


Abstract
DNA sequences determined from ancient organisms have high error rates, primarily due to uracil bases created by cytosine deamination. We use synthetic oligonucleotides, as well as DNA extracted from mammoth and Neandertal remains, to show that treatment with uracil–DNA–glycosylase and endonuclease VIII removes uracil residues from ancient DNA and repairs most of the resulting abasic sites, leaving undamaged parts of the DNA fragments intact. Neandertal DNA sequences determined with this protocol have greatly increased accuracy. In addition, our results demonstrate that Neandertal DNA retains in vivo patterns of CpG methylation, potentially allowing future studies of gene inactivation and imprinting in ancient organisms.
INTRODUCTION
Under favourable conditions, DNA can survive in tissue remains for several millennia and in some cases over 100 000 years (1). Over such long time periods, DNA is invariably affected by degradation and modifications. A ubiquitous feature of ancient DNA is the presence of miscoding lesions that causes incorrect nucleotides to be incorporated during DNA amplification. The identities and underlying biochemical processes of nucleotide misincorporations in ancient DNA have been the subject of some debate (2–5). However, it has now been established (4,6,7) that the vast majority of damage-derived errors in ancient DNA sequences are caused by hydrolytic deamination of cytosine to uracil or possibly hydroxyuracil, which leads to apparent C→T or G→A substitutions in the DNA sequences determined.
Uracil–DNA glycosylase (UDG), which removes uracil residues from DNA to leave abasic sites (8), has been shown to reduce C/G→T/A misincorporations from ancient DNA (4). However, abasic sites prevent replication by Taq polymerase used in the polymerase chain reaction (PCR) (9). Consequently, the use of UDG has not been widely adopted in ancient DNA studies since template molecules in ancient DNA extracts are often limited in number such that UDG treatment may destroy all amplifiable templates available in an extract (4). This is particularly the case when direct PCR is used to retrieve DNA sequences since comparatively long templates are needed to accommodate two primers; furthermore, investigators often target the longest possible templates by PCR in order to reduce work and costs. UDG treatment is then particularly likely to be detrimental since longer template molecules are especially rare in ancient DNA (10) and are more likely than short molecules to contain at least one uracil residue.
High-throughput direct sequencing (11,12), which sequences DNA molecules of all lengths, has opened new possibilities to analyze ancient DNA (13–15). In this approach, the ends of ancient DNA fragments are made amenable to ligation by treatment with T4 DNA polymerase and T4 polynucleotide kinase (PNK). Subsequently, DNA adaptors are ligated to the fragment ends and then used to amplify individual molecules and initiate sequencing reactions on a highly parallelized platform. In theory, UDG treatment would be less harmful with this method than with direct PCR, since most ancient DNA molecules are very short and so are less likely to contain uracil than the few longer molecules accessible to direct PCR. However, direct sequencing of ancient DNA molecules generates many C/G→T/A errors at fragment ends (6). These misincorporations probably derive from single-stranded overhangs of a few bases in the ancient DNA (6,7), where cytosine deamination is much faster than in double-stranded DNA (16). Thus, in some extracts, up to ∼60% of all endogenous DNA fragments are estimated to contain at least one uracil (Supplementary Table S1) and would be excluded from sequencing if the template was treated with UDG. It would therefore be valuable if DNA fragments could be repaired following the removal of uracils by UDG.
Here, we demonstrate that a simple modification to high-throughput sequencing library preparation removes uracil residues from ancient DNA and subsequently repairs the DNA fragments, greatly increasing the accuracy of the DNA sequences determined while maintaining DNA sequence yield from precious DNA sources. In addition, we show that remaining C/G→T/A misincorporations are due to in vivo methylation of cytosine in Neandertal DNA, demonstrating the survival of this epigenetic modification in DNA over 38 000 years old and thus potentially allowing its study in ancient organisms.
MATERIALS AND METHODS
Synthetic oligonucleotides
Oligonucleotide design
Oligonucleotides designed to simulate typical ancient DNA fragments were ordered from Sigma-Aldrich. Each ∼60 bp double-stranded oligonucleotide (A–D, Figure 2) was created by mixing the constituent ssDNA oligonucleotides together to a concentration of 50 μM each, heating to 95°C for 10 s and ramp cooling at 0.5°C s−1 to 25°C. Oligonucleotides were diluted to 5 μM and purified with QIAGEN MinElute spin columns to remove salts left over from oligonucleotide synthesis.
Figure 2.

Demonstration of UDG/endoVIII repair on synthetic oligonucleotides. 1 μg of each of four synthetic double-stranded oligos A-D (top) was subjected to 454 adaptor ligation after enzymatic repair in four conditions: (1) Incubation with PNK followed by addition of T4 polymerase; (2) PNK and UDG followed by T4 polymerase; (3) PNK, UDG and endoVIII followed by T4 polymerase; (4) UDG and endoVIII followed by T4 polymerase (i.e. no PNK). Products were first visualized on agarose gels (middle). The first lane on each gel after the ladder is the untreated oligo. Major bands in the other lanes correspond to the unligated oligos (62–67 bp), the oligos plus one 44-base adaptor and the oligos plus two 44-base adaptors. For some products higher weight bands are visible that probably indicate end-to-end chimeras of the oligos and adaptors. The cause of the faint, diffuse bands seen between 150 and 200 bp in the untreated oligos are unknown but may be artifacts of oligo synthesis. Ligated products were also quantified by qPCR without (dark brown) or with (light brown) prior incubation with UDG. The products marked a and b were sequenced directly on the 454 platform (bottom).
Repair
Each oligo of 500 ng was added to a 50 μl repair reaction containing 1× NE Buffer 2, 0.1 mg ml−1 bovine serum albumen (BSA), 1 mM ATP, 300 μM each of dATP, dCTP, dGTP and dTTP, and one of the following four enzyme repair combinations: (i) 20 U T4 PNK (New England Biolabs); (ii) 20 U PNK, 5 U Escherichia coli UDG (New England Biolabs); (iii) 20 U PNK, 3 U USER enzyme (New England Biolabs). USER enzyme is a proprietary-ratio mixture of UDG and endonuclease VIII (endoVIII) produced by New England Biolabs. Five units of USER enzyme perform similarly in this protocol to a self-made mixture of 5 U UDG and 20 U endoVIII (Supplementary Figure S1). After an incubation period of 3 h at 37°C, 6 U T4 DNA polymerase were added to every tube followed by incubation at 25°C for 30 min. Products were purified with QIAGEN MinElute spin columns and eluted in 14 μl buffer EB (Qiagen).
Ligation
Each purified repair product of 12 μl was mixed with 1 μl 454 adaptors (20 μM each adaptor) (12). The mixture was added to 27 μl ligation mix, making a 40 μl reaction containing 1× Quick Ligation buffer (New England Biolabs) and 1 μl Quick ligase (New England Biolabs). The mixture was incubated at 25°C for 15 min, after which products were purified with QIAGEN MinElute columns and eluted in 14 μl buffer EB.
Adaptor fill-in
Each purified ligation product of 12 μl was added to a 40 μl adaptor fill-in reaction containing 1× Thermopol buffer (New England Biolabs), 300 μM each of dATP, dCTP, dGTP and dTTP, and 16 U Bst DNA polymerase (New England Biolabs). The reaction was incubated for 30 min at 37°C. Note that unlike previous studies (17) we performed fill-in in solution and did not use streptavidin beads, as they are unnecessary (18) and likely to cause some loss of material.
Product visualization, quantification and sequencing
Twenty microliters of products were visualized on a 2.5% agarose gel (Figure 2). In order to quantify ligation success and to measure the extent to which uracils had been successfully removed from the templates, all products were quantified by quantitative (q) PCR using 454 adaptor primers, in the presence or absence of 1 U UDG. Apart from the addition of UDG and a 30 min, 37°C incubation step at the start of the qPCR, conditions for the qPCR were exactly as described (19). Two of the products (Figure 2 and Supplementary Figure S2) were subjected to emulsion PCR and sequenced on a 16th lane of the 454/Roche FLX platform according to the manufacturer’s instructions.
Mammoth DNA
DNA was extracted from 280 mg of a ∼43 000-year-old mammoth bone from Siberia (20) as described (21). The DNA extract was prepared for 454 adaptor ligation and sequencing under different conditions as follows.
Standard blunt end repair
To reproduce the currently published library preparation protocol, 1 μl mammoth DNA extract (out of the 100 μl total) was incubated in a 50 μl reaction containing 1× NEBuffer 2, 1 mM ATP, 0.1 mg ml−1 BSA, 300 uM each of dATP, dCTP, dGTP and dTTP, 20 U PNK and 6 U T4 DNA polymerase. Two replicate reactions were made up, and incubated for 30 min at 25°C. Products were purified with QIAGEN MinElute columns, eluting in 14 μl buffer EB.
Damage repair, no CIP treatment
To test the new damage repair protocol, 50 μl reactions were set up each containing: 1 μl of the mammoth DNA extract, 1× NE Buffer 2, 1 mM ATP, 0.1 mg ml−1 BSA, 300 uM each of dATP, dCTP, dGTP and dTTP, 20 U μl−1 PNK and one of the following three repair enzyme conditions: (i) no repair enzyme; (ii) 5 U UDG; (iii) 3 U USER enzyme (equivalent to 5 U UDG and 20 U endoVIII; see Supplementary Figure S1). Two replicates were performed for each condition. Samples were incubated for 3 h at 37°C, then 6 U T4 DNA polymerase were added to each tube, followed by 30 min incubation at 25°C. Products were purified with QIAGEN MinElute spin columns and eluted in 14 μl buffer EB.
Damage repair, CIP-treatment
Ten microliter of the mammoth DNA extract was included in a 100 μl reaction containing 1× NE Buffer 3, 0.1 mg ml−1 BSA and 20 U CIP(New England Biolabs), incubating for 30 minu at 37°C. The product was purified in one QIAGEN MinElute column and eluted in 30 μl EB. The dephosphorylated product was split equally into six 50 μl reactions, each containing 1× NE Buffer 2, 1 mM ATP, 0.1 mg ml−1 BSA, 300 uM each of dATP, dCTP, dGTP and dTTP, and one of the following three repair enzyme conditions: (i) no repair enzyme; (ii) 5 U UDG; (iii) 3 U USER enzyme. Two replicates were performed per condition. Samples were incubated for 3 h at 37°C, then 6 U T4 DNA polymerase were added to each tube, followed by 30-min incubation at 25°C. Products were purified with QIAGEN MinElute spin columns and eluted in 14 μl buffer EB.
Ligation
All purified products of the standard blunt end repair, no-CIP-treatment damage repair and CIP-treatment damage repair reactions were subjected to the same 454 adaptor ligation and fill-in procedure as follows: 12 μl of each purified repair product was mixed with 1 μl 454 A and B adaptor mix (12), diluted 1:10 relative to the manufacturer’s instructions due to the low amounts of template DNA in this experiment. The mixture was added to 27 μl ligation mix, making a 40 μl reaction containing 1× NEB Quick Ligation buffer and 1 μl NEB Quick ligase. The mixture was incubated at 25°C for 15 min, after which products were purified with QIAGEN MinElute columns and eluted in 15 μl buffer EB.
Adaptor fill-in
Each purified ligation product of 12 μl was added to a 40 μl adaptor fill-in reaction containing 1× NEB Thermopol buffer, 300 μM each of dATP, dCTP, dGTP and dTTP, and 16 U Bst DNA polymerase (New England Biolabs). The reaction was incubated for 30 min at 37°C followed by 20 min at 80°C to inactivate the Bst polymerase. As with the synthetic oligonucleotide experiment described above, streptavidin beads were not used in this step.
Quantification and sequencing
Each product of 1 μl was quantified directly using qPCR as described (19). Products were then subjected to emPCR at 2 copies per bead, and each sequenced on one 16th lane of the 454/Roche FLX platform. Sequences were aligned to the draft elephant genome (Loxafr2.0, AAGU00000000.2/GI:202071911) using a custom mapper, ANFO (22) (freely available for download at http://bioinf.eva.mpg.de/anfo/).
Neandertal DNA
DNA was extracted from 150 mg of a ∼38 000-year-old Neandertal bone from Vindija cave, Croatia (23) as described (21). Thirty-two microliters of the 100 μl total extract was split equally into four 50 μl reactions containing 1× NE Buffer 2, 1 mM ATP, 0.1 mg ml−1 BSA, 250 uM each of dATP, dCTP, dGTP and dTTP, 20 U PNK, and either (1) no repair enzyme or (2) 5 U UDG and 20 U endoVIII. Reactions were incubated for 3 h at 37°C, before 6 U T4 DNA polymerase was added, followed by 30 min at 25°C. Products were purified with QIAGEN MinElute columns and eluted in 14 μl EB. Ligation and fill-in were performed as described in (17), including use of a modified 454 adaptor containing a Neandertal project-specific key (6). Note that in this experiment, streptavidin beads were used in the fill-in step unlike in the synthetic oligonucleotide and mammoth DNA experiments; this was because we had not switched at that point to the more efficient no-beads protocol (18). To make use of the greater parallel sequencing capacity of the Illumina Genome Analyzers (GA) platform relative to 454/Roche, the library was first universally amplified for 5 PCR cycles using the protocol described in (24). The amplified product was subsequently converted into an Illumina-amenable library by a 7 cycle re-amplification under similar conditions except that tailed PCR primers were used that attach the Illumina P5 and P7 grafting sequences outside the 454 adaptor sequences (sequences available on request). This allowed subsequent bridge amplification and 2 × 51 bp paired end sequencing runs to be performed on the GAII platform, following to the manufacturer’s instructions except that sequencing primers were used that anneal to the 454 adaptor sequences. The sequencing run was analyzed starting from raw images using the Illumina GA pipeline 1.3.2. To overcome issues introduced by identical key sequences at the beginning of the first read, for cluster identification the first five sequencing cycles were used. The Ibis basecalling program was used (25). Raw sequences for the two paired end reads of each sequencing cluster were merged by checking for a minimum 11nt overlap between the first and the second read. For bases in the overlapping sequence, a consensus was called by considering the base with the higher quality score or, in case of agreement, summing up the quality scores observed. For further analysis, only successfully merged sequences were considered and aligned to the human genome (NCBI 36.1/hg18), all CpG islands annotated by UCSC (http://genome.ucsc.edu/cgi-bin/hgTables; table: cpgIslandExt) and the mtDNA sequence of this individual (AM948965) using a custom mapper, ANFO (22). Custom scripts were used to count alignment mismatch frequencies.
RESULTS
A UDG/endoVIII repair scheme
In library preparation protocols for high-throughput sequencing of ancient DNA, PNK and T4 DNA polymerase are used to generate ends amenable to DNA ligation (17). This is achieved by phosphorylation of non-phosphorylated 5′-ends by PNK, and removal of 3′-overhangs and fill-in of 5′-overhangs by T4 polymerase, producing blunt ended, 5′-phosphorylated molecules. A large proportion of nucleotide misincorporations generated from ancient DNA libraries are caused by uracils present in short 5′-overhangs in ancient DNA fragments, which are filled in during this end-repair reaction (6). We reasoned that if DNA is incubated with UDG and endoVIII prior to T4 DNA polymerase treatment, this would result in the removal of uracil residues from DNA by UDG, and cleavage on the 5′- and 3′-sides of the resulting abasic sites by endoVIII. PNK and T4 polymerase would then remove 3′-phosphate groups and generate blunt ends (Figure 1), thus avoiding the loss of these molecules from the library.
Figure 1.
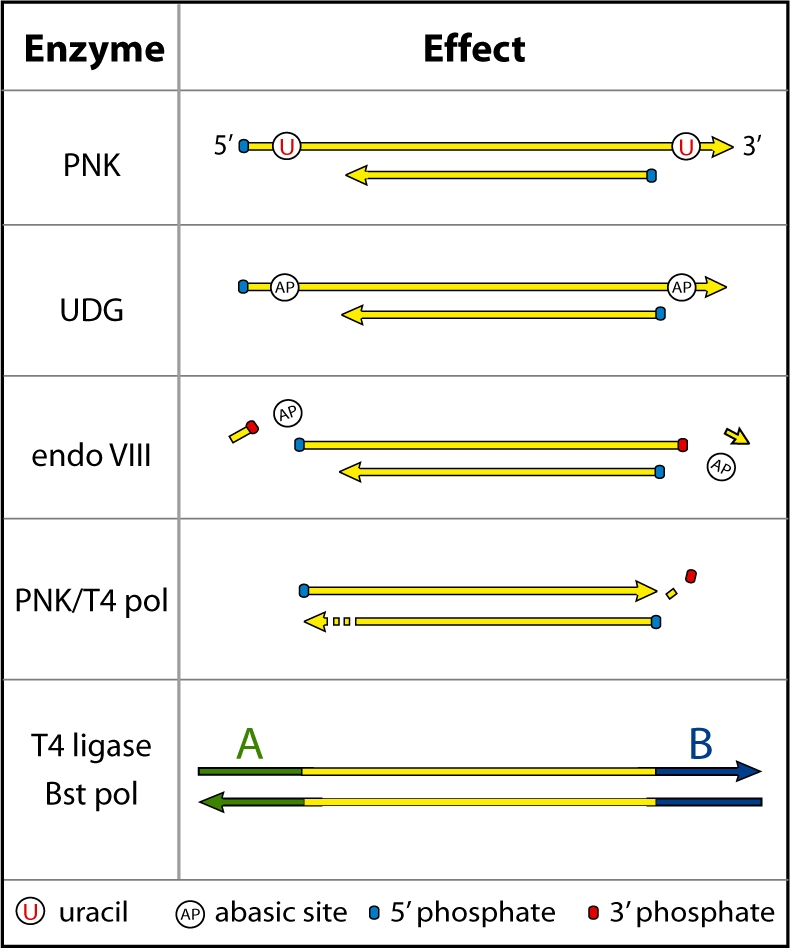
Predicted activity of UDG and endoVIII in 454 library preparation of ancient DNA. T4 PNK phosphorylates 5′-ends leaving 5′-phosphate groups. UDG removes uracils, which are concentrated in short 5′- and 3′-overhangs in ancient DNA, leaving abasic sites. EndoVIII then cleaves on both sides of the abasic sites, leaving 5′- and 3′-phosphate groups. T4 polymerase fills in remaining 5′-overhangs and chews back 3′-overhangs, possibly aided by the 3′-phosphatase activity of PNK. Blunt-end ligation and fill-in of sequencing adaptors can then take place.
Repair of oligonucleotides
In order to test the ability of UDG and endoVIII to repair ancient DNA, we designed four double-stranded oligonucleotides that carry short overhanging ends (Figure 2). Whereas oligos A and B contain exclusively the four natural DNA bases, C and D carry uracil bases in their overhanging ends as predicted to frequently occur in ancient DNA (6). Oligos A and C carry 5′-overhangs whereas oligos B and D carry 3′-overhangs. Oligo C carries a uracil base in the second position of the 5′-overhangs and is therefore the type of fragment that the UDG/endoVIII protocol is designed to repair. Oligo D carries a uracil base in the second position of the 3′-overhangs. In 3′-overhangs, deaminated cytosine should not lead to nucleotide misincorporations in ancient DNA sequences because T4 polymerase removes 3′-overhangs before adaptor ligation. However, we wanted to investigate if T4 polymerase activity may be blocked by the abasic sites left after UDG treatment or the 3′-phosphates left by endoVIII treatment (26) (Figure 1).
The three oligonucleotides were treated in four different ways: (i) With PNK to phosphorylate 5′-ends, followed by T4 polymerase to generate blunt ends; (ii) as 1 with the addition of UDG together with PNK; (iii) as 2 with the addition of endoVIII with the UDG and PNK and (iv) as 3 except that PNK was not used. Subsequently, 454 sequencing adaptors were ligated to the oligos. The ligation reactions were visualized on ethidium-stained agarose gels, and analyzed by quantitative (q) PCR with primers specific for the 454 adaptors, either with or without prior treatment with UDG to gauge the amount of uracil residues in the ligation product (Figure 2).
For oligos A and B, which contain no uracils, gel and qPCR results show that adaptor ligation was similarly efficient for treatments 1–3 (Figure 2). This indicates that UDG and endoVIII do not interfere with the generation of blunt ends in non-damaged DNA. The observed level of between-treatment variation is expected given that two spin column purification steps were performed during the procedure.
Oligo C, which contains uracil residues in 5′-overhanging ends, showed high ligation efficiency following treatment 1. However, the ligated product showed greatly reduced copy number in qPCR if treated with UDG prior to amplification. This is expected given that the ligated product should contain one uracil on each strand. Ligation efficiency was very low after treatment 2, consistent with UDG creating abasic sites and preventing T4 polymerase fill-in of ends. After treatment 3, ligation efficiency was high, and in this case the product was insensitive to UDG treatment prior to qPCR. This indicates that the uracils had been removed prior to ligation, as predicted by the scheme in Figure 1.
Oligo D, which contains uracil residues in 3′-overhanging ends, showed a pattern similar to oligos A, B and C in that ligation efficiency was high after treatments 1–3. This indicates that uracil-containing 3′-overhangs can be successfully removed by T4 polymerase after UDG and endoVIII treatment. The products were largely insensitive to UDG treatment prior to qPCR, as expected since uracils in 3′-overhangs should be removed before ligation. The apparent ∼40% sensitivity to UDG after treatment 1 is surprising, but may be due to uracil misincorporation during manufacture of this oligo. Since oligos are synthesized in the 3′- to 5′-direction, uracil was added in the first coupling step and carry over to later synthesis steps would lead to UDG sensitivity of the ligated product.
When PNK is omitted (treatment 4), only oligo C shows a full-length ligation product. This demonstrates that the UDG/endoVIII repair reaction generates ligatable phosphorylated 5′-ends after removal of the uracils and cleavage of phosphodiester bonds by endoVIII (Figure 1).
To confirm that UDG/endoVIII repair of oligo C removes uracil-derived errors from DNA sequences as predicted (Figure 1), we sequenced oligo C products of treatments 1 and 3. As expected, after treatment 1, the oligo C product is full length and carries nucleotide misincorporations where uracil residues have been replaced by thymine residues. In contrast, after treatment 3, the oligo C product has been shortened by two bases at both ends, removing the uracil positions from the sequence (Figure 2).
Repair of mammoth DNA
We next tested the UDG/endoVIII protocol on DNA extracted from a ∼43 000-year-old woolly mammoth bone. The extract was subjected to various repair reactions in duplicate, followed by 454 adaptor ligation, qPCR quantification and determination of between 783 and 20 935 sequences per sample on the 454 platform. The results were analyzed with respect to numbers of molecules in the 454 libraries generated as well as the percent of sequences in the libraries that show similarity to the elephant genome and are thus assumed to be of mammoth origin. In most ancient remains, only a small proportion of sequences retrieved are from the organism under study. For this extract, it is ∼3%, the rest presumably stemming from microorganisms that have colonized the sample after the death of the individual. If the endogenous mammoth DNA carried more DNA modifications than the environmental DNA, the relative amount of mammoth DNA might be changed by the repair reactions. Other relevant aspects such as the length of the mammoth sequences and the frequency and type of nucleotide misincorporations were also analyzed.
One issue that potentially complicates the application of the UDG/endoVIII protocol to ancient DNA extracts is that DNA modifications other than deaminated cytosines may be present (27,28). Therefore, we first tested if mammoth DNA retrieval was affected by the longer incubation (3 h) at a higher temperature (37°C) that differs from the standard library preparation (30 min at 25°C) and thus could conceivably cause DNA degradation by affecting some unknown modifications. Neither in terms of numbers of molecules in the resultant 454 library, the percent of mammoth sequences, the length of mammoth sequences or patterns of misincorporations did the incubation conditions strongly influence the results (Supplementary Figure S3).
We next tested the three treatments previously performed on the oligonucleotides, i.e. (i) PNK + T4 polymerase; (ii) PNK + T4 polymerase + UDG; (iii) PNK + T4 polymerase + UDG + endoVIII. In a second series of experiments, the mammoth DNA extract was dephosphorylated with CIP, purified, and then subjected to the same three treatments except that PNK was never used. Dephosphorylation should prevent the DNA from successful 454 adaptor ligation except in cases where UDG and endoVIII generates phosphorylated 5′-ends where uracil bases exist close to the 5′-ends (c.f. oligo B in Figure 2). Thus, these experiments tested if endoVIII is active on the mammoth DNA.
Total library yield, which includes mammoth and microbial sequences, is reduced by ∼30% upon UDG treatment (Figure 3a), consistent with uracil removal from damaged DNA fragments and blockage of T4 polymerase fill-in or Taq polymerase amplification by the resulting abasic sites. When endoVIII is added, library copy numbers increase by ∼40% relative to the standard condition. This is unexpected since endoVIII is expected to rescue molecules carrying abasic sites generated by UDG but not to increase copy numbers relative to the standard treatment. We repeated this experiment with more replicates per condition, and again observed a reduction in yield upon UDG treatment but an increase in yield after UDG/endoVIII treatment only to approximately the no repair condition (data not shown). We conclude that UDG reduces copy number of a mammoth DNA library relative to the no repair condition, while UDG together with endoVIII does not, suggesting the repair of uracil-containing DNA fragments. None of the treatments differed in the percent of mammoth DNA sequence, suggesting that uracil or any other unknown modifications that might be affected by the treatments did not differ substantially in frequency between the mammoth DNA and the environmental DNA.
Figure 3.

The UDG/endoVIII repair protocol applied to mammoth DNA. (a) A mammoth DNA extract was subjected to 454 adaptor ligation after enzymatic repair in replicates in three conditions: (1) Incubation with PNK followed by T4 polymerase; (2) PNK and UDG followed by T4 polymerase; (3) PNK, UDG and endoVIII followed by T4 polymerase. The resulting libraries were quantified by qPCR. The same reactions without PNK were applied to mammoth DNA that had first been dephosphorylated by CIP (treatments 4–6). (b) All libraries were sequenced on 454. The proportions of sequences that aligned to the elephant genome are shown. (c) For the sequences resulting from treatments 1–3 and 6 the base composition of aligned elephant sequences is shown for 10 bases either side of the 5′-start point of mammoth sequences.
When the DNA was dephosphorylated prior to the experiments, a small amount of amplifiable material was present after treatments 1 and 2, probably representing both incomplete dephosphorylation of naturally phosphorylated 5′-ends by CIP and 454 adaptor artifacts. This is supported by the observation that only a very small proportion of the sequences determined after this treatment are of mammoth origin (Figure 3b). In contrast, treatment 3 yielded ∼3.5 times more amplifiable library molecules than treatments 1 and 2, consistent with direct selection of DNA fragments that had uracil near the 5′-ends of both strands. Notably, after treatment 3, the proportion of mammoth sequences was 10–12%, four times higher than in the libraries made from non-CIP treated mammoth DNA in the presence of PNK. This suggests that the mammoth DNA carried more uracil residues on average than the environmental DNA, although this is probably a modest difference because selecting for damage at both fragment ends will multiply the effect of any difference in damage between sequence populations. Thus, the observed ∼4-fold enrichment of mammoth DNA suggests a maximum 2-fold difference between the fraction of mammoth DNA strands and environmental DNA strands that carry uracil residues close to their 5′-ends. This is a small enough effect to be compatible with the fact that no obvious differences in mammoth DNA percentage were observed among the non-CIP-treated libraries where PNK was used.
The 5′-start points of the mammoth sequences represent the terminal bases of the ancient DNA fragments at the point of adaptor ligation (6). We plotted the base composition of the elephant reference sequence aligned to the mammoth sequences for 10 bases on either side of the mammoth 5′-ends. UDG/endoVIII treatment should lead to an increase in cytosine frequency at the base position immediately preceding 5′-ends (i.e. the –1 position in Figure 3c), as DNA fragments containing deaminated cytosine will be truncated to the 3′-side of such positions (Figure 1). Consistent with this prediction, we observed in the non-CIP-treated libraries an increased frequency of cytosine at position –1 in the UDG/endoVIII condition relative to the no-repair and UDG-treated conditions. This effect was much stronger in the CIP-treated, UDG/endoVIII repaired sample, where 84% of 5′-ends are preceded by cytosines. This is predicted since this library should predominantly contain fragments where ligatable ends were generated by excision and repair at positions of deaminated cytosine.
The average length of mammoth sequences is around 70–80 nt (Supplementary Figure S4), as is typical of ancient DNA (29). After UDG treatment, fragments are ∼10 nt shorter. This is expected since longer fragments are more likely to contain uracil and thus to be left with an abasic site after UDG treatment. When endoVIII is added, length seems to be intermediate, perhaps representing rescue of some longer fragments by endoVIII treatment. If the DNA is dephosphorylated before UDG and endoVIII treatment, length is ∼10 nt shorter than after UDG and endoVIII treatment without CIP-treatment, as expected if all of the ligated fragments in this condition have been truncated at both ends due to UDG/endoVIII repair, as opposed to the non-CIP-treated libraries where only a fraction of the fragments will have been truncated.
We investigated the effect of the repair protocol on nucleotide misincorporations, by plotting for each library the frequency of each of the 12 possible mismatches observed in the mammoth-to-elephant alignments. Ancient DNA sequences generally show an excess of C→T and G→A substitutions due to uracil-derived nucleotide misincorporations (4,6,7). As expected, this pattern is seen when the sample is not subjected to UDG treatment (Supplementary Figure S4). By taking the excess of C/G→T/A substitutions relative to T/A→C/G substitutions as an estimate of the amount of uracil-derived misincorporations in each sample, we found that uracil-derived misincorporations were reduced 4- to 10-fold in samples where UDG was used (Supplementary Figure S4).
In summary, UDG–endoVIII treatment allows more mammoth DNA sequences to be retrieved than UDG treatment alone, while generating much lower rates of nucleotide misincorporations than if UDG is not used.
Repair of Neandertal DNA
Mammoth and African elephant DNA diverged from each other more than ∼7 million years ago (30,31). Therefore genuine sequence differences will dominate in pairwise alignments, reducing the ability to study patterns of nucleotide misincorporations. In contrast, Neandertals and humans diverged much more recently. Furthermore, in contrast to the elephant genome the human genome is of finished quality allowing better resolution of nucleotide misincorporations. We therefore applied the UDG/endoVIII repair protocol to DNA extracted from a 38 000-year-old Neandertal bone from Croatia and performed deep sequencing of the libraries. An additional advantage of this specimen is that its complete mitochondrial (mt) sequence is known (23), allowing detailed analysis of sequence errors in mtDNA. This may be particularly interesting as mtDNA does not carry 5-methylcytosine (5′-m-C), a naturally occurring modification in vertebrate nuclear DNA which when deaminated is converted to thymine (32). Since UDG will not remove thymine from DNA, deamination-derived misincorporations may still occur at sites of cytosine methylation after UDG treatment of the ancient DNA.
Between 10 and 11 million raw DNA sequence reads were generated on the Illumina GAII platform from four Neandertal DNA libraries prepared in the standard condition or with the UDG/endoVIII protocol (each in two replicates). In order to improve sequencing accuracy, which decreases substantially toward the 3′-end of Illumina reads (25), we sequenced into both ends of each molecules and merged the paired reads, requiring overlaps of at least 11 bases, and discarded all sequences that could not be merged to their paired read. Since the single read length was 48 bases, this creates a maximum read length of 85 bp. However, previous work has shown that the majority of Neandertal fragments in this bone are shorter than this (33).
Sequences were aligned to the previously published complete mtDNA sequence of this Neandertal (23), and to the human nuclear genome (hg18) using a custom mapping program (ANFO) (22). In each library, 0.007–0.009% of sequences aligned well to the mtDNA and 2.0–2.2% to the nuclear genome. Variation in these proportions was as high within as between treatments. Data from replicates were then pooled for each treatment and patterns of nucleotide mismatches between the sequences and the references were analyzed.
When analyzing Neandertal DNA sequences, contamination of experiments with contemporary human DNA is a potential problem (10,34). However, the level of such contamination in a Neandertal DNA library can be assessed by counting the ratio of Neandertal versus contaminant fragments at nucleotide positions where Neandertals differ from all or almost all present-day humans (33). The mtDNA of this Neandertal carries 133 such diagnostic positions (23). The ‘no repair’ dataset yielded 139 mtDNA fragments that overlapped such positions; 138 carried the Neandertal base while one matched modern human mtDNA. The UDG/endoVIII treated dataset yielded 128 informative fragments, of which all were the Neandertal type. Thus, the mtDNA in all libraries was almost completely free of contamination by modern human mtDNA, even after treatment with UDG and endoVIII. Since the ratio of mitochondrial to nuclear DNA may differ between the contaminating and the Neandertal DNA, this estimate is strictly applicable only to the mtDNA (33). However, the estimate of mtDNA contamination in these libraries is low enough that within even a few-fold variation in mtDNA:nuclear DNA ratios between the Neandertal and contaminating DNA, sequences aligning to the human nuclear genome will be predominantly of Neandertal origin.
To analyze patterns of nucleotide misincorporations, we plotted the frequency of each of the 12 possible nucleotide differences in alignments as a function of distance from the ends of DNA fragments for each dataset (Figure 4, top panels). In the ‘no repair’ results, the transitions C→T and G→A are drastically elevated toward the 5′- and 3′-ends ends of fragments, respectively, with up to 40–50% error frequencies at the fragment ends. This occurs in both mtDNA and nuclear DNA fragments, as shown previously (6,23). The G→A substitutions at 3′-ends of fragments originate during the fill-in of 5′-overhangs containing deaminated cytosines (6,7). After UDG/endoVIII treatment C/G→T/A differences were drastically reduced, consistent with the removal of uracils by UDG. In mtDNA this removal seems to be almost complete, whereas a small amount remains in nuclear DNA (see below).
Figure 4.

Effect of UDG/endoVIII treatment on all nucleotide misincorporation rates and on C→T rates at CpN dinucleotides along Neandertal DNA sequences. DNA from a Neandertal bone was subjected to 454 adaptor ligation after enzymatic repair with PNK and T4 polymerase (‘no repair’) as well as those enzymes plus UDG and endoVIII. Approximately 12 million sequences were generated on the Illumina GAII platform for each condition, and sequences were identified that aligned best to the complete mtDNA sequence of this bone AM948969 (947 and 1084 sequences for the no repair and UDG/endoVIII conditions, respectively, left panels) and to the human nuclear genome sequence hg18 (242 070 and 286 737 sequences, right panels). (a) All 12-nt mismatch frequencies are plotted as a function of position along the aligned fragments for the no repair and UDG/endoVIII conditions. (b) The rate of C→T misincorporations along DNA fragments is shown separately for the four dinucleotides that contain cytosine in the 5′-position.
Neandertal CpG methylation
In vertebrates, DNA methylation occurs at the 5′-position of cytosine residues at ∼80% of CpG dinucleotides in somatic tissues (35). When 5′-m-C is deaminated it becomes a thymine residue that is not recognized as an unnatural base by UDG in vertebrate cells. As a consequence, CpG dinucleotides are preferentially lost from vertebrate genomes with the result that the frequency with which CpG dinucleotides occur genome wide is ∼1%, while 4–5% would be expected from the base composition of the genome (36). In some regions of the genome, CpG content is less reduced and can be 60% or more of the statistically expected frequency. Such ‘CpG islands’ represent areas where methylation is reduced or absent. About half of CpG islands overlap transcriptional start sites where their methylation in a tissue is positively correlated with suppression of transcription.
We noted that after UDG/endoVIII treatment ∼5% of C/G→T/A substitutions remained at the first and last positions of Neandertal DNA fragments in nuclear DNA whereas this did not seem to be the case in the mtDNA (Figure 4). Since cytosine methylation does not occur in mtDNA we speculated that this might be due to cytosine methylation at CpG dinucleotides. We therefore analyzed the rates of C→T mismatches seen along the DNA molecules separately for the four dinucleotides that carry cytosine at their first position (Figure 4, lower panels). Without UDG/endoVIII treatment, C→T differences to the human nuclear genome are common in all four dinucleotide contexts, but slightly more common in CpG dinucleotides (e.g. 52.4% at position 1 of DNA fragments) than in the other dinucleotides (38%–40% at position 1). This is likely due to the higher deamination susceptibility of 5′-m-C relative to unmethylated cytosine (32). With UDG/endoVIII treatment, C→T substitutions in the three non-CpG dinucleotides are greatly reduced (to 2–5% at position 1 of DNA fragments), whereas they remain abundant in CpG dinucleotides (40.6% at position 1 of DNA fragments). Thus, at CpG sites, ∼80% of C→T substitutions are resistant to UDG/endoVIII treatment, consistent with findings that ∼80% of CpG sites are methylated (35). At CpG sites in mtDNA, no such resistance of substitutions to UDG/endoVIII treatment is seen. This is consistent with the absence of CpG methylation in mitochondria.
In order to further investigate if the UDG/endoVIII protocol can be used to detect CpG methylation present in the Neandertal individual when alive, we analyzed CpG→TpG mismatch rates in CpG islands, where methylation is reduced relative to the genome-wide average (37) (Figure 5). As described, across the genome 52.4% of CpGs at 5′-ends of fragments are read as TpG without UDG/endoVIII treatment, and this is reduced to 40.6% upon UDG/endoVIII treatment. At CpG islands, 51.7% of 5′-terminal CpGs are read as TpG without UDG/endoVIII treatment and this is reduced to 23.4% with UDG/endoVIII treatment. Thus, deaminated CpG sites in CpG islands are more susceptible to UDG/endoVIII treatment than deaminated CpG sites elsewhere in the genome. This is consistent with a reduced frequency of cytosine methylation in CpG islands. Together with the observation that no resistance to UDG treatment is observed at deaminated CpG sites in mtDNA, we conclude that in vivo Neandertal DNA methylation patterns have survived over 38 000 years in the bone, and can be detected by the UDG/endoVIII protocol.
Figure 5.

CpG→TpG misincorporation rate along Neandertal DNA molecules, genome-wide and inside CpG islands. DNA from a Neandertal bone was subjected to 454 adaptor ligation after enzymatic repair with PNK and T4 polymerase (‘no repair’) as well as those enzymes plus UDG and endoVIII. Approximately 12 million sequences were generated on the Illumina GAII platform for each condition, and sequences were identified that aligned best to the human nuclear genome sequence hg18 (left panels) or specifically to CpG islands in the human genome (right panels) (35). The rates of CpG→TpG mismatches are shown for the first 30 bases of DNA fragments.
Neandertal DNA sequence accuracy
Although UDG/endoVIII treatment of ancient DNA clearly reduces nucleotide misincorporations, a remaining limitation to obtaining maximal sequence accuracy from ancient DNA molecules is the high rate of machine sequencing errors of current high-throughput platforms. The ‘merged-paired-end’ sequencing approach used here helps to decrease machine errors by requiring that the 3′-ends of fragments, where errors are most common (25), are sequenced in two directions. However, near-elimination of machine sequencing errors should be possible if libraries are first amplified (24,29), as they were here, and sequenced deeply enough so that multiple copies of each original molecule are sequenced, allowing a consensus to be generated from such replicate sequences, which can be identified by their strand orientation, start and end points (6,13).
To investigate the accuracy, that can be achieved when combining a deep sequencing approach with UDG/endoVIII repair, we determined the overall rates of each Neandertal nucleotide misassignment in (i) all sequences from the ‘no-repair’ dataset; (ii) all sequences from the UDG/endoVIII dataset; and (iii) all ‘multi-pass’ sequences from the UDG/endoVIII dataset, which we generated by taking consensus sequences of all distinct fragments that were sequenced more than once. Figure 6 shows that for mitochondrial sequences, overall error rate per base (Figure 6) was 2.20% for ‘no repair’ sequences; 0.40% for UDG/endoVIII sequences and 0.09% for multi-pass UDG/endoVIII-treated sequences. Thus, UDG/endoVIII treatment alone results in a 5.5-fold reduction in error rates while deep sequencing results in an additional 4.4-fold reduction. In combination this results in a 22-fold reduction in errors. In nuclear DNA, for which we removed CpG sites from the analysis due to the effect of methylation described above, a similar pattern is observed (Figure 6) although the true error rate at these low levels cannot be accurately calculated due to genuine sequence differences between this Neandertal and the human reference.
Figure 6.

Effects of UDG/endoVIII treatment and multiple-pass sequencing on ancient DNA sequence error rate. DNA from a Neandertal bone was subjected to 454 adaptor ligation after enzymatic repair with PNK and T4 polymerase (no repair) as well as those enzymes plus UDG and endoVIII. Approximately 12 million sequences were generated on the Illumina GAII platform for each condition, and sequences were aligned to the complete mtDNA sequence of this bone and to the human nuclear genome (hg18). Overall rates of each alignment mismatch type, and total mismatch rates, are shown for sequences from each condition, plus the result achieved when only fragments that were sequenced more than once (‘multi-pass’) are considered. For nuclear DNA, positions of CpG in the reference were excluded from the analysis since misincorporations remain frequent at these sites even after UDG/endoVIII treatment (see Figure 4).
DISCUSSION
The damaged state of DNA recovered from ancient remains causes two major problems for the determination of ancient DNA sequences. First, the DNA quantities recovered are often extremely low. Second, chemical modification of bases, in particular deamination of cytosine to uracil, leads to frequent sequence errors. Because even in highly damaged DNA, only a minority of cytosines are deaminated (4,6,7), and deaminated cytosines are largely randomly distributed in ancient DNA sequences (4), deamination-derived errors can usually be excluded at positions where three or more overlapping molecules are sequenced, and a consensus is used (4,38). However, in sequencing libraries made from ancient DNA, sequence coverage is often so low that deamination-derived errors represent a problem.
When nuclear DNA sequences in ancient diploid organisms are determined, a further problem is caused by the need to accurately determine both alleles at heterozygous positions, or to select one correctly determined allele in an unbiased way. When sequencing ancient nuclear DNA without removing uracil, it will be hard to distinguish true C/T or G/A heterozygote sites from sites of cytosine deamination. Since homozygote sites greatly outnumber heterozygote sites [∼1000:1 in present-day humans (39)], events where multiple damaged and undamaged templates are sequenced at a true homozygote site may be more common than the occurrence of true heterozygote sites. This could create a high proportion of false-positive heterozygote calls. If just one allele is required to be accurately called, errors could in theory be avoided by always taking the C or G allele at apparently C/T- or G/A-heterozygote sites. However, this will introduce a severe bias in population genetic analyses by systematically calling only one possible allele at heterozygote sites. Thus, it would be very valuable to reduce error rates when ancient nuclear DNA sequences are determined.
The UDG/endoVIII protocol presented here removes the vast majority of errors caused by miscoding lesions. When combined with machine error removal by sequencing multiple copies of each original molecule, the overall per-base error rate is reduced over 20-fold relative to single-pass sequencing of unrepaired DNA, at least for Neandertal mtDNA. We are unable to measure this reduction accurately for nuclear DNA due to genuine sequence differences between the Neandertal and the human reference sequence. However, outside CpG sites the error rate in nuclear DNA is presumably similar to that in mtDNA, at roughly 1 error per 1000 bases in UDG/endoVIII treated, multi-pass sequences.
It has been previously reported (4) that UDG treatment removes C/G→T/A errors from ancient DNA sequences. However, since UDG treatment causes molecules to be lost, use of the enzyme has not been widely adopted, with some exceptions (40). UDG is particularly problematic for direct PCR studies, which usually target the longest possible template molecules; longer templates are more likely to contain at least one uracil and so be destroyed by UDG treatment. Fortunately, the introduction of direct high-throughput sequencing of ancient DNA has allowed very short (<100 bp) DNA fragments, which make up the vast majority of ancient DNA, to be efficiently sequenced in large quantities. Since uracil bases are concentrated close to the 5′- and 3′-ends in single-stranded overhangs (6,23,29), endoVIII will rescue many of the fragments that would have been excluded from sequencing after UDG treatment alone (Figure 1). The UDG/endoVIII protocol also repairs sites of uracil in double-stranded parts of molecules, although in our hands this repair is only ∼20% efficient (data not shown). Further work exploring the effects of other repair enzymes on ancient DNA library yield and sequence accuracy may be useful. However, it will be desirable for future repair approaches to avoid any extra purification steps, as these inevitably cause loss of material.
The UDG/endoVIII protocol has the additional advantage that any remaining C/G→T/A misincorporations can be attributed to deamination of methylated cytosine residues (provided that UDG treatment is complete). The observation that C→T substitutions in CpG dinucleotides, but not in other dinucleotides, are resistant to repair by UDG and that this pattern does not occur in mtDNA but in nuclear DNA where it is reduced in CpG islands, strongly suggests that the signal stems from in vivo Neandertal methylation as opposed to postmortem DNA modifications. In order to determine the methylation status of any particular CpG site by this approach, very high sequence coverage would obviously be needed. However, the methylation status of a region such as a particular gene or CpG island will be measurable with more moderate sequence coverage. Furthermore, emerging techniques such as bisulphite treatment of small DNA quantities (e.g. Genetic Signatures’ MethylEasy Xceed kit) or single-molecule sequencing technologies that can distinguish 5′-m-C from C (41) may soon allow high-resolution analysis of methylation in ancient DNA. It might therefore become possible to investigate the activity of genes and phenomena such as X chromosomal inactivation and genetic imprinting in extinct species, provided that the signals are manifested in cells present in bones.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
We thank the Presidential Innovation Fund of the Max Planck Society for funding. Funding for open access charge: Max Planck Society.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Hernan Burbano, Paul Czechowski, Ed Green, Gregory Hannon, Emily Hodges, Tomislav Maricic, Philip Johnson, Pavao Rudan, Dejana Brajković, Željko Kućan and Ivan Gušić and the Croatian Academy of Sciences and Arts for collaboration and help.
REFERENCES
- 1.Orlando L, Darlu P, Toussaint M, Bonjean D, Otte M, Hanni C. Revisiting Neandertal diversity with a 100,000 year old mtDNA sequence. Curr Biol. 2006;16:R400–R402. doi: 10.1016/j.cub.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 2.Gilbert MT, Binladen J, Miller W, Wiuf C, Willerslev E, Poinar H, Carlson JE, Leebens-Mack JH, Schuster SC. Recharacterization of ancient DNA miscoding lesions: insights in the era of sequencing-by-synthesis. Nucleic Acids Res. 2007;35:1–10. doi: 10.1093/nar/gkl483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilbert MT, Hansen AJ, Willerslev E, Rudbeck L, Barnes I, Lynnerup N, Cooper A. Characterization of genetic miscoding lesions caused by postmortem damage. Am. J. Hum. Genet. 2003;72:48–61. doi: 10.1086/345379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hofreiter M, Jaenicke V, Serre D, Haeseler Av A, Paabo S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001;29:4793–4799. doi: 10.1093/nar/29.23.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stiller M, Green RE, Ronan M, Simons JF, Du L, He W, Egholm M, Rothberg JM, Keates SG, Ovodov ND, et al. Patterns of nucleotide misincorporations during enzymatic amplification and direct large-scale sequencing of ancient DNA. Proc Natl Acad. Sci. USA. 2006;103:13578–13584. doi: 10.1073/pnas.0605327103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Briggs AW, Stenzel U, Johnson PL, Green RE, Kelso J, Prufer K, Meyer M, Krause J, Ronan MT, Lachmann M, et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl Acad. Sci. USA. 2007;104:14616–14621. doi: 10.1073/pnas.0704665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brotherton P, Endicott P, Sanchez JJ, Beaumont M, Barnett R, Austin J, Cooper A. Novel high-resolution characterization of ancient DNA reveals C > U-type base modification events as the sole cause of post mortem miscoding lesions. Nucleic Acids Res. 2007;35:5717–5728. doi: 10.1093/nar/gkm588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindahl T, Ljungquist S, Siegert W, Nyberg B, Sperens B. DNA N-glycosidases: properties of uracil-DNA glycosidase from Escherichia coli. J. Biol. Chem. 1977;252:3286–3294. [PubMed] [Google Scholar]
- 9.McDonald JP, Hall A, Gasparutto D, Cadet J, Ballantyne J, Woodgate R. Novel thermostable Y-family polymerases: applications for the PCR amplification of damaged or ancient DNAs. Nucleic Acids Res. 2006;34:1102–1111. doi: 10.1093/nar/gkj512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paabo S, Poinar H, Serre D, Jaenicke-Despres V, Hebler J, Rohland N, Kuch M, Krause J, Vigilant L, Hofreiter M. Genetic analyses from ancient DNA. Annu. Rev. Genet. 2004;38:645–679. doi: 10.1146/annurev.genet.37.110801.143214. [DOI] [PubMed] [Google Scholar]
- 11.Bennett S. Solexa Ltd. Pharmacogenomics. 2004;5:433–438. doi: 10.1517/14622416.5.4.433. [DOI] [PubMed] [Google Scholar]
- 12.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green RE, Krause J, Ptak SE, Briggs AW, Ronan MT, Simons JF, Du L, Egholm M, Rothberg JM, Paunovic M, et al. Analysis of one million base pairs of Neanderthal DNA. Nature. 2006;444:330–336. doi: 10.1038/nature05336. [DOI] [PubMed] [Google Scholar]
- 14.Noonan JP, Coop G, Kudaravalli S, Smith D, Krause J, Alessi J, Chen F, Platt D, Paabo S, Pritchard JK, et al. Sequencing and analysis of Neanderthal genomic DNA. Science. 2006;314:1113–1118. doi: 10.1126/science.1131412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poinar HN, Schwarz C, Qi J, Shapiro B, Macphee RD, Buigues B, Tikhonov A, Huson DH, Tomsho LP, Auch A, et al. Metagenomics to paleogenomics: large-scale sequencing of mammoth DNA. Science. 2006;311:392–394. doi: 10.1126/science.1123360. [DOI] [PubMed] [Google Scholar]
- 16.Lindahl T, Nyberg B. Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry. 1974;13:3405–3410. doi: 10.1021/bi00713a035. [DOI] [PubMed] [Google Scholar]
- 17.Maricic T, Pääbo S. Optimization of 454 sequencing library preparation from small amounts of DNA permits sequence determination of both DNA strands. Biotechniques. 2009;46:51–57. doi: 10.2144/000113042. [DOI] [PubMed] [Google Scholar]
- 18.Wiley G, Macmil S, Qu C, Wang P, Xing Y, White D, Li J, White JD, Domingo A, Roe BA. Methods for generating shotgun and mixed shotgun/paired-end libraries for the 454 DNA sequencer. Curr. Protoc. Hum. Genet. 2009 doi: 10.1002/0471142905.hg1801s61. Chapter 18, Unit18 11. [DOI] [PubMed] [Google Scholar]
- 19.Meyer M, Briggs AW, Maricic T, Hober B, Hoffner B, Krause J, Weihmann A, Paabo S, Hofreiter M. From micrograms to picograms: quantitative PCR reduces the material demands of high-throughput sequencing. Nucleic Acids Res. 2007;36:e5. doi: 10.1093/nar/gkm1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rompler H, Rohland N, Lalueza-Fox C, Willerslev E, Kuznetsova T, Rabeder G, Bertranpetit J, Schoneberg T, Hofreiter M. Nuclear gene indicates coat-color polymorphism in mammoths. Science. 2006;313:62. doi: 10.1126/science.1128994. [DOI] [PubMed] [Google Scholar]
- 21.Rohland N, Hofreiter M. Ancient DNA extraction from bones and teeth. Nat Protoc. 2007;2:1756–1762. doi: 10.1038/nprot.2007.247. [DOI] [PubMed] [Google Scholar]
- 22.Stenzel U. German Conference on Bioinformatics. Germany: Halle; 2009. [Google Scholar]
- 23.Green RE, Malaspinas AS, Krause J, Briggs AW, Johnson PL, Uhler C, Meyer M, Good JM, Maricic T, Stenzel U, et al. A complete neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell. 2008;134:416–426. doi: 10.1016/j.cell.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Briggs AW, Good JM, Green RE, Krause J, Maricic T, Stenzel U, Paabo S. Primer extension capture: targeted sequence retrieval from heavily degraded DNA sources. J. Vis. Exp. 2009;1573 doi: 10.3791/1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kircher M, Stenzel U, Kelso J. Improved base calling for the Illumina Genome Analyzer using machine learning strategies. Genome. Biol. 2009;10:R83. doi: 10.1186/gb-2009-10-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang D, Hatahet Z, Melamede RJ, Kow YW, Wallace SS. Characterization of Escherichia coli endonuclease VIII. J. Biol. Chem. 1997;272:32230–32239. doi: 10.1074/jbc.272.51.32230. [DOI] [PubMed] [Google Scholar]
- 27.Hoss M, Jaruga P, Zastawny TH, Dizdaroglu M, Paabo S. DNA damage and DNA sequence retrieval from ancient tissues. Nucleic Acids Res. 1996;24:1304–1307. doi: 10.1093/nar/24.7.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paabo S. Ancient DNA: extraction, characterization, molecular cloning, and enzymatic amplification. Proc. Natl Acad. Sci. USA. 1989;86:1939–1943. doi: 10.1073/pnas.86.6.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Briggs AW, Good JM, Green RE, Krause J, Maricic T, Stenzel U, Lalueza-Fox C, Rudan P, Brajkovic D, Kucan Z, et al. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science. 2009;325:318–321. doi: 10.1126/science.1174462. [DOI] [PubMed] [Google Scholar]
- 30.Rohland N, Malaspinas AS, Pollack JL, Slatkin M, Matheus P, Hofreiter M. Proboscidean mitogenomics: chronology and mode of elephant evolution using mastodon as outgroup. PLoS Biol. 2007;5:e207. doi: 10.1371/journal.pbio.0050207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krause J, Dear PH, Pollack JL, Slatkin M, Spriggs H, Barnes I, Lister AM, Ebersberger I, Paabo S, Hofreiter M. Multiplex amplification of the mammoth mitochondrial genome and the evolution of Elephantidae. Nature. 2006;439:724–727. doi: 10.1038/nature04432. [DOI] [PubMed] [Google Scholar]
- 32.Shen JC, Rideout W.M., 3rd, Jones PA. The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 1994;22:972–976. doi: 10.1093/nar/22.6.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Green RE, Briggs AW, Krause J, Prufer K, Burbano HA, Siebauer M, Lachmann M, Paabo S. The Neandertal genome and ancient DNA authenticity. EMBO J. 2009;28:2494–2502. doi: 10.1038/emboj.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wall JD, Kim SK. Inconsistencies in Neanderthal genomic DNA sequences. PLoS Genet. 2007;3:1862–1866. doi: 10.1371/journal.pgen.0030175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 37.Illingworth R, Kerr A, Desousa D, Jorgensen H, Ellis P, Stalker J, Jackson D, Clee C, Plumb R, Rogers J, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6:e22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gilbert MT, Tomsho LP, Rendulic S, Packard M, Drautz DI, Sher A, Tikhonov A, Dalen L, Kuznetsova T, Kosintsev P, et al. Whole-genome shotgun sequencing of mitochondria from ancient hair shafts. Science. 2007;317:1927–1930. doi: 10.1126/science.1146971. [DOI] [PubMed] [Google Scholar]
- 39.Nachman MW. Single nucleotide polymorphisms and recombination rate in humans. Trends Genet. 2001;17:481–485. doi: 10.1016/s0168-9525(01)02409-x. [DOI] [PubMed] [Google Scholar]
- 40.Pruvost M, Grange T, Geigl EM. Minimizing DNA contamination by using UNG-coupled quantitative real-time PCR on degraded DNA samples: application to ancient DNA studies. Biotechniques. 2005;38:569–575. doi: 10.2144/05384ST03. [DOI] [PubMed] [Google Scholar]
- 41.Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009;4:265–270. doi: 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.