for the analysis of the diverse META-PHOR species, than the UMAN method [L1] which was optimised for high-throughput analysis of yeast media (O’Hagan et al. 2005). The research warrants a further comparison of splitless and split injection methodologies for these sample types in future experimentation, although the repeatability of data between the laboratories on the whole was impressive.
To include a measure of the reproducibility of the data mining methods applied to our ring-experiment Fig. 4 was generated. In Fig. 4a the raw data from instrument method [L3] (Table 2) were mined with several data processing methods, comprising the use of peak area evaluation [M7], peak deconvolution [M6], different base-line correction algorithms [M1], [M2], [M3], and [M4], different peak height picking algorithms [M1], [M2], [M3], and [M4], as well as employing restrictions with regard to different expected peak widths [M1] and [M5] (Tables 3, 4). For the 12 rice samples from the META-PHOR ring-experiment the maximum normalised response of the deuterated internal standard succinic-d4 acid 3TMS is shown in Fig. 4a and c reflecting the alternative data-mining possibilities. In Fig. 4b and c the corresponding information allowing comparison between the laboratories instrumental methods [L1] to [L3] (Table 2) is represented, a method as similar as possible to [M1] was used (Table 3).
Fig. 4.
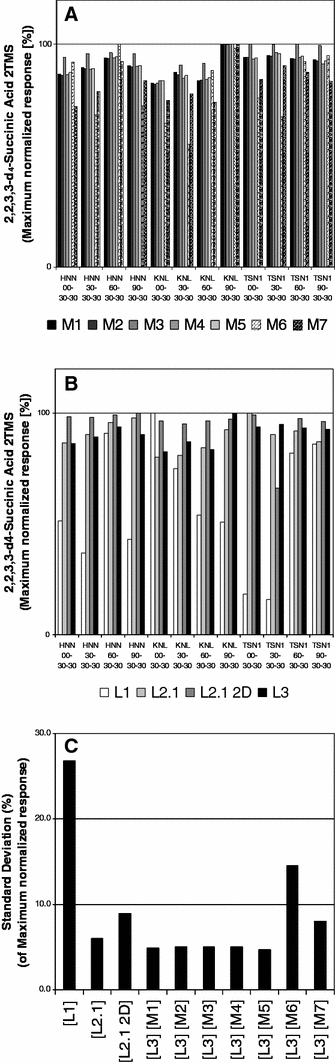
Technical reproducibility evaluated by the internal standard, d4-succinic acid (2TMS). Response data were maximum normalised for comparison of the data-mining methods [M1] to [M7] using exemplary [L3] data (a). b compares maximum normalised d4-succinic acid (2TMS) response between laboratories [L1] to [L3] using processing method [M1]. The respective standard deviations of each of the previous calculations are reported in (c) with laboratory and method combinations indicated
The highest deviations in reproducibility can be observed in the split mode faster-GC based dataset from UMAN [L1]. However, this variability which became apparent through the internal standard compound can be effectively corrected. When applying the common normalisation method for matrix metabolites the fast-GC based dataset exhibits similar reproducibility to the other methods (Fig. 5). The increased standard deviation observed for instrument method [L2.1 2D] (Table 2) may not be attributed to represent a technological feature of GC×GC–TOF/MS, but is currently the result of the non-optimised fingerprinting of high intensity GC×GC–TOF/MS peaks which are split among several subsequent 2nd dimension modulations. Using the information of Lu et al. (2008) we can now demonstrate the improved quality of data based on peak-picking strategies from TagFinder (Lüdemann et al. 2008) and MetAlign (de Vos et al. 2007; Lommen et al. 2007, 2009).
Fig. 5.
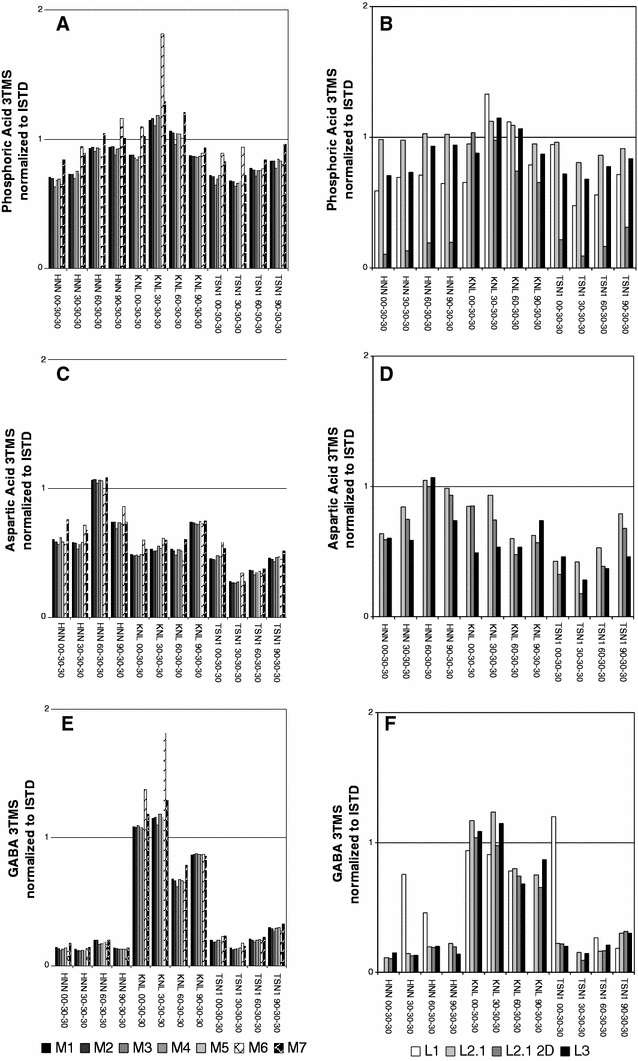
Comparisons of endogenous metabolite levels using responses normalised to the d4-succinic acid internal standard. Metabolites were chosen to represent the borderline of potential distinctive features, such as a, b phosphoric acid (3TMS) and c, d aspartic acid (3TMS), as well as clear differences between sample groups, e.g. e, f GABA, 4-aminobutyric acid (3TMS). Variation of processing methods [M1] to [M7] of an identical data set [L3] (a, c, e) is compared to variations between laboratories [L1] to [L3] with processing fixed to [M1] (b, d, f). Abbreviations HNN, KNL and TSN1 represent rice cultivars, numbers encode nitrogen regimes (Sect. 2.1.)
Reproducibility of data for representative metabolites between variations in GC–EI–TOF/MS analytical methodology and data processing strategy
After normalisation to the internal standard the responses of three representative metabolites were analysed and are visualised in Fig. 5a, c and e (based upon instrument method [L3] and data processing methods [M1] to [M7]) and Fig. 5b, d and f (based upon instrument methods [L1] to [L3] and data processing method [M1]) (Tables 2, 3, 4). Of course, both the error propagation from the internal standard values and the inherent variability of the data processing methods must be kept in mind. In the case of phosphoric acid 3TMS the comparability of data from the different laboratories and data-mining methods is shown in Fig. 5a and b. The metabolite aspartic acids’ corresponding analyte aspartic acid 3TMS is missing in the UMAN dataset [L1] (Table 2), which is possibly due to discrimination of the analyte based on the split-injection of the derivate compared to splitless injections in the other instrument methods [L2] and [L3] (Fig. 5c, d). However, it must be noted that the UMAN [L1] on-column volume was almost 1/10th that of the LECO [L2] and MPIMP [L3] (Table 2) methods (0.11 μl [L1] in comparison to 1 μl [L2] and [L3]).
Gama-Aminobutyric acid (GABA) 3TMS represents a metabolite in the rice experiment showing a specific increase associated with the KNL cultivar (Fig. 5e, f). A lower precision was observed for GABA 3TMS in the UMAN instrument method [L1], GABA is not commonly detected in yeast footprint media and so the method optimisation did not account for it (O’Hagan et al. 2005), however this observation may also result from the split or different injection system employed by UMAN [L1] (Table 2). In the case of GABA 3TMS, the analyte concentration is still above the detection-limit for some conditions but erroneous due to noise in others. Dealing with fragment intensities of a signal-to-noise of 2.0 and higher, results in details within the low level detection region of noisy data remaining in the chromatogram after baseline-correction (Fig. 2).
Caution is also necessary when comparing data from different software-versions or algorithms, e.g. data processing method [M2] used ChromaTOF 2.22 which leaves noise of ~25 units after baseline-correction while data processing method [M1] used ChromaTOF 3.34 which leaves noise of ~100 units. When using ChromaTOF 2.22 the operator defines the smoothing factor manually whereas ChromaTOF 3.34 has the option to select the smoothing factor automatically. The automatic smoothing also takes into account the data acquisition rate to ensure that 18–20 data points are present across the chromatographic peaks. Additionally, standardisation of baseline cutting parameters (above the noise, mid-way and at the noise) in all data processing methods is necessary and must not be over looked. This is important since there is potential for the generation of different results and therefore data cannot be compared without applying the correct standardisation.
GC×GC offers enhanced resolution and depth of data over conventional GC
For an overview of the complexity of the evaluated rice matrix, and to also assess the resolution and depth of data gained through GC×GC–EI–TOF/MS compared to conventional GC–EI–TOF/MS, data from the same derivate samples obtained on both instruments were compared. Figure 6a is based on LECO instrument method [L2.1] and Fig. 6b on [L2.1 2D] for comparison between 1D and 2D GC–EI–TOF/MS. As can be seen from the two chromatograms represented in Fig. 6a and b, the two dimensional GC×GC–EI–TOF/MS chromatogram shows a significantly greater wealth of information and enhanced level of resolution, which at the current state of automated data pre-processing is not fully accessed. Thus, a strong incentive is given to improve on the development of automated metabolite targeted and non-targeted multi-parallel fingerprinting analyses of these 4-dimensional data rich files.
Fig. 6.
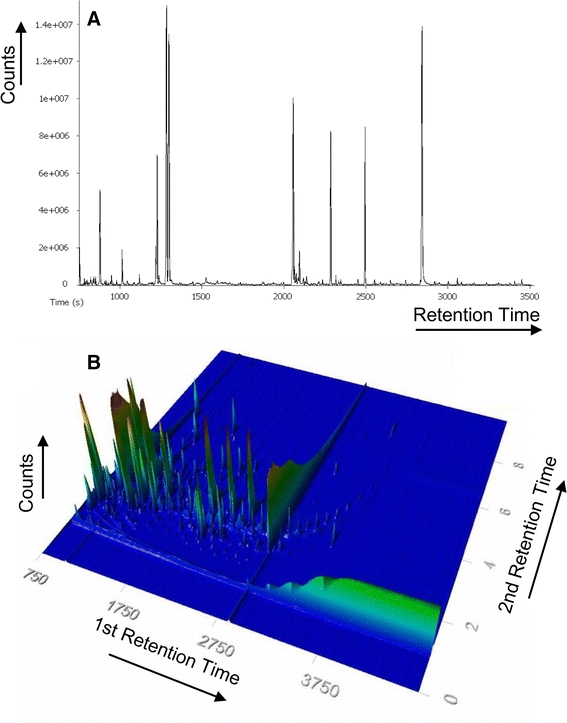
GC×GC–TOF/MS is expected to enhance routine metabolite profiling. An exemplary GC–TOF/MS Chromatogram (a) of the evaluated rice samples (Figs. 4, 5) is compared to the corresponding GC×GC–TOF/MS analysis (b). Total ion count (TIC) is plotted
Ring experiment “take homes” and improvements for future laboratory-to-laboratory comparisons
Despite the excellent reproducibility illustrated between the different laboratories analytical methodologies, further improvement could be made by using identical analytical setup and chromatographic methods. The differences between split and splitless injection methods (and to a lesser extent the different injection systems used) and on-column volumes has potentially been shown to influence results. For future assessments of split based GC methods it will be crucial to also test for matrix dependent discrimination effects. The chromatography generated from the melon extracts for all laboratories suffered greatly from monosaccharide overloading. In future it may be of benefit to perform a two-stage GC–EI–TOF/MS analysis of melon, where a whole melon extracts polar phase is used for the analysis of sugars and highly concentrated bulk metabolites, and a second sample is prepared via subjecting the same polar phase to a solid phase extraction (SPE) for the removal of free sugars (Suzuki et al. 2002), prior to being analysed for trace metabolites. Of course analysis of extraction solvents subjected to SPE would also be required to identify artefacts resulting from the process. To further enhance sample stability during future experiments the authors recommend that samples are best sealed dry under inert gas and shipped upon excessive amounts of dry ice. It is also recommended that a minimum of one backup sample set per laboratory be held in storage by the laboratory responsible for extract preparation as a means for testing unexpected artefact laboratory-to-laboratory deviations.
In our hands relative quantification based upon peak area worked impressively well especially for peaks giving high responses. In contrast the comparison of data generated between several laboratories in a short-term experiment for relative quantification based upon peak height was found to be more feasible as automated peak height retrieval is a simple process compared to the required area decomposition of multiple co-eluting metabolic components. Thus peak height will be employed as the future preferred method for META-PHOR experimentation until robust peak area calculations may become available. It must be taken into account that for long term experimental comparisons (months–years) employment of peak area may be more appropriate, since changing consumables such as the injection liners influence peak shape and thus peak height more than peak area.
Ring experiment precedents from across all disciplines of “omic” research
Precedents for the assessment of inter-laboratory reproducibility from the alternative ‘omic’ fields of proteomics and transcriptomics can be found. In the transcriptomics field, reproducible and highly overlapping results based upon the independent treatment of rats with bromobenzene and microarray analyses have been reported. This was despite the two laboratories using alternative routes of bromobenzene administration and differing in-house constructed microarray chips (Heijne et al. 2003, 2004). Further, in a more recent study a large consortium of transcriptomics laboratories tested standardised operating procedures (SOPs) for the processing and analysis of a common set of sample material, again resulting in highly overlapping datasets (Pennie et al. 2004). Unsurprisingly, the inter-laboratory reproducibility of proteomics is lower than transcriptomics. One study produced three technical replicate 2D gels per each biological sample and reported that variability between the gels was very high to such an extent that statistical analysis could only confirm changes in the levels of 24 proteins, despite having a high number of changes that were not technically reproducible between gels (Heijne et al. 2003). Notwithstanding, good reproducibility has been demonstrated for the MS analysis of proteins and mass fingerprinting of peptide digests (Verhoeckx et al. 2004). It is currently a major and ongoing focus of all three ‘omics’ fields to develop robust and standardised high-throughput operating procedures.
Many previous studies can be found where GC–MS ring experiments have been conducted, however these were not non-targeted metabolomic studies but tended to focus upon the analysis of soils (Karstensen et al. 1998) and water samples (Hoogerbrugge et al. 1999) for the detection of specific contaminants during quality testing. Through non-targeted metabolomics literature searches only one previous study could be found where GC–MS results from two laboratories were compared for biological quality assurance purposes, here the authors did not focus on the inter-laboratory reproducibility, but more on the biological significance of the data (Catchpole et al. 2005; Beckmann et al. 2007). Catchpole, Beckman, and colleagues, performed a comparison of GM potato lines generated from the Désirée cultivar using a combination of flow infusion (FI)MS, LC–MS and GC–MS. However, in that study extracts were prepared independently by different technicians and run on various manufacturers and models of instrument (Catchpole et al. 2005; Beckmann et al. 2007). By contrast, for the present META-PHOR ring experiment a common set of extracts was prepared by a single technician, aliquoted and distributed for parallel runs on the same model of instrument (Agilent 6890N GC with LECO Pegasus III TOF–MS) in different locations, though with different injector systems (Agilent 7673 and CTC CombiPAL).
Concluding remarks
In conclusion, the work reported here provides an unbiased assessment of the inter-laboratory repeatability of GC–EI–TOF/MS taking into consideration the different analytical method variants, and the suitability of a range of data processing and statistical analysis routines. The major metabolite features generated in the different META-PHOR laboratories proved to be highly reproducible indicating great promise for the future generation of global metabolomics databases. We suggest that further ring experiments tuned to the specific approaches and properties of fingerprinting and profiling studies be performed to monitor and document the future advances of the ongoing standardisation process in the metabolomic field of qualitative and quantitative food and health related analyses.
Acknowledgements
This study was funded by the EU as part of the Frame work VI initiative within the plant metabolomics project META-PHOR (FOOD-CT-2006-036220). RG and WBD are also grateful to the BBSRC for financial support of the MCISB (Manchester Centre for Integrative Systems Biology). We very gratefully thank our project collaborators at INRA Bordeaux, The Volcani Centre Israel, Plant Research International Holland, IRRI and Laos NARC, for provision and processing of plant materials for extraction and analysis.
Footnotes
J. William Allwood and Alexander Erban have Equally contributed to this study.
Contributor Information
J. William Allwood, Email: William.Allwood@manchester.ac.uk.
Alexander Erban, Email: Erban@mpimp-golm.mpg.de.
References
- Beckmann M, Enot DP, Overy DP, Draper J. Representation, comparison, and interpretation of metabolome fingerprint data for total composition analysis and quality trait investigation in potato cultivars. Journal of Agricultural and Food Chemistry. 2007;55(9):3444–3451. doi: 10.1021/jf0701842. [DOI] [PubMed] [Google Scholar]
- Biais B, Allwood JW, Deborde C, Xu Y, Maucort M, Beauvoit B, Dunn WB, Jacob D, Goodacre R, Rolin D, Moing A. 1H-NMR, GC-EI-TOF/MS, and dataset correlation for fruit metabolomics: Application to spatial metabolite analysis in melon. Analytical Chemistry. 2009;81(8):2884–2894. doi: 10.1021/ac9001996. [DOI] [PubMed] [Google Scholar]
- Catchpole GS, Beckman M, Enot DP, Mondhe M, Zywicki B, Taylor J, Hardy N, Smith A, King RD, Kell DB, Fiehn O, Draper J. Hierarchical metabolomics demonstrates substantial compositional similarity between genetically modified and conventional potato crops. Proceedings of the National Academy of Sciences. 2005;102:14458–14462. doi: 10.1073/pnas.0503955102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub CO, Kloska S, Selbig J. MetaGeneAlyse: Analysis of integrated transcriptional and metabolite data. Bioinformatics. 2003;17:2332–2333. doi: 10.1093/bioinformatics/btg321. [DOI] [PubMed] [Google Scholar]
- Vos CHR, Moco S, Lommen A, Keurentjes JJB, Bino RJ, Hall RD. Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nature Protocols. 2007;2(4):778–791. doi: 10.1038/nprot.2007.95. [DOI] [PubMed] [Google Scholar]
- Dixon RA, Gang DR, Charlton AJ, Fiehn O, Kuiper H, Reynolds TL, Tjeerdema RS, Jeffery EH, German JB, Ridley WP, Seiber JN. Applications of metabolomics in agriculture. Journal of Agricultural and Food Chemistry. 2006;54:8984–8994. doi: 10.1021/jf061218t. [DOI] [PubMed] [Google Scholar]
- Dunn, W. B. (2008). Current trends and future requirements for the mass spectrometric investigation of microbial, mammalian and plant metabolomes. Physical Biology, 5(1), 011001. [DOI] [PubMed]
- Erban A, Schauer N, Fernie AR, Kopka J. Non-supervised construction and application of mass spectral and retention time index libraries from time-of-flight gas chromatography-mass spectrometry metabolite profiles. Methods in Molecular Biology. 2007;358:19–38. doi: 10.1007/978-1-59745-244-1_2. [DOI] [PubMed] [Google Scholar]
- Fernie AR, Trethewey RN, Krotzky AJ, Willmitzer L. Metabolite profiling: From molecular diagnostics to systems biology. Nature Reviews Molecular Cell Biology. 2004;5(9):763–769. doi: 10.1038/nrm1451. [DOI] [PubMed] [Google Scholar]
- Fiehn O, Kopka J, Dormann P, Altmann T, Trethewey RN, Willmitzer L. Metabolite profiling for plant functional genomics. Nature Biotechnology. 2000;18:1157–1161. doi: 10.1038/81137. [DOI] [PubMed] [Google Scholar]
- Fiehn O, Kopka J, Trethewey RN, Willmitzer L. Identification of uncommon plant metabolites based on calculation of elemental composition using gas chromatography and quadrupole mass spectrometry. Analytical Chemistry. 2000;72:3573–3580. doi: 10.1021/ac991142i. [DOI] [PubMed] [Google Scholar]
- Fiehn O, Robertson D, Griffin J, derf Werf M, Nikolau B, Morrison N, Sumner LW, Goodacre R, Hardy NW, Taylor C, Fostel J, Kristal B, Kaddurah-Daouk R, Mendes P, Ommen BV, Lindon JC, Sansone S-A. The metabolomics standards initiative (MSI) Metabolomics. 2007;3(3):175–178. doi: 10.1007/s11306-007-0070-6. [DOI] [Google Scholar]
- Fiehn O, Sumner LW, Rhee SY, Ward J, Dickerson J, Lange BM, Lane G, Roessner U, Last R, Nikolau B. Minimum reporting standards for plant biology context information in metabolomic studies. Metabolomics. 2007;3(3):195–201. doi: 10.1007/s11306-007-0068-0. [DOI] [Google Scholar]
- Fiehn O, Wohlgemuth G, Scholz M, Kind T, Lee D-Y, Lu Y, Moon S, Nikolau B. Quality control for plant metabolomics: Reporting MSI-compliant studies. The Plant Journal. 2008;53:691–704. doi: 10.1111/j.1365-313X.2007.03387.x. [DOI] [PubMed] [Google Scholar]
- Gao Z, Petreikov M, Zamski E, Schaffer AA. Carbohydrate metabolism during early fruit development of sweet melon (Cucumis melo) Physiologia Plantarum. 1999;106:1–8. doi: 10.1034/j.1399-3054.1999.106101.x. [DOI] [Google Scholar]
- Goodacre R, Broadhurst D, Smilde AK, Kristal BS, Baker JD, Beger R, Bessant C, Connor S, Capuani G, Craig A, Ebbels T, Kell DB, Manetti C, Newton J, Paternostro G, Somorjai J, Sjöström M, Trygg J, Wulfert F. Proposed minimum reporting standards for data analysis in metabolomics. Metabolomics. 2007;3(3):231–241. doi: 10.1007/s11306-007-0081-3. [DOI] [Google Scholar]
- Hall RD. Plant metabolomics: From holistic hope, to hype, to hot topic. New Phytologist. 2006;169:453–468. doi: 10.1111/j.1469-8137.2005.01632.x. [DOI] [PubMed] [Google Scholar]
- Hall RD. Food metabolomics: META-PHOR. A new European research initiative. Agro Food Industry Hi-Tech. 2007;18:14–16. [Google Scholar]
- Hall RD, Brouwer ID, Fitzgerald MA. Plant metabolomics and its potential application for human nutrition. Physiologia Plantarum. 2008;132:2162–2175. doi: 10.1111/j.1399-3054.2007.00989.x. [DOI] [PubMed] [Google Scholar]
- Hardy NW, Taylor CF. A roadmap for the establishment of standard data exchange structures for metabolomics. Metabolomics. 2007;3(3):243–248. doi: 10.1007/s11306-007-0071-5. [DOI] [Google Scholar]
- Heijne WH, Slitt AL, Bladeren PJ, Groten JP, Klaassen CD, Stierum RH, Ommen B. Bromobenzene induced hepatotoxicity at the transcriptome level. Toxicological Sciences. 2004;79:411–422. doi: 10.1093/toxsci/kfh128. [DOI] [PubMed] [Google Scholar]
- Heijne WM, Stierum RH, Slijper M, Bladeren PJ, Ommen B. Toxicogenomics of bromobenzene hepatotoxicity: A combined transcriptomics and proteomics approach. Biochemical Pharmacology. 2003;65:857–875. doi: 10.1016/S0006-2952(02)01613-1. [DOI] [PubMed] [Google Scholar]
- Hoogerbrugge, R., Gort, S. M., van der Velde, E. G., & Van Zoonen, P. (1999). Multi- and univariate interpretation of the inter-laboratory validation of PrEN 12673; GC determination of polyphenols in water. Analytica Chima Acta, 388, 119–135.
- Horning EC. Use of combined gas–liquid chromatography and mass spectrometry for clinical problems. Clinical Chemistry. 1968;14:777. [Google Scholar]
- Jansen JJ, Allwood JW, Marsden-Edwards E, Putten WH, Goodacre R, Dam NM. Metabolomic analysis of the interaction between plants and herbivores. Metabolomics. 2008;5(1):150–161. doi: 10.1007/s11306-008-0124-4. [DOI] [Google Scholar]
- Jenkins H, Hardy N, Beckmann M, Draper J, Smith AR, Taylor J, Fiehn O, Goodacre R, Bino RJ, Hall RD, Kopka J, Lane GA, Lange BM, Liu JR, Mendes P, Nikolau BJ, Oliver SG, Paton NW, Rhee S, Roessner-Tunali U, Saito K, Smedsgaard J, Sumner LW, Wang T, Walsh S, Wurtele ES, Kell DB. A proposed framework for the description of plant metabolomics experiments and their results. Nature Biotechnology. 2004;22:1601–1606. doi: 10.1038/nbt1041. [DOI] [PubMed] [Google Scholar]
- Jolliffe, I. T. (1986). Principal components analysis. New York: Springer-Verlag.
- Karstensen KH, Ringstad O, Rustad I, Kalevi K, Jørgensen K, Nylund K, Alsberg T, Ólafsdóttir K, Heidenstam O, Solberg H. Methods for chemical analysis of contaminated soil samples-tests of their reproducibility between Nordic laboratories. Talanta. 1998;46:423–437. doi: 10.1016/S0039-9140(97)00401-3. [DOI] [PubMed] [Google Scholar]
- Kopka J, Schauer N, Krueger S, Birkemeyer C, Usadel B, Bergmueller E, Doermann P, Weckwerth W, Gibon Y, Stitt M, Willmitzer L, Fernie AR, Steinhauser D. GMD@CSB.DB: The Golm Metabolome Database. Bioinformatics. 2005;21:1635–1638. doi: 10.1093/bioinformatics/bti236. [DOI] [PubMed] [Google Scholar]
- Lisec J, Schauer N, Kopka J, Willmitzer L, Fernie AR. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nature Protocols. 2006;1:387–396. doi: 10.1038/nprot.2006.59. [DOI] [PubMed] [Google Scholar]
- Lommen, A. (2009). MetAlign: An interface-driven, versatile metabolomics tool for hyphenated full-scan MS data pre-processing. Analytical Chemistry, 81(8), 3079–3086. [DOI] [PubMed]
- Lommen A, Weg G, Engelen MC, Bor G, Hoogenboom LAP, Nielen MWF. An untargeted metabolomics approach to contaminant analysis––pinpointing potential unknown compounds. Analytica Chimica Acta. 2007;584:43–49. doi: 10.1016/j.aca.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Lu H, Dunn WB, Shen H, Kell DB, Liang Y. Comparative evaluation of software for deconvolution of metabolomics data based on GC-TOF-MS. Trends in Analytical Chemistry. 2008;27(3):215–227. doi: 10.1016/j.trac.2007.11.004. [DOI] [Google Scholar]
- Lüdemann A, Strassburg K, Erban A, Kopka J. TagFinder for the quantitative analysis of gas chromatography-mass spectrometry (GC-MS)-based metabolite profiling experiments. Bioinformatics. 2008;24:732–737. doi: 10.1093/bioinformatics/btn023. [DOI] [PubMed] [Google Scholar]
- Noctor G, Bergot GL, Mauve C, Thominet D, Lelarge-Trouverie C, Prioul JL. A comparative study of amino acid measurement in leaf extracts by gas chromatography-time of flight-mass spectrometry and high performance liquid chromatography with fluorescence detection. Metabolomics. 2007;3(2):161–174. doi: 10.1007/s11306-007-0057-3. [DOI] [Google Scholar]
- O’Hagan S, Dunn WB, Brown M, Knowles JD, Kell DB. Closed loop, multiobjective optimization of analytical instrumentation: Gas chromatography/time-of-flight mass spectrometry of the metabolomes of human serum and of yeast fermentations. Analytical Chemistry. 2005;77:290–303. doi: 10.1021/ac049146x. [DOI] [PubMed] [Google Scholar]
- Pauling L, Robinson AB, Teranishi R, Cary P. Quantitative analysis of urine vapor and breath by gas-liquid partition chromatography. Proceedings of the National Academy of Sciences. 1971;68:2374–2376. doi: 10.1073/pnas.68.10.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennie W, Pettit SD, Lord PG. Toxicogenomics in risk assessment: An overview of an HESI collaborative research programme. Environmental Health Perspectives. 2004;112:417–419. doi: 10.1289/ehp.6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongsuwan W, Fukusaki E, Bamba T, Yonetani T, Yamahara T, Kobayashi A. Prediction of Japanese green tea ranking by gas chromatography/mass spectrometry-based hydrophilic metabolite fingerprinting. Journal of Agricultural and Food Chemistry. 2007;55:231–236. doi: 10.1021/jf062330u. [DOI] [PubMed] [Google Scholar]
- Roessner U, Wagner C, Kopka J, Tretheway RN, Willmitzer L. Simultaneous analysis of metabolites in potato tuber by gas chromatography-mass spectrometry. The Plant Journal. 2000;23(1):131–142. doi: 10.1046/j.1365-313x.2000.00774.x. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Hagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li JW, Thiagarajan M, White JA, Quackenbush J. TM4: Microarray software suite. Methods in Enzymology. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Rylt-sov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: A free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- Sauter, H., Lauer, M., & Fritsch, H. (1988). Metabolic profiling of plants a new diagnostic technique. In D. R. Baker, J. G. Fenyes, & W. K. Moberg (Eds.), Abstracts of papers of the American chemical society (Vol. 195, p. 129). Washington DC, US: American Chemical Society, NW.
- Schauer, N., & Fernie, A. R. (2006). Plant metabolomics: Towards biological function and mechanism. Trends in Plant Science,11, 508–516. [DOI] [PubMed]
- Scholz M, Gatzek S, Sterling A, Fiehn O, Selbig J. Metabolite fingerprinting: Detecting biological features by independent component analysis. Bioinformatics. 2004;20:2447–2454. doi: 10.1093/bioinformatics/bth270. [DOI] [PubMed] [Google Scholar]
- Scholz M, Kaplan F, Guy CL, Kopka J, Selbig J. Non-linear PCA: A missing data approach. Bioinformatics. 2005;21:3887–3895. doi: 10.1093/bioinformatics/bti634. [DOI] [PubMed] [Google Scholar]
- Stepansky A, Kovalski I, Schaffer AA, Perl-Treves R. Variation in sugar levels and invertase activity in mature fruit representing a broad spectrum of Cucumis melo genotypes. Genetic Resources and Crop Evolution. 1999;46:53–62. doi: 10.1023/A:1008636732481. [DOI] [Google Scholar]
- Stone JV. Independent component analysis: An introduction. Trends in Cognitive Sciences. 2002;6:59–64. doi: 10.1016/S1364-6613(00)01813-1. [DOI] [PubMed] [Google Scholar]
- Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TW-M, Fiehn O, Goodacre R, Griffin JL, Hankemeier T, Hardy N, Harnly J, Higashi R, Kopka J, Lane AN, Lindon JC, Marriott P, Nicholls AW, Reily MD, Thaden JJ, Viant MR. Proposed minimum reporting standards for chemical analysis. Metabolomics. 2007;3(3):211–221. doi: 10.1007/s11306-007-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Achnine L, Xu R, Matsuda SPT, Dixon RA. A genomics approach to the early stages of saponin biosynthesis in Medicago truncatula. The Plant Journal. 2002;32:1033–1048. doi: 10.1046/j.1365-313X.2002.01497.x. [DOI] [PubMed] [Google Scholar]
- Trygg, J., Gullberg, J., Johansson, A. I., Jonsson, P., & Moritz, T. (2006). Chemometrics in metabolomics—an introduction. In T. Nagata, H. Lörz, & J. M. Widholm (Series Eds.), K. Saito, R. A. Dixon, & L. Willmitzer (Volume Eds.), Biotechnology in agriculture and forestry, Vol. 57: Plant metabolomics. (pp. 117–128). Springer-Verlag.
- Verhoeckx KC, Bijlsma S, Groene EM, Witkamp RF, Greef J, Rodenburg RJT. A combination of proteomics, principal component analysis and transcriptomics is a powerful tool for the identification of biomarkers for macrophage maturation in the U937 cell line. Proteomics. 2004;4:1014–1028. doi: 10.1002/pmic.200300669. [DOI] [PubMed] [Google Scholar]