


Abstract
We found that both tetramethylammonium chloride (TMA-Cl) and tetra-ethylammonium chloride (TEA-Cl), which are used as monovalent cations for northern hybridization, drastically destabilized the tertiary structures of tRNAs and enhanced the formation of tRNA•oligoDNA hybrids. These effects are of great advantage for the hybridization-based method for purification of specific tRNAs from unfractionated tRNA mixtures through the use of an immobilized oligoDNA complementary to the target tRNA. Replacement of NaCl by TMA-Cl or TEA-Cl in the hybridization buffer greatly improved the recovery of a specific tRNA, even from unfractionated tRNAs derived from a thermophile. Since TEA-Cl destabilized tRNAs more strongly than TMA-Cl, it was necessary to lower the hybridization temperature at the sacrifice of the purity of the recovered tRNA when using TEA-Cl. Therefore, we propose two alternative protocols, depending on the desired properties of the tRNA to be purified. When the total recovery of the tRNA is important, hybridization should be carried out in the presence of TEA-Cl. However, if the purity of the recovered tRNA is important, TMA-Cl should be used for the hybridization. In principle, this procedure for tRNA purification should be applicable to any small-size RNA whose gene sequence is already known.
INTRODUCTION
tRNAs function as adapter molecules between mRNAs and proteins, and contribute to the accuracy of protein biosynthesis (1). To ensure this accuracy, a tRNA must definitely accept only its cognate amino acid onto its CCA terminus and recognize the cognate codon of mRNA complementary to its anticodon. We know that the in vitro transcription method using T7 RNA polymerase developed by Sampson and Uhlenbeck (2) has greatly contributed to studies on the structure–function relationships of tRNAs, but this method is rather powerless for the studies of tRNAs, especially in such a case that modified nucleotides affect the accuracy of aminoacylation and/or codon recognition (3,4). Therefore, analyses of modified nucleotides and measurements of biological activities of the natural tRNAs are very important for studies in the field of translation. For such experiments, purifying considerable amounts of the target tRNAs from cells is an inevitable process. However, purification of a specific tRNA from unfractionated tRNAs by using conventional column-chromatographic steps is usually more laborious than the purification process for a protein, because the chemical properties and molecular weights of tRNAs are quite similar. Therefore, the development of an efficient method to purify any natural tRNA of special interest is eagerly awaited.
Among the methods for tRNA purification, the selective hybridization method using solid-phase DNA probes (5,6) may be the most promising procedure in this genomic era because the gene for the tRNA of interest has already been sequenced in most cases. The principles of this method are briefly described below. First, unfractionated tRNA mixtures are heat-denatured and subjected to hybridization with an immobilized oligoDNA complementary to part of the target tRNA. Non-hybridized tRNAs are then easily washed out from the oligoDNA-immobilized resin. Finally, the target tRNA hybridized with the oligoDNA is eluted from the resin by heating. To date, this method has often been used to purify specific mitochondrial tRNAs from the overwhelming majority of cytoplasmic tRNAs (5,7–11). Suzuki and Suzuki (12) succeeded in purifying all the individual mitochondrial tRNAs at once by connecting 22 columns of immobilized oligoDNA resins, which they called ‘chaplet columns’. However, the applications of this method have remained to be limited because it is not very effective for purifying cytoplasmic tRNAs. The difference in purification between mitochondrial and cytoplasmic tRNAs is probably related to their thermal stabilities.
Most mitochondrial tRNAs from higher eukaryotes lack the conserved nucleotides that are thought to be important for the formation of 3D interactions, such as the GG sequence in the D-loop and the ΨC sequence in the T-loop. Therefore, their melting temperatures are much lower (about 55°C) than those of most cytoplasmic tRNAs (about 75°C), probably owing to weak tertiary interactions (13). Cytoplasmic tRNAs may be too stable to undergo denaturation or hybrid formation with oligoDNAs. Miyauchi et al. (14) tried to overcome this problem by heating the tRNA solution up to 68°C and succeeded in preparing any desired species of tRNA from Escherichia coli cells with a sophisticated but complex system called ‘reciprocal circulating chromatography’. Nevertheless, there must be additional problems when preparing more thermostable tRNAs, such as those from thermophilic bacteria. It is likely that the use of denaturants such as urea to destabilize the cytoplasmic tRNAs may be the first choice to be considered, but this intrinsically includes a paradox that destabilization simultaneously causes lower hybridization. Therefore, an ideal denaturation reagent that would only destabilize tRNAs and not influence any tRNA•oligoDNA hybrids is desired.
Among the so-called ‘chaotropic’ salts, tetramethylammonium chloride (TMA-Cl) and tetra-ethylammonium chloride (TEA-Cl) are especially known as ‘magical’ salts because they melt duplex DNAs (15) or RNAs (16) with every GC content at the same temperature at high concentrations (3 M TMA-Cl or 2.4 M TEA-Cl). Tsurui et al. (6) have already tried to use 2.4 M TEA-Cl solution for hybridization between tRNAs and oligoDNA-immobilized resins to purify a specific tRNA. Their results indicated that 2.4 M TEA-Cl functions as an effective denaturation reagent, but the hybridization must be kept at very low temperatures (10–15°C) because the tRNA•oligoDNA hybrids are also destabilized.
To the best of our knowledge, no studies have assessed how tetra-alkylammonium salts influence the melting profiles of tRNAs or tRNA•oligoDNA hybrids. Here, we present the melting profiles of tRNAs and tRNA•oligoDNA hybrids in aqueous solutions containing various monovalent cations. Moreover, applying the data of these melting profiles, a more sophisticated version of the hybridization-based method to purify a specific tRNA is demonstrated.
MATERIALS AND METHODS
Materials and buffers
Biotinylated oligoDNAs (Figure 1) (Oligo-EfMet: 5′-TTATGAGCCCGACGAGCTACCAGGCT-Biotin-3′; OligoGC-YPhe: 5′-TTCAGTCTGGCGCTCTCCCAACTGAG-Biotin-3′; and OligoGT-YPhe: 5′-TTCAGTCTGGTGCTCTCCCAACTGAG-Biotin-3′) were obtained from Operon Biotechnologies. All chemicals not otherwise specified were obtained from Wako Pure Chemicals.
Figure 1.

Secondary structures of tRNAs and biotinylated oligoDNAs used in this study. Red lines and characters indicate oligoDNAs complementary to target tRNAs. (A) Escherichia coli
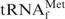 and its oligoDNA, Oligo-EfMet. (B) Yeast tRNAPhe and its oligoDNAs, OligoGC-YPhe and OligoGT-YPhe.
and its oligoDNA, Oligo-EfMet. (B) Yeast tRNAPhe and its oligoDNAs, OligoGC-YPhe and OligoGT-YPhe.
The buffers used in this study were as follows: 2× hybridization buffer Na+: 20 mM Tris–HCl (pH 7.6), 1.8 M NaCl, 0.2 mM EDTA; 2× hybridization buffer TMA+: 20 mM Tris–HCl (pH 7.6), 1.8 M TMA-Cl, 0.2 mM EDTA; 2× hybridization buffer TEA+: 20 mM Tris–HCl (pH 7.6), 1.8 M TEA-Cl, 0.2 mM EDTA; and 2× Tm buffer: 20 mM Tris–HCl (pH 7.6), 0.4 M NaCl, 20 mM MgCl2.
Preparation of tRNAs
Unfractionated tRNAs (tRNAMix) from E. coli and yeast were prepared essentially as described by Zubay (17) and Holley (18), respectively. Yeast tRNAPhe was purified according to the method described by Wimmer et al. (19). Escherichia coli
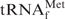 was prepared as follows. The PCR-amplified
was prepared as follows. The PCR-amplified 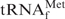 gene (metW) from E. coli was inserted into the site between XbaI and HindIII of pGEMEX-1 (Promega) and the resulting vector was designated pGEMEX_MetW. The sequence of the tRNA gene was confirmed by dideoxy sequencing methods with Thermo Sequenase Primer Cycle Sequencing Kit according to the manufacturer’s protocol (GE Healthcare). The
gene (metW) from E. coli was inserted into the site between XbaI and HindIII of pGEMEX-1 (Promega) and the resulting vector was designated pGEMEX_MetW. The sequence of the tRNA gene was confirmed by dideoxy sequencing methods with Thermo Sequenase Primer Cycle Sequencing Kit according to the manufacturer’s protocol (GE Healthcare). The 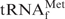 was overproduced in E. coli BL21(DE3) transfected with pGEMEX_MetW, and purified by ion-exchange column chromatographies using a similar procedure to that described for purifying tRNATyr (20).
was overproduced in E. coli BL21(DE3) transfected with pGEMEX_MetW, and purified by ion-exchange column chromatographies using a similar procedure to that described for purifying tRNATyr (20).
Melting profiles of tRNAs and tRNA•oligoDNA hybrids
A UV-visible spectrophotometer (Model UV-2450; Shimadzu) and the LabSolutions software (Shimadzu) were used to plot the melting curves of tRNAs and tRNA•oligoDNA hybrids.
To measure the hyperchromicities, the initial temperature of measurement and rate of increase in temperature were set at 25°C and 0.5°C/min, respectively, with a temperature controller (Model TMSPC-8; Shimadzu). Samples for measurements were prepared as follows. For tRNAs, a degassed aqueous solution (80 µl) of purified tRNA (2 µM) or unfractionated E. coli tRNAMix (1 A260 unit/ml) was mixed with an equal volume (80 µl) of degassed 2× hybridization buffer (Na+, TMA+ or TEA+) or 2× Tm buffer. For tRNA•oligoDNA hybrids, an aqueous solution (80 µl) containing 2 µM E. coli
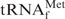 and 3 µM oligoDNA (Oligo-EfMet) was mixed with 80 µl of 2× hybridization buffer TMA+ and the mixture was incubated at 65°C for 10 min. After cooling to room temperature, the buffer was exchanged for 10 mM Tris–HCl (pH 7.6) by repeated ultrafiltration with a Microcon (molecular weight cut-off: 10 000; Millipore) and the final volume was made up to 160 µl with degassed 10 mM Tris–HCl (pH 7.6).
and 3 µM oligoDNA (Oligo-EfMet) was mixed with 80 µl of 2× hybridization buffer TMA+ and the mixture was incubated at 65°C for 10 min. After cooling to room temperature, the buffer was exchanged for 10 mM Tris–HCl (pH 7.6) by repeated ultrafiltration with a Microcon (molecular weight cut-off: 10 000; Millipore) and the final volume was made up to 160 µl with degassed 10 mM Tris–HCl (pH 7.6).
To measure the hypochromicities, the initial temperature of measurement and rate of decrease in temperature were set at 95°C and 0.5°C/min, respectively. A mixture of degassed 2 µM tRNA with or without 2 µM oligoDNA solution (80 µl) and degassed 2× hybridization buffer (Na+, TMA+ or TEA+) (80 µl) was used as the sample for measurement.
Procedure for the selective hybridization method using a solid-phase DNA probe
OligoDNA-immobilized resins were prepared as follows. A suspension of Streptavidin Sepharose High Performance (GE Healthcare) resin (200 µl) was poured into the upper cup of an Ultrafree-MC (0.22 µm; Millipore) and equilibrated with 10 mM Tris–HCl (pH 7.6). After complete removal of the buffer by centrifugation at 10 000 r.p.m. for 10 s, 400 µl of 7.5 µM biotinylated oligoDNA solution was mixed with the resin and incubated for 10 min at room temperature. Agitation of the resin is not necessary. The amount of the oligoDNA bound to the resin was estimated from the concentration of the unbound oligoDNA, after complete removal of the unbound oligoDNA solution by centrifugation. The resin was washed with 10 mM Tris–HCl (pH 7.6) and suspended in an adequate volume of 10 mM Tris–HCl (pH 7.6). In the case of Oligo-EfMet, 2.5 nmol of Oligo-EfMet was bound to the resin. Although the amount of the bound Oligo-EfMet is considerably less than the value stated by the manufacturer, it is within the range of empirically predicted value probably because the repulsion among the phosphates of oligoDNAs would hinder the coupling. The Oligo-EfMet-immobilized resin was suspended in up to 250 µl of 10 mM Tris–HCl (pH 7.6).
The resin suspension (50 µl, corresponding to 500 pmol of immobilized oligoDNA) was placed in a new upper cup for the Ultrafree-MC and the suspension buffer [10 mM Tris–HCl (pH 7.6)] was completely removed by centrifugation. An aqueous solution (50 µl) of unfractionated tRNAMix (5 A260 units) was mixed with 50 µl of 2× hybridization buffer. The resulting mixture was loaded onto the oligoDNA-immobilized resin and the suspension was incubated at various temperatures (25°C, 35°C, 45°C, 55°C or 65°C) for 10 min. Agitation of the resin is not necessary during this hybridization step. Unbound tRNAs were removed by centrifugation and the resin was repeatedly washed with 400 µl each of 10 mM Tris–HCl (pH 7.6) at room temperature until the absorbance at 260 nm of the washed solution was <0.01 A260 unit/ml. Finally, the target tRNA was detached from the resin by heating at 65°C for 5 min in 200 µl of 10 mM Tris–HCl (pH 7.6) and recovered by quick centrifugation to avoid cooling the solution. When the eluted tRNA was to be used for aminoacylation reaction, 2 µl of 1 M MgCl2 was put in the bottom of lower tube of the Ultrafree-MC in advance. In many cases, this immediate addition of MgCl2 improved the aminoacylation capacity of the eluted target tRNA probably by helping the proper tRNA folding. This elution step was repeated twice. The recovery of the target tRNA was estimated from its UV spectrum and its purity was checked by 10% PAGE containing 7 M urea. In addition, the proportion of the target tRNA remaining in the unbound tRNA fraction was estimated by northern hybridization.
RESULTS AND DISCUSSION
Designing an oligoDNA complementary to the target tRNA
Our policy for designing a biotinylated oligoDNA complementary to a target tRNA is as follows. First, a biotin should be introduced into the 3′-end. Since the oligoDNA is chemically synthesized from 3′ to 5′, the efficiency of the biotin modification would be highest at the 3′ terminus. Second, the oligoDNA itself should not form a strong secondary structure that would lead to loss of the hybridization efficiency (21). Third, the oligoDNA should be longer than 25 nt so that sufficient specificity can be ensured. Fourth, the oligoDNA should be shorter than 30 nt so that the bound tRNA can be fully detached from the tRNA•oligoDNA hybrid by heating. Considering these prerequisites, there remain two regions of tRNA. One is the region around the anticodon loop and D-arm, and the other is the region from the accepter stem to the valuable loop of each tRNA. We usually select the region around the anticodon loop and D-arm as the first choice for appropriate oligoDNAs, because this region is empirically more likely to be successful (21–23) (Figure 1).
Melting profiles of E. coli
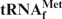 in buffers of various compositions
in buffers of various compositions
As the target for detailed investigations of the denaturing process of tRNAs, we initially selected E. coli
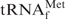 , because this tRNA was reported to have the highest melting temperature (Tm) among the E. coli tRNAs (24). We rationalized that, if it was possible to purify
, because this tRNA was reported to have the highest melting temperature (Tm) among the E. coli tRNAs (24). We rationalized that, if it was possible to purify 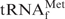 by using the hybridization-based method, any other specific E. coli tRNAs could be purified satisfactorily. Figure 2 shows the melting profiles of E. coli
by using the hybridization-based method, any other specific E. coli tRNAs could be purified satisfactorily. Figure 2 shows the melting profiles of E. coli
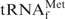 in buffers of various compositions. In the presence of 0.2 M NaCl and 10 mM MgCl2 (1× Tm buffer), E. coli
in buffers of various compositions. In the presence of 0.2 M NaCl and 10 mM MgCl2 (1× Tm buffer), E. coli
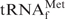 actually had a high melting temperature (Tm = 82.5°C) and its melting profile showed typical cooperativity (Figure 2, blue line). The removal of Mg2+ (1× hybridization buffer Na+) caused multistep denaturation of E. coli
actually had a high melting temperature (Tm = 82.5°C) and its melting profile showed typical cooperativity (Figure 2, blue line). The removal of Mg2+ (1× hybridization buffer Na+) caused multistep denaturation of E. coli
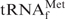 (Figure 2, black line). This non-cooperative profile may indicate that the melting of E. coli
(Figure 2, black line). This non-cooperative profile may indicate that the melting of E. coli
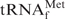 starts at its tertiary structure level and is followed by gradual destruction of the secondary structure. When TMA-Cl was substituted for NaCl (1× hybridization buffer TMA+), restoration of the cooperativity was observed in the melting profile of E. coli
starts at its tertiary structure level and is followed by gradual destruction of the secondary structure. When TMA-Cl was substituted for NaCl (1× hybridization buffer TMA+), restoration of the cooperativity was observed in the melting profile of E. coli
 concomitant with a drastic decrease in the melting temperature (Tm = 65.8°C; Figure 2, red line). In addition, the initial absorbance at 260 nm of the tRNA solution at 25°C showed an increase of about 16% compared with that in 0.2 M NaCl and 10 mM MgCl2 (1× Tm buffer). This hyperchromicity suggests that TMA-Cl partially denatures the tRNA structure even at 25°C. It is likely that when TMA ions bind to the intricate networks of phosphates at the hinge region, for example, of the tRNA, the bulkiness of the ions may somehow distort the tertiary structure of the tRNA. It is noteworthy that the melting profile of E. coli
concomitant with a drastic decrease in the melting temperature (Tm = 65.8°C; Figure 2, red line). In addition, the initial absorbance at 260 nm of the tRNA solution at 25°C showed an increase of about 16% compared with that in 0.2 M NaCl and 10 mM MgCl2 (1× Tm buffer). This hyperchromicity suggests that TMA-Cl partially denatures the tRNA structure even at 25°C. It is likely that when TMA ions bind to the intricate networks of phosphates at the hinge region, for example, of the tRNA, the bulkiness of the ions may somehow distort the tertiary structure of the tRNA. It is noteworthy that the melting profile of E. coli
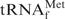 in TMA-Cl solution is very similar to that of bovine mitochondrial tRNAPhe, which is known to have a very weak interaction between the D- and T-loops (25). This similarity also implies that TMA-Cl denatures the tertiary structure of tRNAs. TEA-Cl, containing the bulkier TEA ion compared with the TMA ion, showed a stronger denaturing effect than TMA-Cl (Figure 2, green line). A further decrement of about 20°C was observed in the melting temperature (Tm = 45.2°C). Since the TEA+ ions are too bulky to bind in an orderly manner in the minor grooves of RNA duplexes (16), not only the tertiary structure but also a part of the secondary structure of the tRNA may be disrupted, even at room temperature.
in TMA-Cl solution is very similar to that of bovine mitochondrial tRNAPhe, which is known to have a very weak interaction between the D- and T-loops (25). This similarity also implies that TMA-Cl denatures the tertiary structure of tRNAs. TEA-Cl, containing the bulkier TEA ion compared with the TMA ion, showed a stronger denaturing effect than TMA-Cl (Figure 2, green line). A further decrement of about 20°C was observed in the melting temperature (Tm = 45.2°C). Since the TEA+ ions are too bulky to bind in an orderly manner in the minor grooves of RNA duplexes (16), not only the tertiary structure but also a part of the secondary structure of the tRNA may be disrupted, even at room temperature.
Figure 2.

Melting profiles of E. coli
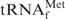 in buffers of various compositions. The blue line (Tm) shows the melting profile of 1 µM E. coli
in buffers of various compositions. The blue line (Tm) shows the melting profile of 1 µM E. coli
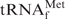 in Tm buffer [10 mM Tris–HCl (pH 7.6), 0.2 M NaCl, 10 mM MgCl2], which is often used for the measurement of melting temperatures. The black line (Na+) indicates the melting profile of the same tRNA in hybridization buffer Na+ [10 mM Tris–HCl (pH 7.6), 0.9 M NaCl, 0.1 mM EDTA], which is generally used for hybridization. The red (TMA+) and green (TEA+) lines show the melting profiles in the hybridization buffers in which NaCl was replaced with TMA–Cl and TEA–Cl, respectively. The arrows and figures indicate the inflection points of the melting curves and the temperatures at those points, respectively. No apparent inflection point was identified in the melting profile in the buffer Na+ (black line).
in Tm buffer [10 mM Tris–HCl (pH 7.6), 0.2 M NaCl, 10 mM MgCl2], which is often used for the measurement of melting temperatures. The black line (Na+) indicates the melting profile of the same tRNA in hybridization buffer Na+ [10 mM Tris–HCl (pH 7.6), 0.9 M NaCl, 0.1 mM EDTA], which is generally used for hybridization. The red (TMA+) and green (TEA+) lines show the melting profiles in the hybridization buffers in which NaCl was replaced with TMA–Cl and TEA–Cl, respectively. The arrows and figures indicate the inflection points of the melting curves and the temperatures at those points, respectively. No apparent inflection point was identified in the melting profile in the buffer Na+ (black line).
Hypochromicities of E. coli
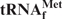 in solutions containing various monovalent cations in the presence or absence of its complementary oligoDNA
in solutions containing various monovalent cations in the presence or absence of its complementary oligoDNA
The hypochromicities of denatured E. coli
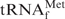 in the presence or absence of its complementary 3′ biotinylated oligoDNA (Oligo-EfMet) were measured in solutions containing Na+, TMA+ or TEA+, in order to examine the effects of these monovalent cations on E. coli
in the presence or absence of its complementary 3′ biotinylated oligoDNA (Oligo-EfMet) were measured in solutions containing Na+, TMA+ or TEA+, in order to examine the effects of these monovalent cations on E. coli
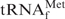 •Oligo-EfMet hybrids. The profiles are shown in Figure 3. Compared with the melting profiles in Figure 2, the hypochromicity profiles of E. coli
•Oligo-EfMet hybrids. The profiles are shown in Figure 3. Compared with the melting profiles in Figure 2, the hypochromicity profiles of E. coli
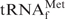 alone (Figure 3, dotted lines) were very consistent with their corresponding melting profiles. Therefore, the denaturation and renaturation steps of E. coli
alone (Figure 3, dotted lines) were very consistent with their corresponding melting profiles. Therefore, the denaturation and renaturation steps of E. coli
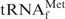 are essentially reversible. In the presence of an equal concentration of the oligoDNA (Oligo-EfMet), the shapes of the profiles were obviously different (Figure 3, solid lines). Although the presence or absence of Oligo-EfMet caused only subtle differences in the hypochromicity profiles when measured in 1× hybridization buffer Na+ (Figure 3, black lines), remarkable differences were observed when the profiles were measured in 1× hybridization buffer TMA+ and TEA+ (Figure 3, red and green lines, respectively). The hypochromicity of E. coli
are essentially reversible. In the presence of an equal concentration of the oligoDNA (Oligo-EfMet), the shapes of the profiles were obviously different (Figure 3, solid lines). Although the presence or absence of Oligo-EfMet caused only subtle differences in the hypochromicity profiles when measured in 1× hybridization buffer Na+ (Figure 3, black lines), remarkable differences were observed when the profiles were measured in 1× hybridization buffer TMA+ and TEA+ (Figure 3, red and green lines, respectively). The hypochromicity of E. coli
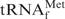 in the presence of Oligo-EfMet (solid lines) declined more steeply than that of E. coli
in the presence of Oligo-EfMet (solid lines) declined more steeply than that of E. coli
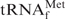 alone (dotted lines) and the transition point shifted to much higher temperatures. These results may indicate that E. coli
alone (dotted lines) and the transition point shifted to much higher temperatures. These results may indicate that E. coli
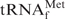 is preferably renatured irrespective of the presence of Oligo-EfMet in 1× hybridization buffer Na+, but is prone to form hybrids with Oligo-EfMet in 1× hybridization buffer TMA+ or TEA+. Therefore, a gradual decline in the solution temperature is not necessary for hybridization of E. coli
is preferably renatured irrespective of the presence of Oligo-EfMet in 1× hybridization buffer Na+, but is prone to form hybrids with Oligo-EfMet in 1× hybridization buffer TMA+ or TEA+. Therefore, a gradual decline in the solution temperature is not necessary for hybridization of E. coli
 with Oligo-EfMet. When E. coli
with Oligo-EfMet. When E. coli
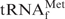 and Oligo-EfMet were incubated at 65°C for 10 min in 1× hybridization buffer TMA+, almost all of the E. coli
and Oligo-EfMet were incubated at 65°C for 10 min in 1× hybridization buffer TMA+, almost all of the E. coli
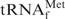 was found to form hybrids with Oligo-EfMet, as evaluated by 10% native PAGE (data not shown).
was found to form hybrids with Oligo-EfMet, as evaluated by 10% native PAGE (data not shown).
Figure 3.

Hypochromicity profiles of E. coli
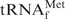 in the presence or absence of the complementary oligoDNA, Oligo-EfMet, in buffers containing various monovalent cations, Na+, TMA+ or TEA+. The dotted and solid lines show the hypochromicity profiles of E. coli
in the presence or absence of the complementary oligoDNA, Oligo-EfMet, in buffers containing various monovalent cations, Na+, TMA+ or TEA+. The dotted and solid lines show the hypochromicity profiles of E. coli
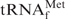 alone and the same tRNA with its complementary oligoDNA, Oligo-EfMet, respectively.
alone and the same tRNA with its complementary oligoDNA, Oligo-EfMet, respectively.
Thermostability of E. coli
 •Oligo-EfMet hybrids under low-salt conditions
•Oligo-EfMet hybrids under low-salt conditions
The hydrogen bonds between bases of nucleic acids are known to be strengthened in high concentrations of monovalent cation solutions. Therefore, hybridization experiments are usually performed in the presence of 0.9 M NaCl. Here, we found that TMA+ or TEA+ ions selectively destabilized the tertiary structure of E. coli
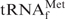 and effectively converted the
and effectively converted the 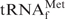 stand-alone form to the
stand-alone form to the 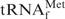 •Oligo-EfMet hybrid form. Incidentally, TMA-Cl and TEA-Cl should be properly removed prior to ethanol precipitation because their hydrophobic natures hinder easy handling of tRNAs. However, there was a concern that the hybrids might be destabilized under the low-salt conditions after the effective removal of TMA-Cl and TEA-Cl. Consequently, the melting profile of the E. coli
•Oligo-EfMet hybrid form. Incidentally, TMA-Cl and TEA-Cl should be properly removed prior to ethanol precipitation because their hydrophobic natures hinder easy handling of tRNAs. However, there was a concern that the hybrids might be destabilized under the low-salt conditions after the effective removal of TMA-Cl and TEA-Cl. Consequently, the melting profile of the E. coli
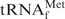 •Oligo-EfMet hybrid was measured in 10 mM Tris–HCl (pH 7.6) (Figure 4). The hybrid was stable at room temperature even under the ‘almost no salt’ condition and showed cooperative melting with a melting temperature of 54.1°C. The hybrid seemed to be completely denatured at temperatures >60°C.
•Oligo-EfMet hybrid was measured in 10 mM Tris–HCl (pH 7.6) (Figure 4). The hybrid was stable at room temperature even under the ‘almost no salt’ condition and showed cooperative melting with a melting temperature of 54.1°C. The hybrid seemed to be completely denatured at temperatures >60°C.
Figure 4.

Melting profile of the E. coli
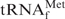 •Oligo-EfMet hybrid in a low-salt buffer [10 mM Tris–HCl (pH 7.6)]. The arrow and figure indicate the melting temperature of the hybrid in 10 mM Tris–HCl (pH 7.6).
•Oligo-EfMet hybrid in a low-salt buffer [10 mM Tris–HCl (pH 7.6)]. The arrow and figure indicate the melting temperature of the hybrid in 10 mM Tris–HCl (pH 7.6).
Preparation of E. coli
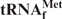 from E. coli tRNAMix using immobilized Oligo-EfMet
from E. coli tRNAMix using immobilized Oligo-EfMet
Strictly speaking, the hybridization conditions described above only refer to the purified tRNA and free biotinylated oligoDNA. For practical usage, we therefore examined whether these conditions could be adopted for E. coli tRNAMix and immobilized Oligo-EfMet. Resins (∼20 µl of the resin) containing 500 pmol of immobilized Oligo-EfMet were mixed with 5 A260 units (100 µl of 50 A260 unit/ml tRNAMix in 1× hybridization buffer) of E. coli tRNAMix. After incubation at various temperatures for 10 min, the resins were repeatedly washed with 400 µl of 10 mM Tris–HCl (pH 7.6) at room temperature. Finally, the tRNAs hybridized to the Oligo-EfMet resins were detached by incubation at 65°C for 5 min in 200 µl of 10 mM Tris–HCl (pH 7.6) and eluted twice. The amounts of the recovered tRNA from 5 A260 units of E. coli tRNAMix were plotted in a graph (Figure 5A). When Na+ was used as the monovalent cation, the recoveries of the tRNA were quite low except at the point where hybridization was performed at 65°C. At temperatures of 55°C or lower, it is likely that E. coli
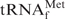 may not have been sufficiently denatured to form a hybrid with the immobilized oligoDNA. When TMA+ was used as the monovalent cation, the recoveries of tRNA gradually increased as the hybridization temperature increased. When TEA+ was used as the monovalent cation, the recoveries of tRNA showed a distinct peak for the maximum recovery around the hybridization temperature of 35°C.
may not have been sufficiently denatured to form a hybrid with the immobilized oligoDNA. When TMA+ was used as the monovalent cation, the recoveries of tRNA gradually increased as the hybridization temperature increased. When TEA+ was used as the monovalent cation, the recoveries of tRNA showed a distinct peak for the maximum recovery around the hybridization temperature of 35°C.
Figure 5.

Quantities and qualities of the recovered E. coli
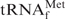 under various hybridization conditions. (A) Recovered amounts of the tRNA from 5 A260 units of unfractionated E. coli tRNAs at various hybridization temperatures. (B) Analysis by 10% PAGE containing 7 M urea. Lane mix: 0.1 A260 units of unfractionated E. coli tRNAs. Lane Au: 0.01 A260 units of the authentic E. coli
under various hybridization conditions. (A) Recovered amounts of the tRNA from 5 A260 units of unfractionated E. coli tRNAs at various hybridization temperatures. (B) Analysis by 10% PAGE containing 7 M urea. Lane mix: 0.1 A260 units of unfractionated E. coli tRNAs. Lane Au: 0.01 A260 units of the authentic E. coli
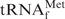 . Lanes Na+, TMA+ and TEA+: 0.01 A260 units of the recovered E. coli
. Lanes Na+, TMA+ and TEA+: 0.01 A260 units of the recovered E. coli
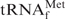 with hybridization buffer Na+ (0.9 M NaCl, hybridized at 65°C), hybridization buffer TMA+ (TMA-Cl, hybridized at 65°C) and hybridization buffer TEA+ (TEA-Cl, hybridized at 35°C), respectively. The gel was stained with methylene blue. The asterisk shows a contaminant band found in the recovered E. coli
with hybridization buffer Na+ (0.9 M NaCl, hybridized at 65°C), hybridization buffer TMA+ (TMA-Cl, hybridized at 65°C) and hybridization buffer TEA+ (TEA-Cl, hybridized at 35°C), respectively. The gel was stained with methylene blue. The asterisk shows a contaminant band found in the recovered E. coli
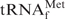 . (C) Northern hybridization to detect E. coli
. (C) Northern hybridization to detect E. coli
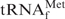 in the tRNA fractions unbound to the resin. (Left) Lane mix: 0.1 A260 units of unfractionated E. coli tRNAs. Lanes Na+, TMA+ and TEA+: 0.1 A260 units of the tRNA fractions unbound to the resin using hybridization buffer Na+ (hybridized at 65°C), hybridization buffer TMA+ (hybridized at 65°C) and hybridization buffer TEA+ (hybridized at 35°C), respectively. The gel was stained with methylene blue. (Right) Northern hybridization analysis with Oligo-EfMet. Biotins on the nylon membrane were detected with streptavidin, bitotinyl alkaline phosphatase and BCIP/NBT substrate.
in the tRNA fractions unbound to the resin. (Left) Lane mix: 0.1 A260 units of unfractionated E. coli tRNAs. Lanes Na+, TMA+ and TEA+: 0.1 A260 units of the tRNA fractions unbound to the resin using hybridization buffer Na+ (hybridized at 65°C), hybridization buffer TMA+ (hybridized at 65°C) and hybridization buffer TEA+ (hybridized at 35°C), respectively. The gel was stained with methylene blue. (Right) Northern hybridization analysis with Oligo-EfMet. Biotins on the nylon membrane were detected with streptavidin, bitotinyl alkaline phosphatase and BCIP/NBT substrate.
The quality of the recovered tRNA was evaluated by 10% PAGE containing 7 M urea (Figure 5B). All the recovered tRNAs were sufficiently pure and mainly contained E. coli
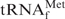 . Since the methionine acceptance of the recovered tRNA was over 1100 pmol/A260 unit, the activity of the recovered tRNA was sufficient (data not shown). In addition, the results of northern hybridization allowed us to estimate that ∼85% of the E. coli
. Since the methionine acceptance of the recovered tRNA was over 1100 pmol/A260 unit, the activity of the recovered tRNA was sufficient (data not shown). In addition, the results of northern hybridization allowed us to estimate that ∼85% of the E. coli
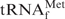 in E. coli tRNAMix was recovered by using the hybridization buffer containing TEA-Cl (Figure 5C). Therefore, this method for purifying any target tRNA from unfractionated tRNAs in TMA-Cl or TEA-Cl aqueous solutions should be very useful for preparing thermostable tRNAs from thermophiles. In fact, we have succeeded in purifying tRNACys from Aquifex aeolicus (23) by the same procedure using TMA-Cl as described above. However, it should be noted that the tRNAs recovered by this method did contain slight but obvious contaminants (Figure 5B, asterisks). Since the data reported by Jacobs et al. (26) showed that tetraalkylammonium ions (TMA-Cl and TEA-Cl) slightly strengthen mismatched base pairs, any tRNA with a similar sequence to
in E. coli tRNAMix was recovered by using the hybridization buffer containing TEA-Cl (Figure 5C). Therefore, this method for purifying any target tRNA from unfractionated tRNAs in TMA-Cl or TEA-Cl aqueous solutions should be very useful for preparing thermostable tRNAs from thermophiles. In fact, we have succeeded in purifying tRNACys from Aquifex aeolicus (23) by the same procedure using TMA-Cl as described above. However, it should be noted that the tRNAs recovered by this method did contain slight but obvious contaminants (Figure 5B, asterisks). Since the data reported by Jacobs et al. (26) showed that tetraalkylammonium ions (TMA-Cl and TEA-Cl) slightly strengthen mismatched base pairs, any tRNA with a similar sequence to 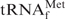 may also be hybridized to the immobilized oligoDNA. Consequently, we should pay attention to the quality of the recovered tRNA, especially when a minor tRNA species is to be prepared. However, we noticed that the contaminant band was more prominent when using TEA-Cl than using TMA-Cl. This difference was probably due to the hybridization temperature (35°C for TEA-Cl and 65°C for TMA-Cl). Higher temperature in the hybridization step could raise the specificity of hybridization.
may also be hybridized to the immobilized oligoDNA. Consequently, we should pay attention to the quality of the recovered tRNA, especially when a minor tRNA species is to be prepared. However, we noticed that the contaminant band was more prominent when using TEA-Cl than using TMA-Cl. This difference was probably due to the hybridization temperature (35°C for TEA-Cl and 65°C for TMA-Cl). Higher temperature in the hybridization step could raise the specificity of hybridization.
Differences in the melting and hypochromicity profiles between E. coli
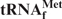 and yeast tRNAPhe
and yeast tRNAPhe
To assess whether the procedure described above is also applicable to other tRNAs, we selected yeast tRNAPhe, whose melting temperature is rather lower (24) than that of E. coli
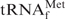 , as our next target. The melting profiles of both tRNAs are shown in Figure 6. When measured in 1× hybridization buffer Na+, the shapes of the melting profiles were notably different (Figure 6, thin and thick black lines). These findings may reflect the difference in the stabilities of the tertiary structures of the two tRNAs. However, when measured in 1× hybridization buffer TMA+ or TEA+, the melting profiles as a whole shifted to lower temperatures but the cooperativities of the melting profiles were similar (Figure 6; thin and thick red and green lines). We deduced that the tertiary structures of both tRNAs were already denatured in the presence of TMA-Cl or TEA-Cl, and the lower melting temperature of yeast tRNAPhe is thought to reflect the fact that yeast tRNAPhe has fewer G-C pairs in its secondary structure than E. coli
, as our next target. The melting profiles of both tRNAs are shown in Figure 6. When measured in 1× hybridization buffer Na+, the shapes of the melting profiles were notably different (Figure 6, thin and thick black lines). These findings may reflect the difference in the stabilities of the tertiary structures of the two tRNAs. However, when measured in 1× hybridization buffer TMA+ or TEA+, the melting profiles as a whole shifted to lower temperatures but the cooperativities of the melting profiles were similar (Figure 6; thin and thick red and green lines). We deduced that the tertiary structures of both tRNAs were already denatured in the presence of TMA-Cl or TEA-Cl, and the lower melting temperature of yeast tRNAPhe is thought to reflect the fact that yeast tRNAPhe has fewer G-C pairs in its secondary structure than E. coli
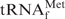 . Next, the hypochromicity profiles of yeast tRNAPhe in the presence of the complementary oligoDNA, OligoGC-YPhe, were measured (Figure 7, thin lines). Steep declines in the hypochromicity of E. coli
. Next, the hypochromicity profiles of yeast tRNAPhe in the presence of the complementary oligoDNA, OligoGC-YPhe, were measured (Figure 7, thin lines). Steep declines in the hypochromicity of E. coli
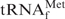 and Oligo-EfMet (Figure 7, thick lines) were again observed for yeast tRNAPhe and OligoGC-YPhe. Unexpectedly, a shift to a lower temperature of the transition profile was also observed. Since the GC contents of Oligo-EfMet and OligoGC-YPhe were the same (57%), we rationalized that this difference may be due to the presence of modified nucleotides. Specifically, yeast tRNAPhe contains N2,N2-dimethylguanosine (
and Oligo-EfMet (Figure 7, thick lines) were again observed for yeast tRNAPhe and OligoGC-YPhe. Unexpectedly, a shift to a lower temperature of the transition profile was also observed. Since the GC contents of Oligo-EfMet and OligoGC-YPhe were the same (57%), we rationalized that this difference may be due to the presence of modified nucleotides. Specifically, yeast tRNAPhe contains N2,N2-dimethylguanosine (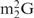 ) at the 26th position of the tRNA. Since
) at the 26th position of the tRNA. Since 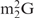 cannot form a Watson–Crick-type base-pair with cytidine, an alternative oligoDNA, OligoGT-YPhe (Figure 1B), was synthesized with the expectation of improving the stability of the yeast tRNAPhe•oligoDNA hybrid.
cannot form a Watson–Crick-type base-pair with cytidine, an alternative oligoDNA, OligoGT-YPhe (Figure 1B), was synthesized with the expectation of improving the stability of the yeast tRNAPhe•oligoDNA hybrid.
Figure 6.

Differences in the melting profiles between E. coli
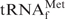 and yeast tRNAPhe in buffers containing various monovalent cations, Na+, TMA+ or TEA+. The thick and thin lines show the melting profiles of E. coli
and yeast tRNAPhe in buffers containing various monovalent cations, Na+, TMA+ or TEA+. The thick and thin lines show the melting profiles of E. coli
 and yeast tRNAPhe, respectively. The black lines (Na+), red lines (TMA+) and green lines (TEA+) show the profiles with hybridization buffer Na+, TMA+ or TEA+, respectively.
and yeast tRNAPhe, respectively. The black lines (Na+), red lines (TMA+) and green lines (TEA+) show the profiles with hybridization buffer Na+, TMA+ or TEA+, respectively.
Figure 7.

Differences in the hypochromicity profiles between E. coli
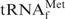 and yeast tRNAPhe with their complementary oligoDNAs in hybridization buffers containing tetra-alkylammonium ions. The thick lines show the hypochromicity profiles of E. coli
and yeast tRNAPhe with their complementary oligoDNAs in hybridization buffers containing tetra-alkylammonium ions. The thick lines show the hypochromicity profiles of E. coli
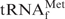 with its complementary oligoDNA, Oligo-EfMet, in hybridization buffer TMA+ or TEA+. The thin lines show the profiles of yeast tRNAPhe with one of its complementary oligoDNAs, OligoGC-YPhe. The red and green lines show the profiles in hybridization buffer TMA+ containing TMA-Cl and hybridization buffer TEA+ containing TEA-Cl, respectively.
with its complementary oligoDNA, Oligo-EfMet, in hybridization buffer TMA+ or TEA+. The thin lines show the profiles of yeast tRNAPhe with one of its complementary oligoDNAs, OligoGC-YPhe. The red and green lines show the profiles in hybridization buffer TMA+ containing TMA-Cl and hybridization buffer TEA+ containing TEA-Cl, respectively.
Effect of a modified nucleotide on the stability of a tRNA•oligoDNA hybrid
Figure 8A shows the hypochromicity profiles of yeast tRNAPhe in the presence or absence of an oligoDNA (OligoGC-YPhe or OligoGT-YPhe). The transition temperatures of yeast tRNAPhe and OligoGT-YPhe (Figure 8A, dashed–dotted red and green lines) were slightly shifted to higher temperatures compared with those of yeast tRNAPhe and OligoGC-YPhe (Figure 8A, solid red and green lines). These findings probably indicate that the destabilizing effect of a single modified nucleotide (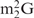 in this case) interfering with the formation of the Watson–Crick-type base-pair in the tRNAPhe•oligoDNA hybrid was partly compensated for by a thymidine at the counter-position in the complementary oligoDNA. If the presence of
in this case) interfering with the formation of the Watson–Crick-type base-pair in the tRNAPhe•oligoDNA hybrid was partly compensated for by a thymidine at the counter-position in the complementary oligoDNA. If the presence of 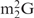 is known in advance, an oligoDNA with a T at the counter-position of
is known in advance, an oligoDNA with a T at the counter-position of 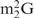 should be made. However, when no information is available about the modified nucleotides in the target tRNA, we cannot be prepared for such compensation as described above. Therefore, we compared the recoveries using OligoGC-YPhe with those using OligoGT-YPhe when purifying yeast tRNAPhe from yeast tRNAMix. At first, we examined the recovery versus hybridization temperature by using the same procedure as conducted with E. coli
should be made. However, when no information is available about the modified nucleotides in the target tRNA, we cannot be prepared for such compensation as described above. Therefore, we compared the recoveries using OligoGC-YPhe with those using OligoGT-YPhe when purifying yeast tRNAPhe from yeast tRNAMix. At first, we examined the recovery versus hybridization temperature by using the same procedure as conducted with E. coli
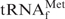 (Figure 5A). When TMA-Cl was used, the optimum recovery was obtained at 55°C, irrespective of the oligoDNA used. This temperature was lower than that of E. coli
(Figure 5A). When TMA-Cl was used, the optimum recovery was obtained at 55°C, irrespective of the oligoDNA used. This temperature was lower than that of E. coli
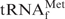 (Figure 5A), probably due to the instability of the yeast tRNAPhe•oligoDNA hybrid observed in Figure 7. However, when TEA-Cl was used, the optimum temperature was the same as that of E. coli
(Figure 5A), probably due to the instability of the yeast tRNAPhe•oligoDNA hybrid observed in Figure 7. However, when TEA-Cl was used, the optimum temperature was the same as that of E. coli
 (35°C), irrespective of the oligoDNA used. Therefore, hybridization in this case was conducted at 35°C in the presence of TEA-Cl. Irrespective of the oligoDNA used, the recoveries of yeast tRNAPhe were very similar (0.15 and 0.14 A260 units recovered from 5 A260 units of yeast tRNAMix using OligoGC-YPhe and OligoGT-YPhe, respectively). The purities of the recovered tRNAs were assessed by 10% PAGE containing 7 M urea (Figure 8B). The purities of the two tRNAPhe preparations were indistinguishable and satisfactory, irrespective of the oligoDNA used in the hybridization step. Although both purified tRNAs appeared to move slightly faster than the authentic yeast tRNAPhe, this occurred because the yeast tRNAMix used as the source was obtained from yeast cells at the stationary phase, in which tRNAs are known to often lack the CCA terminus. This deficiency at the 3′-terminus can be fully repaired by incubating the tRNA with tRNA-nucleotidyl transferase and CTP/ATP. These results suggest that even a complementary oligoDNA based solely on the Watson–Crick-type base pairs would work substantially in this purification procedure. In a practical meaning, this finding would be very helpful for those who plan to purify any tRNA from any organism without any information about the modification status of nucleotides in the tRNA of interest.
(35°C), irrespective of the oligoDNA used. Therefore, hybridization in this case was conducted at 35°C in the presence of TEA-Cl. Irrespective of the oligoDNA used, the recoveries of yeast tRNAPhe were very similar (0.15 and 0.14 A260 units recovered from 5 A260 units of yeast tRNAMix using OligoGC-YPhe and OligoGT-YPhe, respectively). The purities of the recovered tRNAs were assessed by 10% PAGE containing 7 M urea (Figure 8B). The purities of the two tRNAPhe preparations were indistinguishable and satisfactory, irrespective of the oligoDNA used in the hybridization step. Although both purified tRNAs appeared to move slightly faster than the authentic yeast tRNAPhe, this occurred because the yeast tRNAMix used as the source was obtained from yeast cells at the stationary phase, in which tRNAs are known to often lack the CCA terminus. This deficiency at the 3′-terminus can be fully repaired by incubating the tRNA with tRNA-nucleotidyl transferase and CTP/ATP. These results suggest that even a complementary oligoDNA based solely on the Watson–Crick-type base pairs would work substantially in this purification procedure. In a practical meaning, this finding would be very helpful for those who plan to purify any tRNA from any organism without any information about the modification status of nucleotides in the tRNA of interest.
Figure 8.

Influence of a modified nucleotide disturbing the normal Watson–Crick-type base-pair on the hybridization of yeast tRNAPhe and oligoDNAs. (A) Hypochromicity profiles of yeast tRNAPhe in the presence of oligoDNAs, OligoGC-YPhe (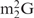 26 faces up to C) or OligoGT-YPhe (
26 faces up to C) or OligoGT-YPhe ( 26 faces up to T). The dotted lines show the hypochromicity profiles of yeast tRNAPhe alone. The solid and dashed-dotted lines show yeast tRNAPhe with OligoGC-YPhe and OligoGT-YPhe, respectively. The red and green lines show the profiles in hybridization buffer TMA+ and TEA+, respectively. (B) Quality check of the recovered yeast tRNAPhe by 10% PAGE containing 7 M urea. Lane mix: 0.1 A260 units of unfractionated yeast tRNAs. Lane Au: 0.01 A260 units of the authentic yeast tRNAPhe. Lanes GC and GT: 0.01 A260 units of the recovered yeast tRNAPhe in the presence of the oligoDNAs OligoGC-YPhe and OligoGT-YPhe, respectively. The gel was stained with methylene blue.
26 faces up to T). The dotted lines show the hypochromicity profiles of yeast tRNAPhe alone. The solid and dashed-dotted lines show yeast tRNAPhe with OligoGC-YPhe and OligoGT-YPhe, respectively. The red and green lines show the profiles in hybridization buffer TMA+ and TEA+, respectively. (B) Quality check of the recovered yeast tRNAPhe by 10% PAGE containing 7 M urea. Lane mix: 0.1 A260 units of unfractionated yeast tRNAs. Lane Au: 0.01 A260 units of the authentic yeast tRNAPhe. Lanes GC and GT: 0.01 A260 units of the recovered yeast tRNAPhe in the presence of the oligoDNAs OligoGC-YPhe and OligoGT-YPhe, respectively. The gel was stained with methylene blue.
Adequate cases for the use of TMA-Cl or TEA-Cl in the preparation of a target tRNA
In a series of experiments, we found that both TMA-Cl and TEA-Cl used as monovalent cations for hybridization drastically denatured the tertiary structures of tRNAs and enhanced the formation of tRNA•oligoDNA hybrids. In other words, the thermal properties of all tRNAs become indistinguishable in the presence of TMA-Cl or TEA-Cl. In fact, the melting profile of unfractionated E. coli tRNAs was indistinguishable from that of E. coli
 in the presence of TEA-Cl (data not shown). This effect could be a big advantage in purifying isoacceptor tRNAs or multiple tRNAs simultaneously by using advanced hybridization-based purification methods, such as ‘chaplet columns’ by Suzuki and Suzuki (12) or ‘reciprocal circulating chromatography’ by Miyauchi et al. (14).
in the presence of TEA-Cl (data not shown). This effect could be a big advantage in purifying isoacceptor tRNAs or multiple tRNAs simultaneously by using advanced hybridization-based purification methods, such as ‘chaplet columns’ by Suzuki and Suzuki (12) or ‘reciprocal circulating chromatography’ by Miyauchi et al. (14).
Although both of TMA-Cl and TEA-Cl are applicable to the hybridization-based purification of tRNAs from unfractionated tRNAs, there are considerable differences in their properties. TEA-Cl denatures tRNA more strongly than TMA-Cl because the bulkiness of TEA-Cl destroys the A-form duplex of the tRNA by binding to the phosphates in the minor grooves (16). On the other hand, TMA-Cl does not destroy the duplex of the tRNA very much. Therefore, the melting temperature of the tRNA in the presence of TEA-Cl was about 20°C lower than that in the presence of TMA-Cl (Figure 2). This lowering effect on the melting temperature of the tRNA must be advantageous for adopting this hybridization-based method to prepare thermostable tRNAs from thermophilic bacteria. However, since RNA•DNA heteroduplexes are also A-form duplexes, it is of concern that the tRNA•oligoDNA hybrids will also be destabilized. In fact, the transition temperature of hybridization for TEA-Cl observed in the hypochromicity profile became lower than that for TMA-Cl (Figure 3). Therefore, when TEA-Cl was used for hybridization, we needed to lower the temperature of the hybridization step from 65°C to 35°C (Figure 5A). Clearly, the lower hybridization temperature caused lower selectivity (Figure 5B, compare asterisks). This is probably a disadvantage for the use of TEA-Cl. Therefore, we propose two alternative protocols for preparing a desired tRNA on a case-by-case basis. When the tRNA to be purified is the major component in the cell or if a slight contaminant can be tolerated, the use of TEA-Cl at a hybridization temperature of 35°C would be the best choice. On the contrary, if the tRNA of interest is a minor component or if even a slight contaminant would cause a big problem, the use of TMA-Cl at the hybridization temperature of 65°C would be recommended.
As mentioned earlier, we have already reported successful purification of tRNACys from a hyperthermophilic bacterium, A. aeolicus, to verify the existence of 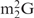 in a native tRNA (23). In addition, we have succeeded in purifying several species of tRNAs by using either of the two protocols described above. Examples of such purified tRNAs are as follows: archaeal tRNAs: tRNAGlu, major and minor tRNAIle and elongator tRNAMet from Methanosarcina acetivorans; eukaryotic tRNAs: tRNATyr and tRNACys from Saccharomyces cerevisiae; and bacterial tRNAs: minor tRNAIle and
in a native tRNA (23). In addition, we have succeeded in purifying several species of tRNAs by using either of the two protocols described above. Examples of such purified tRNAs are as follows: archaeal tRNAs: tRNAGlu, major and minor tRNAIle and elongator tRNAMet from Methanosarcina acetivorans; eukaryotic tRNAs: tRNATyr and tRNACys from Saccharomyces cerevisiae; and bacterial tRNAs: minor tRNAIle and 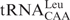 from Bifidobacterium adolescentis. The structural and/or functional analyses of these tRNAs will be described elsewhere. Following are brief overview of the aminoacylation capacities of the above tRNAs purified by this method. As a whole, tRNAs purified from Bacteria or Eucarya showed reasonable activities under our aminoacylation conditions in vitro, while tRNAs from Archaea were generally less active, even when their purities were satisfactory (data not shown). This inactiveness of archaeal tRNAs may probably indicate that the denaturation and renaturation steps of archaeal tRNAs are irreversible. At present, we speculate that the presence or absence of Archaea-specific modification(s), such as archaeosine, might cause their irreversibility. Anyway, further experiments are necessary to prepare archaeal tRNAs in a biologically active state.
from Bifidobacterium adolescentis. The structural and/or functional analyses of these tRNAs will be described elsewhere. Following are brief overview of the aminoacylation capacities of the above tRNAs purified by this method. As a whole, tRNAs purified from Bacteria or Eucarya showed reasonable activities under our aminoacylation conditions in vitro, while tRNAs from Archaea were generally less active, even when their purities were satisfactory (data not shown). This inactiveness of archaeal tRNAs may probably indicate that the denaturation and renaturation steps of archaeal tRNAs are irreversible. At present, we speculate that the presence or absence of Archaea-specific modification(s), such as archaeosine, might cause their irreversibility. Anyway, further experiments are necessary to prepare archaeal tRNAs in a biologically active state.
Finally, it should be emphasized that the protocols described here are specialized for small-scale preparation of specific tRNAs of interest. In the case that large-scale purification of specific tRNAs is required, there remain some problems originating from the instability of the immobilized DNA because the connection between the resin and the oligoDNA used in this protocol is based on the biotin-avidin interaction. To overcome such a defect, it will be necessary to make covalently-immobilized oligoDNAs at a reasonable cost. Since this method is, in principle, applicable for the preparation of any RNA as long as the gene sequence is already known, overcoming the issue of the instability of the immobilized DNA will make the use of this method widespread.
FUNDING
Scientific Research (B) (20350075) from the Japan Society for the Promotion of Science (to T.Y.). Funding for open access charge: Scientific Research (B) (20350075) from Japan Society for the Promotion of Science (to T.Y.).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Dr Y. Ueno of Gifu University for the measurements of the tRNA melting profiles. We also thank Dr H. Hori of Ehime University for useful comments on the design of the hybridization probe for the modified nucleotide.
REFERENCES
- 1.Söll D, RajBhandary UL, editors. tRNA: Structure, Biosynthesis, and Function. Washington, DC: ASM Press; 1995. [Google Scholar]
- 2.Sampson JR, Uhlenbeck OC. Biochemical and physical characterization of an unmodified yeast phenylalanine transfer RNA transcribed in vitro. Proc. Natl Acad. Sci. USA. 1988;85:1033–1037. doi: 10.1073/pnas.85.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giegé R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26:5017–5035. doi: 10.1093/nar/26.22.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agris PF, Vendeix FA, Graham WD. tRNA's; wobble decoding of the genome: 40 years of modification. J. Mol. Biol. 2007;366:1–13. doi: 10.1016/j.jmb.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 5.Wakita K, Watanabe Y, Yokogawa T, Kumazawa Y, Nakamura S, Ueda T, Watanabe K, Nishikawa K. Higher-order structure of bovine mitochondrial tRNAPhe lacking the ‘conserved’ GG and T psi CG sequences as inferred by enzymatic and chemical probing. Nucleic Acids Res. 1994;22:347–353. doi: 10.1093/nar/22.3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsurui H, Kumazawa Y, Sanokawa R, Watanabe Y, Kuroda T, Wada A, Watanabe K, Shirai T. Batchwise purification of specific tRNAs by a solid-phase DNA probe. Anal. Biochem. 1994;221:166–172. doi: 10.1006/abio.1994.1393. [DOI] [PubMed] [Google Scholar]
- 7.Tomita K, Ueda T, Watanabe K. The presence of pseudouridine in the anticodon alters the genetic code: a possible mechanism for assignment of the AAA lysine codon as asparagine in echinoderm mitochondria. Nucleic Acids Res. 1999;27:1683–1689. doi: 10.1093/nar/27.7.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomita K, Ueda T, Ishiwa S, Crain PF, McCloskey JA, Watanabe K. Codon reading patterns in Drosophila melanogaster mitochondria based on their tRNA sequences: a unique wobble rule in animal mitochondria. Nucleic Acids Res. 1999;27:4291–4297. doi: 10.1093/nar/27.21.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki T, Suzuki T, Wada T, Saigo K, Watanabe K. Taurine as a constituent of mitochondrial tRNAs: new insights into the functions of taurine and human mitochondrial diseases. EMBO J. 2002;21:6581–6589. doi: 10.1093/emboj/cdf656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaneko T, Suzuki T, Kapushoc ST, Rubio MA, Ghazvini J, Watanabe K, Simpson L, Suzuki T. Wobble modification differences and subcellular localization of tRNAs in Leishmania tarentolae: implication for tRNA sorting mechanism. EMBO J. 2003;22:657–667. doi: 10.1093/emboj/cdg066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirino Y, Yasukawa T, Ohta S, Akira S, Ishihara K, Watanabe K, Suzuki T. Codon-specific translational defect caused by a wobble modification deficiency in mutant tRNA from a human mitochondrial disease. Proc. Natl Acad. Sci. USA. 2004;101:15070–15075. doi: 10.1073/pnas.0405173101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki T, Suzuki T. Chaplet column chromatography: isolation of a large set of individual RNAs in a single step. Methods Enzymol. 2007;425:231–239. doi: 10.1016/S0076-6879(07)25010-4. [DOI] [PubMed] [Google Scholar]
- 13.Yokogawa T, Kumazawa Y, Miura K, Watanabe K. Purification and characterization of two serine isoacceptor tRNAs from bovine mitochondria by using a hybridization assay method. Nucleic Acids Res. 1989;17:2623–2638. doi: 10.1093/nar/17.7.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyauchi K, Ohara T, Suzuki T. Automated parallel isolation of multiple species of non-coding RNAs by the reciprocal circulating chromatography method. Nucleic Acids Res. 2007;35:e24. doi: 10.1093/nar/gkl1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melchior WB, Von Hippel PH. Alteration of the relative stability of dA-dT and dG-dC base pairs in DNA. Proc. Natl Acad. Sci. USA. 1973;70:298–302. doi: 10.1073/pnas.70.2.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Golaś T, Miller M, Shugar D. The effects of tetraalkylammonium salts on helix-coil transition parameters in natural and synthetic ribo- and deoxyribo-polynucleotides. Chem. Biol. Interact. 1980;30:209–222. doi: 10.1016/0009-2797(80)90127-1. [DOI] [PubMed] [Google Scholar]
- 17.Zubay G. The isolation and fractionation of soluble ribonucleic acid. J. Mol. Biol. 1962;4:347–356. [Google Scholar]
- 18.Holley RW. Large-scale preparation of yeast “soluble” ribonucleic acid. Biochem. Biophys. Res. Commun. 1963;10:186–188. doi: 10.1016/0006-291x(63)90048-2. [DOI] [PubMed] [Google Scholar]
- 19.Wimmer E, Maxwell IH, Tener GM. A simple method for isolating highly purified yeast phenylalanine transfer ribonucleic acid. Biochemistry. 1968;7:2623–2628. doi: 10.1021/bi00847a026. [DOI] [PubMed] [Google Scholar]
- 20.Ohno S, Yokogawa T, Nishikawa K. Changing the amino acid specificity of yeast tyrosyl-tRNA synthetase by genetic engineering. J. Biochem. 2001;130:417–423. doi: 10.1093/oxfordjournals.jbchem.a003001. [DOI] [PubMed] [Google Scholar]
- 21.Kumazawa Y, Yokogawa T, Tsurui H, Miura K, Watanabe K. Effect of the higher-order structure of tRNAs on the stability of hybrids with oligodeoxyribonucleotides: separation of tRNA by an efficient solution hybridization. Nucleic Acids Res. 1992;20:2223–2232. doi: 10.1093/nar/20.9.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hasegawa T, Yokogawa T. Escherichia coli proline tRNA: structure and recognition sites for prolyl-tRNA synthetase. Nucleic Acids Symp. Ser. 2000;44:7–8. doi: 10.1093/nass/44.1.7. [DOI] [PubMed] [Google Scholar]
- 23.Awai T, Kimura S, Tomikawa C, Ochi A, Ihsanawati I, Bessho Y, Yokoyama S, Ohno S, Nishikawa K, Yokogawa T. Aquifex aeolicus tRNA (N2, N2-guanine)-dimethyltransferase (Trm1) catalyzes transfer of methyl groups not only to guanine 26 but also to guanine 27 in tRNA. J. Biol. Chem. 2009;284:20467–20478. doi: 10.1074/jbc.M109.020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watanabe K, Oshima T, Iijima K, Yamaizumi Z, Nishimura S. Purification and thermal stability of several amino acid-specific tRNAs from an extreme thermophile, Thermus thermophilus HB8. J. Biochem. 1980;87:1–13. doi: 10.1093/oxfordjournals.jbchem.a132713. [DOI] [PubMed] [Google Scholar]
- 25.Kumazawa Y, Yokogawa T, Hasegawa E, Miura K, Watanabe K. The aminoacylation of structurally variant phenylalanine tRNAs from mitochondria and various nonmitochondrial sources by bovine mitochondrial phenylalanyl-tRNA synthetase. J. Biol. Chem. 1989;264:13005–13011. [PubMed] [Google Scholar]
- 26.Jacobs KA, Rudersdorf R, Neill SD, Dougherty JP, Brown EL, Fritsch EF. The thermal stability of oligonucleotide duplexes is sequence independent in tetraalkylammonium salt solutions: application to identifying recombinant DNA clones. Nucleic Acids Res. 1988;16:4637–4650. doi: 10.1093/nar/16.10.4637. [DOI] [PMC free article] [PubMed] [Google Scholar]