

Abstract
Harmful algal blooms (HABs) are a global problem that affects both human and ecosystem health. One of the most serious and widespread HAB poisoning syndromes is paralytic shellfish poisoning, commonly caused by Alexandrium spp. dinoflagellates. Like many toxic dinoflagellates, Alexandrium produces resistant resting cysts as part of its life cycle. These cysts play a key role in bloom initiation and decline, as well as dispersal and colonization of new areas. Information on cyst numbers and identity is essential for understanding and predicting blooms, yet comprehensive cyst surveys are extremely time- and labor-intensive. Here we describe the development and validation of a quantitative real-time PCR (qPCR) technique for the enumeration of cysts of A. tamarense of the toxic North American/Group I ribotype. The method uses a cloned fragment of the large subunit ribosomal RNA gene as a standard for cyst quantification, with an experimentally determined conversion factor of 28,402±6152 LSU ribosomal gene copies per cyst. Tests of DNA extraction and PCR efficiency show that mechanical breakage is required for adequate cyst lysis, and that it was necessary to dilute our DNA extracts 50-fold in order to abolish PCR inhibition from compounds co-extracted from the sediment. The resulting assay shows a linear response over 6 orders of magnitude and can reliably quantify ≥10cysts/cc sediment.
For method validation, 129 natural sediment samples were split and analyzed in parallel, using both the qPCR and primulin-staining techniques. Overall, there is a significant correlation (p<0.001) between the cyst abundances determined by the two methods, although the qPCR counts tend to be lower than the primulin values. This underestimation is less pronounced in those samples collected from the top 1 cm of sediment, and more pronounced in those derived from the next 1–3 cm of the core. These differences may be due to the condition of the cysts in the different layers, as the top 1cm contains more recent cysts while those in the next 1–3cm may have been in the sediments for many years. Comparison of the cyst densities obtained by both methods shows that a majority (56.6%) of the values are within a two-fold range of each other and almost all of the samples (96.9%) are within an order of magnitude. Thus, the qPCR method described here represents a promising alternative to primulin-staining for the identification and enumeration of cysts. The qPCR method has a higher throughput, enabling the extraction and assay of 24 samples in the time required to process and count 8–10 samples by primulin staining. Both methods require prior expertise, either in taxonomy or molecular biology. Fewer person-hours per sample are required for qPCR, but primulin staining has lower reagent costs. The qPCR method might be more desirable for large-scale cyst mapping, where large numbers of samples are generated and a higher sample analysis rate is necessary. While the qPCR and primulin-staining methods generate similar data, the choice of counting method may be most influenced by the practical issue of the different relative costs of labor and materials between the two methods.
Keywords: quantitative PCR, ribosomal, toxic dinoflagellate, saxitoxins, algal blooms, red tides
1. Introduction
Harmful algal blooms (HABs) are a global problem that affects both human and ecosystem health. One of the most serious and widespread HAB poisoning syndromes is paralytic shellfish poisoning (PSP), which results from the consumption of shellfish that have fed on toxic dinoflagellates. The dinoflagellate toxins include the potent neurotoxin saxitoxin (STX) and its derivatives, which accumulate in shellfish and other marine organisms, causing human illnesses and death, shellfish quarantines, and the mortality of birds, larval and adult fish, and even marine mammals. The most common cause of PSP outbreaks is dinoflagellates of the genus Alexandrium, comprising 28 species (Balech, 1995), 11 of which are toxic.
Within the Alexandrium genus, the tamarense species complex is frequently associated with PSP outbreaks around the world. This globally distributed species complex includes three toxigenic morphospecies, A. tamarense, A. fundyense, and A. catenella. Molecular phylogenetic analysis has shown that the species complex is composed of several ribotypes, which are defined by their toxicity and geographic origin (Scholin et al., 1994). In the northeastern U.S. and Western Europe, known toxic strains exhibit both the A. tamarense and A. fundyense morphospecies, but all have the same toxic North American (Scholin et al., 1994) or Group I (Lilly et al., 2007) ribotype1.
Over the last three decades, both the frequency and geographic range of Alexandrium blooms have increased, as have HABs in general (Anderson, 1989; Hallegraeff, 1993). Possible explanations for this expansion include increased monitoring and awareness of HABs, eutrophication (Anderson et al., 2002), natural transport (Franks and Anderson, 1992; Vila et al., 2001), and human-assisted dispersal (e.g. (Lilly et al., 2002). A significant factor in this global expansion is that many toxic dinoflagellate species, including Alexandrium, produce resistant resting cysts as part of their life cycle. In the plankton, A. tamarense reproduces via asexual cell division, until changing environmental conditions, e.g. nutrient limitation (Anderson et al., 1984; Turpin et al., 1978), induce sexuality and gamete formation. Gametes then fuse to form planozygotes that become resting cysts (hypnozygotes) that settle to the benthos. The resting cysts can remain dormant for years, until conditions become favorable for excystment and growth. Thus, the resting cysts provide an inoculum of cells to begin a toxic bloom. The resistant cyst stage can also greatly facilitate dispersal (Dale, 1983), thereby enabling cyst-forming species to colonize new areas.
Because the cyst stage of the dinoflagellate life cycle plays an important role in both the initiation and decline of toxic blooms, information regarding the number and identity of cysts in the sediment is crucial to the understanding and prediction of blooms. For example, the numbers of cysts in the sediments appears to be an important determinant of the magnitude of the bloom observed in the following year (He et al., 2008; Stock et al., 2005). Thus, accurate and recent maps of sediment cyst distributions are needed to drive predictive models of bloom dynamics (McGillicuddy et al., 2005; Stock et al., 2005). Cysts also provide an integrated record of the presence of toxic species in an area. If the blooms are small, short-lived, or otherwise go undetected, vegetative cells may not be observed in the water column, but the cysts can be found in the sediments at any time of the year.
While information on cyst density and identity is highly desirable for many research and monitoring purposes, comprehensive cyst surveys are almost prohibitively difficult. Core collection is technically demanding and time-intensive, especially when mapping large areas. Downstream sample processing includes sectioning the cores and separating the cysts from the bulk of the sediment matrix for analysis. The samples then need to be counted by microscopy, which requires taxonomic expertise on the part of the operator in order to distinguish between morphologically similar cysts from different species. Microscopic cyst counts are also challenging because the samples often contain a lot of the sediment matrix, despite the processing, and so it can be difficult to discern the cysts amongst the surrounding particles. This latter problem can be mitigated by staining the cysts with the fluorescent primulin dye (Yamaguchi et al., 1995).
Because the current methods of cyst counting are time- and labor-intensive, we sought to develop a cyst enumeration method that would reduce the time required to generate cyst maps. The method would need to have a higher throughput than microscopic methods, yet still be quantitative and specific to the species of interest. Recently, quantitative real-time PCR (qPCR) methods have been developed for the detection of planktonic cells of a variety of HAB species (Dyhrman et al., 2006; Galluzzi et al., 2004; Hosoi-Tanabe et al., 2004; Moorthi et al., 2006; Popels et al., 2003) and also the cyst stages of A. tamarense and A. catenella (Kamikawa et al., 2007). All of these methods show a good correspondence between cell counts obtained using qPCR or microscopic methods. However, most of the reports examined relatively few field samples, generally less than twenty.
Here we report the development and validation of a qPCR method for the enumeration of cysts of toxic A. tamarense. The assay is adapted from a qPCR method previously described for planktonic cells (Dyhrman et al., 2006) that is specific for the toxic North American/Group I ribotype. The extraction efficiency of different DNA extraction methods was determined, and field DNA samples were extracted and tested to assess the presence of PCR inhibitors in field samples. Standard curves were constructed using a cloned ribosomal RNA gene and calibrated against manually isolated cysts from natural sediments. Lastly, 129 field-collected sediment samples were split and analyzed in parallel, using both the qPCR and primulin staining/microscopy techniques. We present a comparison and analysis of the results from these two approaches, along with a discussion of their relative strengths and weaknesses.
2. Materials and Methods
2.1 Cloning of large subunit ribosomal RNA gene
The D1-D2 hypervariable region of the large subunit (LSU) ribosomal RNA gene was cloned for use as a standard in the qPCR assay. DNA was extracted from A. fundyense strain GTCA28 using a Generation Capture Column kit (Qiagen, USA) per the manufacturer’s protocol. The D1-D2 region was PCR amplified in a 25-μL reaction volume containing 1X reaction buffer, 2 mM of each dNTP (TaKaRa Bio Inc., USA), 0.15 μL of TaKaRa Taq DNA Polymerase (TaKaRa Bio Inc., USA), 5 pmol each of forward primer D1R and reverse primer D2C (Scholin et al., 1994) and 3 μL of DNA. The PCR reaction consisted of an initial denaturing step of 94 °C for 5 min, followed by 40 cycles of 45 s at 94 °C, 45 s at 50 °C and 1 min at 72 °C followed by a final extension step of 7 min at 72 °C. PCR products were visualized using agarose gel electrophoresis, and the single band observed was excised from the gel and purified using a MinElute PCR purification kit (Qiagen, USA). The amplicon was then cloned and chemically transformed into E. coli DH5alpha using a TOPO TA Cloning kit (Invitrogen, USA) according to the manufacturer’s instructions.
Of the resultant transformed E. coli, seven colonies were chosen for analysis. Colony PCR was performed by using a sterile toothpick to transfer some of each colony to 10 μL of sterile water and adding sufficient reagent to yield a 25 μL reaction containing 1X reaction buffer (TaKaRa Bio Inc., USA), 2 mM of each dNTP (TaKaRa Bio Inc., USA), 0.15 μL of TaKaRa Taq DNA Polymerase (TaKaRa Bio Inc., USA), 5 pmol each of primers M13F and M13R. The PCR reaction consisted of an initial denaturing step of 94 °C for 2 min, followed by 25 cycles of 1 min at 94 °C, 1 min at 55 °C and 1 min at 72 °C followed by a final extension step of 7 min at 72 °C.
Of the seven colonies analyzed, two produced single discrete bands of the desired size on the gel. These clones were transferred into tubes containing 5 mL of LB broth with 50 μg/mL ampicillin and cultured overnight at 37 °C in a shaker. The plasmid was then isolated using the Wizard Plus Minipreps DNA Purification System (Promega Corp., USA) per the manufacturer’s protocol. Purified plasmid was sequenced at the Core DNA Sequencing Facility of the University of Texas at Austin using the vector-encoded primers M13F and M13R. The plasmid insert sequences were analysed using Sequencher 4.8 DNA sequence assembly software (Gene Codes Corp., USA), and they were confirmed to be 100 % similar to the Alexandrium tamarense North American/Group I ribotype by BLAST search of the NCBI GenBank database.
The purified plasmid was quantified via a dye-association assay using the DNA-specific fluorescent dye EvaGreen (Biotium, Inc. USA) and a real-time PCR machine (Eppendorf, USA), based on the method of (Wang et al., 2006). Briefly, 1.25 μL Evagreen (25X concentrate) was mixed with 18.75 μL of water and 5 μL of DNA in a 0.2-mL PCR reaction tube. Triplicate reactions of pUC19 plasmid (New England Biolabs, USA) ranging from 20–100 ng were used as standards. The assay was run at 72 °C for 10 minutes, and fluorescence was measured at the end of the incubation. In total, six reactions containing Alexandrium plasmid were analyzed, and the resulting concentration values were averaged. The copy number per uL of plasmid solution was then calculated using the molecular weight of the plasmid plus insert.
2.2 Testing of DNA extraction methods
To determine the best method for lysing cysts prior to qPCR, four different extraction methods were tested. Previous studies had shown that the DNEasy Tissue DNA Extraction kit (Qiagen, Inc. USA) was very effective for purifying DNA from Alexandrium cells, and so it was also tested for use with cysts. The QIAamp Stool DNA Mini kit (Qiagen, Inc. USA) was also tested, as it included reagents designed to remove inhibitors that are associated with some sample matrices. Both of these kits were tested with and without mechanical breakage (bead-beating) of the cysts.
The extraction methods were tested on cysts derived from cultured cells. Cysts were produced by combining two clonal strains of compatible mating type in low nitrogen medium (Anderson et al., 2003). After one month, the resulting cysts were collected from the bottom of the culture tubes and resuspended in nutrient-free seawater. Cyst numbers were counted using light microscopy. The cyst suspension was thoroughly homogenized, and replicate aliquots containing ca. 800 cysts were dispensed onto 25 mm, 5 μm Durapore (Millipore Inc., USA) filters that were immediately frozen at −80°C. Because of the limited number of cultured cysts available for extraction testing, only a single filter was used to test each extraction method. After extraction, the concentration of DNA in the samples was measured using a Nano-Drop spectrophotometer (ThermoScientific, USA), and the recovery of ribosomal RNA genes was assessed via qPCR (Section 2.3).
DNEasy Tissue kit
The DNEasy Tissue kit was used according to the manufacturer’s instructions, with a few modifications. The volumes of buffer ATL and Proteinase K solution were doubled, to 360 μL and 40 μL, respectively. The Proteinase K incubation time was increased to 4 hr at 55°C, before continuing with the remainder of the protocol. For mechanical breakage of cells, 0.5 mm silica-zirconium beads were added to the sample, which was then processed for 3, 50 s cycles in a mini-BeadBeater (BioSpec Products, Inc.) with cooling on ice in between the cycles.
QIAamp Stool DNA Mini Kit
The QIAamp Stool DNA Mini kit was used according to the manufacturer’s protocol, also with a few modifications. After the addition of buffer ASL, the samples were heated at 85°C for 10 min. If bead-beating was used, it was performed after the 85°C heating step, using the cycles described above. Because the entire lysate volume was not used during the QIAamp protocol, volumes were recorded at each step, and the final elution volume was adjusted to provide the same ratio of initial lysate processed:purified DNA volume eluted for both the Tissue and Stool kits.
2.3 Real-time Quantitative PCR
Real-time PCR reactions (25 μL final volume) contained 1X Full Velocity SYBR Green QPCR Master Mix (Stratagene, USA), 3.75 pmol each of the reverse primer AF1 (5′-GCAAGTGCAACACTCCCACCAAGCAA-3′) and the forward primer AlexLSUf2 (5′-GGCATTGGAATGCAAAGTGGGTGG-3′), and 5 μL of DNA template. The cycling conditions consisted of 1 cycle at 95 °C for 3 min, followed by 30 cycles of 95 °C for 10 seconds, 55 °C for 30 s, followed by melt curve analysis. Six plasmid standards were included on each plate, comprising 10-fold serial dilutions of the plasmid ranging from 5.2 × 103 to 5.2 × 107 copies. Each reaction was performed in triplicate on an Eppendorf MasterCycler ep RealPlex, with threshold cycle (Ct) values determined by the software.
2.4 Determination of cyst ribosomal copy number
The average number of LSU ribosomal RNA gene copies per cyst was determined using field-collected cysts from the Gulf of Maine. Sediments were collected from near Penobscot Bay, Maine as described in Section 2.6. Two mL of homogenized sediment was sonicated and sieved as described in Section 2.6. The material retained on the 20μm mesh was rinsed into a 15 mL centrifuge tube and resuspended to 14mL with filtered seawater. The sample was evenly split, and each 7 mL was concentrated on a density gradient of 1.4 g/mL sodium metatungstate monohydrate (Alfa Aesar, USA) (Bolch, 1997). The resulting pellet was resuspended in 1 mL of filtered seawater. A subsample of the material was examined under a light microscope and 40 A. fundyense hypnozygotes were isolated via micropipette and placed onto a Durapore 5 μm pore size membrane filter (Millipore) that was placed in 360 μL of ATL buffer (Qiagen) in a 2 mL screw-capped microcentrifuge tube. This was repeated in triplicate.
Silica-zirconium beads (0.5 mm, BioSpec Products, Inc.) were added to each tube and the cysts were disrupted by vortexing for 1 min. The lysate was transferred to a clean 2-mL microcentrifuge tube and 40 μL of the included Proteinase K solution (Qiagen DNEasy kit) were added. The sample was incubated at 56 °C for 60 min and then processed according to the remainder of the manufacturer’s protocol. The sample was eluted in 200 μL of AE (elution buffer) and stored at −20 °C until analysis. Cyst DNA extracts were analyzed using qPCR (Section 2.3) along with cloned plasmid standards. Cyst ribosomal copy number was determined by comparing the Ct values of the extracted cyst samples with the plasmid copy number standards.
2.5 PCR inhibition assay
Prior to qPCR analysis of the extracted sediment samples, a subset was tested for the presence of PCR inhibitors. Inhibition assays used purified pUC19 plasmid (New England BioLabs, Inc.) as template along with the plasmid-specific primers M13F and M13R. Each reaction contained 3 ng of pUC19 plasmid, along with 5 μL of extracted sediment DNA (to test for inhibition) or 5 μL of sterile nuclease-free water (as a control). The sediment DNA was added at full strength (undiluted), or diluted 1:2, 1:10 or 1:50 with water. Real-time PCR reactions were performed as described in Section 2.3. The same assay was also performed using sediment DNA extracts that were cleaned, post-extraction, using either the Powerclean DNA Clean-up kit (Mo Bio Laboratories, USA) or ethanol precipitation (Sambrook and Russell, 2001).
2.6 Collection and processing of field sediment samples
Cysts were collected during October 2004 on cruise CH1504 on the R/V Cape Hatteras during the ECOHAB-Gulf of Maine program (Anderson et al., 2005b). Samples were collected using a hydraulically damped corer (Craib, 1965) that provides undisturbed sediment samples. The top 3 cm of sediment were extruded from the core and split into two subsamples comprising the top 1 cm (0–1 cm) and the next 2 cm (1–3 cm). Samples were stored at 2°C onboard and processed according to (Anderson et al., 1982) upon return to the laboratory. Briefly, sediment samples were diluted with filtered seawater (FSW) and sonicated on ice for 1 min using a Branson Sonifier 250 at a constant output of 40W. The sample was then sequentially sieved through 80μm and 20μm meshes, and the 20–80 μm fraction was recovered from the 20-μm mesh using FSW (Anderson et al., 2003). This cleaned fraction was further diluted to a standard volume of 24mL with FSW and mixed thoroughly, then a 12\-mL subsample was collected onto a 25-mm diameter, 5-μm pore size Durapore membrane. The filtered sample was immediately stored at −80°C for later analysis by qPCR. The remainder of the resuspended cyst sample was used for microscopic enumeration of cysts using primulin staining. In total, 129 samples were analyzed by both qPCR and primulin staining/microscopy.
2.7 Primuline staining and cyst enumeration
A. fundyense cysts were counted microscopically according to standard methods for cyst identification and enumeration (Anderson et al., 2003) using primulin to stain the cysts (Yamaguchi et al., 1995). Briefly, the processed sediment from Section 2.3 above was preserved in 1% paraformaldehyde at 2–4°C for at least 30 min. The cysts were then collected by centrifugation, the overlying seawater was removed by aspiration, and the pelleted cysts were resuspended in 10 mL of cold methanol and stored at 2–4°C for at least 48 hr. The sample was centrifuged and aspirated as before, and resuspended in 9 mL of deionized water and 1 mL of primulin stain (1 mg/mL). After staining for 30 min the sample was centrifuged and aspirated to remove excess stain and resuspended in 5 mL of deionized water. A 1-mL subsample of each final, stained sample were counted in a Sedgwick-Rafter slide on a Zeiss Axioskop epifluorescence microscope with a chlorophyll filter set (band pass 450–490 nm excitation, long pass 520 nm emission). Stained cysts were evident from their yellow-green fluorescence.
2.8 Extraction and analysis of field-collected cyst samples
The frozen, processed sediment samples were removed from −80°C, and 360 μL of buffer ATL (Qiagen, Inc.) was added to each tube. DNA was extracted as described in Section 2.4 and eluted in 200 μL of buffer AE (Qiagen, Inc.). In the majority of cases some sediment material was visible in the eluted sample. Therefore, prior to analysis, the sample was centrifuged at 16,000 rpm for 30 min at 4 °C to pellet this material. The top 150 μL of supernatant were then decanted into a clean tube and stored at −20 °C until analysis. Real-time PCR was performed as described in Section 2.3. Each analysis plate contained 24 extracted sediment samples, in triplicate, along with a set of five plasmid standards ranging from 5.2 ×102 to 5.2 × 107 copies. Regression analysis was used to assess the relationship between the primulin-staining/microscopy and qPCR counts.
3. Results and Discussion
3.1 Development of plasmid standards and determination of ribosomal gene copy number
In the last five years, a number of laboratories have reported the development of quantitative real-time PCR (qPCR) assays for the determination of algal cell numbers. In general, these assays rely on the inclusion of a standard curve in each qPCR run, so that unknown cell densities can be calculated by comparison. Standards for cell quantification assays consist of either DNA extracts from known numbers of cells (usually cultured cells, e.g. Dyhrman et al., 2006) or a serial dilution of a purified plasmid containing the target gene (e.g. Galluzzi et al., 2004). Extracted cell standards have the advantage of being more like the actual samples, as they are subject to the same procedures such as filtration and DNA extraction, and presumably incorporate any variability associated with that processing. However, extracted cell standards require a stable and abundant source of the target organism – either “wild” or cultured. In the case of Alexandrium cysts, it is often difficult to obtain sufficient quantities of pure cysts for standards. Cysts can be made in the laboratory (e.g. Anderson et al., 1984; Kamikawa et al., 2007) but those preparations can be contaminated with vegetative cells. Even after sonication, cell fragments can remain and this can contaminate the cyst preparations with DNA from vegetative cells. Natural sediments do not have significant numbers of vegetative cells, but cyst densities are generally not very high. Large quantities of sediment have to be processed to obtain the >10,000 cysts needed for each standard curve, and there is so much sedimentary material left behind that it makes it difficult to count the samples in order to collect known numbers of cysts for standards. For these reasons, we chose to use a cloned plasmid standard curve for the cyst qPCR assay. The plasmid contains a fragment of the large subunit (LSU) ribosomal DNA gene containing the D1-D2 hypervariable regions that was PCR amplified from an A. fundyense isolate from the northeastern U.S. The cloned fragment was confirmed by sequencing to be of the North American/Group I ribotype. Once cloned and transformed into E. coli, this plasmid provides a stable and renewable source of our target gene.
To use a plasmid standard for cell quantification, the number of ribosomal gene copies in the genome of the target organism needs to be known, in order to accurately convert between copies per sample and cells per sample. This is complicated by the potential variation in ribosomal gene copy number between different strains or species of an organism. For example, in the Gulf of Maine there are different strains of both A. fundyense and A. tamarense. To mitigate the problem of copy number variation, we used natural Alexandrium cysts to determine ribosomal copy number, on the assumption that by “pooling” wild cysts we would average over the range of copy numbers present in natural samples. Quadruplicate analyses of manually isolated A. fundyense cysts yielded a value of 28,402±6152 LSU ribosomal gene copies per cyst.
This cyst rDNA copy number for A. fundyense is much smaller than the ~460,000 copies per cell determined for the vegetative haploid cells of A. catenella, a member of the same species complex (Penna et al., 2006). Similar high values (ca. 500,000 to 1,000,000 copies per cell) have also been found for vegetative cells of A. fundyense (D. Erdner, unpublished data; Brosnahan et al., 2010). However, there appears to be considerable variation within the genus Alexandrium, as vegetative cells of A. minutum have been shown to contain ~1100 rDNA gene copies per cell (Galluzzi et al., 2004). The high numbers of rDNA copies in A. fundyense and A. catenella may initially seem somewhat surprising, especially in light of the lower number reported for the congeneric A. minutum.
Comparative analyses, however, have demonstrated a positive relationship between rDNA copy number and genome size in eukaryotes, both in plants and animals (Prokopowich et al., 2003). A. minutum contains ~11 pg of DNA per cell (Galluzzi et al., 2004), whereas A. tamarense contains much more: ~100 pg DNA/cell (LaJeunesse et al., 2005). If we assume that A. tamarense, A. fundyense, and A. catenella (all members of the same species complex) have similar genome sizes, then we would expect them to have many more rDNA copies as compared to A. minutum. Prokopowich et al. (2003) examined a variety of plant species within genome sizes ranging between 0.12 and 54.7 pg DNA/cell, and the highest rDNA copy number they found was 26,048. A. tamarense has more DNA per cell than the largest plant genome analyzed, thus it would be expected to have more rDNA copies as well.
While vegetative cells have rDNA copy numbers that seem commensurate with their genome size, A. fundyense cysts have ca. 10-fold fewer rDNA copies than vegetative cells, despite the fact that they result from the fusion of two vegetative cells. Based on these results, it appears that Alexandrium may modulate its ribosomal DNA copy number throughout its life cycle. It has recently been demonstrated that the ribosomal RNA gene copy number of the haptophyte alga Emiliania huxleyi changes with physiological condition; copy number declines as the cultures become older (Nejstgaard et al., 2008). Thus it is not surprising that the ribosomal gene copy number in A. tamarense varies between life cycle stages, especially considering that resting cysts are thought to be physiologically inactive as compared to vegetative cells.
3.2 Choice of DNA extraction methods
Dinoflagellate resting cysts are highly resistant to environmental conditions, and they can remain viable in sediments for many years. The thick cyst wall also makes it more difficult to break cysts open to extract DNA, especially in a quantitative fashion. We tested two different DNA extraction kits, with and without mechanical breakage, to determine which method was most suitable for extracting DNA from Alexandrium cysts. The DNEasy Tissue kit and the silica-zirconium beads were chosen because they are very effective in extracting DNA from Alexandrium vegetative cells (D. Erdner, unpublished data). The QIAmp Stool DNA MiniKit was used because it includes reagents designed to remove PCR inhibitors from difficult DNA samples. Due to the limited amount of clean, cultured cysts available for testing, we were able to run only a single sample with each of the methods.
The lowest Ct values (highest number of target copies) were obtained when silica-zirconium beads were used in addition to the extraction kits (Fig. 1B). The Ct values of both kits with and without beads differed about 3-fold, which corresponds to an almost 10-fold difference in starting copy number. The DNEasy Tissue kit with beads produced the greatest apparent DNA yields as measured by spectrophotometry (Fig. 1A). However, this may merely reflect the presence of greater amounts of UV-absorbing substances in the Tissue kit extract, as the Stool kit includes reagents that remove inhibitory substances, many of which absorb UV light. These substances, if present, do not appear to inhibit the PCR, as both the Tissue and Stool kits performed similarly with regard to Ct. We opted to use the Tissue kit for DNA extractions because it requires fewer processing steps than the Stool kit. In addition, the entire lysate volume is carried through the whole procedure with the Tissue kit, whereas the Stool kit protocol uses only part of the initial lysate and includes centrifugation steps, both of which make it more difficult to quantitatively extract the DNA.
Fig. 1.
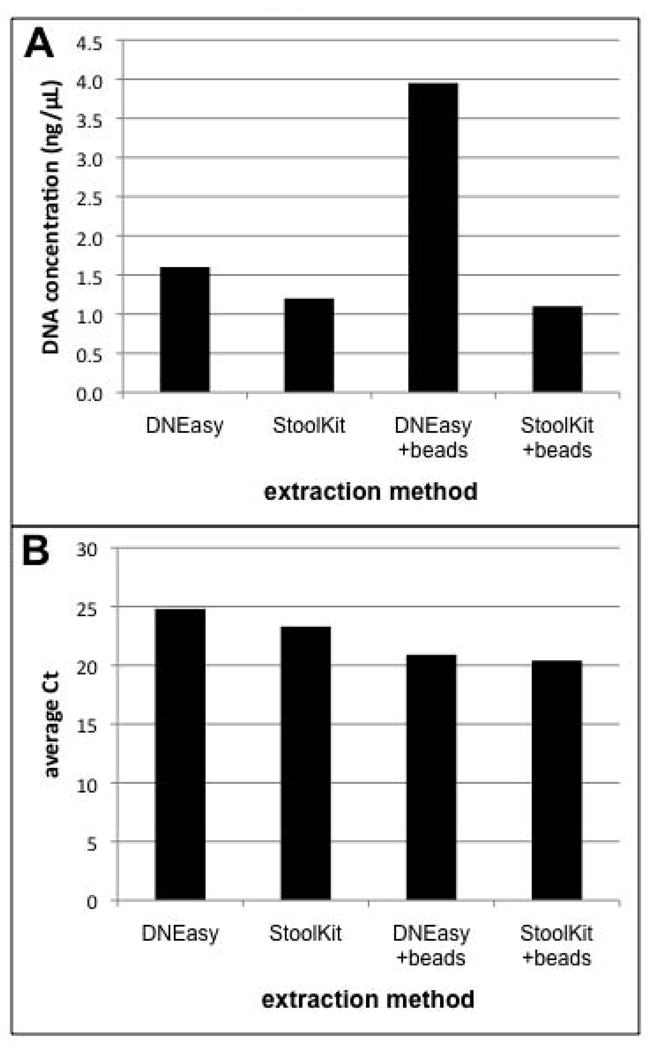
Comparison of different methods for cyst DNA extraction. Replicate samples of approximately 800 cysts were extracted using the DNEasy Tissue Kit or QIAmp Stool DNA kit, with or without mechanical breakage (bead-beating). Extraction efficiency was assessed based on (A) the total amount of DNA recovered and (B) the extent of amplification by real-time PCR (as a proxy for the number of ribosomal RNA gene copies recovered).
3.3 Inhibition of PCR by sediment extracts
Sediments are known to contain many substances that inhibit PCR amplification by DNA polymerases (Porteous and Armstrong, 1991). The presence of PCR inhibitors in our cyst DNA extracts was tested via the addition of a control DNA (pUC plasmid) to full strength and diluted cyst DNA extracts. In addition, we used a commercial DNA cleanup kit or ethanol precipitation to try to remove inhibitors from the extracts. Figure 2 shows the effect of cyst DNA extracts on the amplification of the pUC control target. Inhibition of PCR amplification is evident from the increased Ct values of extracts diluted 20-fold or less, even when cleaned with ethanol or the MoBio kit. To abolish the PCR inhibition, it was necessary to dilute the cyst DNA extracts at least 50-fold. This result is similar to the findings of Galluzzi et al. (2004) with Alexandrium vegetative cells and Kamikawa et al. (2007) with Alexandrium cysts; both report diluting their DNA extracts by 50-fold to control for PCR inhibition.
Fig. 2.

Inhibition of PCR by sediment DNA extracts. DNA was extracted from field-collected sediment samples, and added to a qPCR reaction containing pUC plasmid DNA as a target. Sample DNA was added at (A) full strength, 2-fold, 5-fold, or 10-fold dilution and (B) 20-fold, 50-fold or 100-fold dilution. In (B), “mb” indicates that the extract was cleaned using the MoBio cleanup kit, and “etoh” indicates that the extract was ethanol precipitated before the assay. PCR inhibition was indicated by an increase in reaction Ct relative to the pUC only control, which had no added sample DNA.
3.4 Real-time Quantitative PCR
The qPCR primers and amplification protocol used here are based on a method developed for Alexandrium vegetative cells of the North American/Group I ribotype (Dyhrman et al., 2006). Dyhrman et al. (2006) used extracted cells as standards; thus it was necessary to characterize the assay performance using the cloned plasmid standard. The method showed a linear response over 6 orders of magnitude (Fig. 3) with an efficiency of 86–100% over all of the runs performed. The lowest target copy number tested was 520 copies per reaction. As each reaction contains 0.05% of the total cyst DNA extract (2.5% of the total extract volume, diluted 50-fold), 520 copies per reaction correspond to about 36 cysts per extracted sample. For our standard sample size of 4 cc of sediment, the method can reliably quantify as few as 10 cysts/cc sediment. This is similar to the results of Kamikawa et al. (2007), who found a detection limit of 10 cysts for Japanese A. tamarense and A. catenella from natural samples. This detection limit is roughly equivalent to that of primulin staining and microscopy, which can detect as few as 3–10 cysts/cc, depending on the final resuspension volume. As more sediment material is processed, in order to lower the detection limit, the amount of residual sediment material becomes too dense for accurate cyst counting. The detection limit of the qPCR method suffers because of the need to dilute the samples 50-fold to abolish PCR inhibition by co-extracted substances. Nonetheless, 10 cysts/cc would be considered a low value for cyst densities, and as such is still a useful detection level for most cyst studies.
Fig. 3.

Representative qPCR standard curve. Standard curves were constructed from serial dilutions of a plasmid containing a cloned fragment of the large subunit ribosomal RNA gene of A. tamarense. Reaction efficiencies ranged from 86–100% over all runs. The lowest target copy number tested was 520 copies per reaction, which corresponds to a detection limit of 10 cysts/cc in a standard sample volume of 4cc.
3.5 Comparison of PCR and primulin staining methods for cyst enumeration
Overall, there is a significant correlation (p<0.001) between the cyst abundances determined by qPCR and those determined by primulin staining/microscopy (Fig. 4). The slope of the relationship is 0.75±0.11 which indicates that the counts obtained by primulin staining/microscopy tend to be generally higher than those obtained from qPCR. This is consistent with the results of Kamikawa et al. (2007) who also observed that qPCR cyst counts tended to be slightly lower than those from the primulin-staining method. Although their values (n=10) are presented in a table, regression analysis of the data yields a slope of 0.58±0.15 (p<0.05) and an r2 of 0.68. Kamikawa et al. (2007) attribute the underestimation by qPCR to the inclusion of excysted and/or empty cysts in the primulin counts; empty cysts have no DNA and would not be detected by qPCR. However, Anderson et al. (2005a) contend that empty cysts do not contribute significantly to primulin counts, as empty cysts are easily deformed during processing, thereby destroying the characteristic morphology which is one basis for identifying cysts during the counting procedure.
Fig. 4.
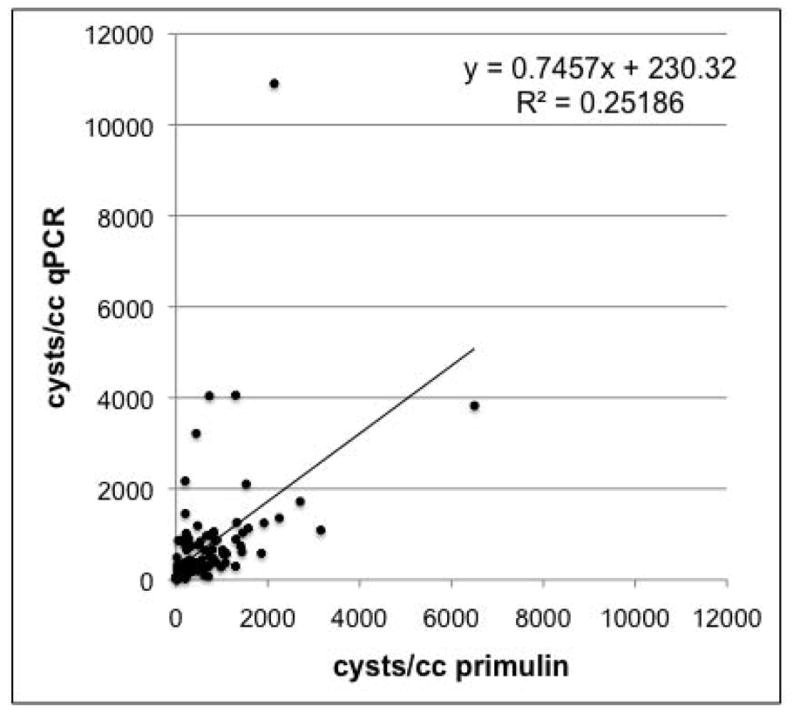
Comparison of PCR and microscope methods for cyst enumeration. A total of 129 field-collected sediment samples were processed, split, and analyzed in parallel using both the qPCR and primulin staining/microscopy methods. The solid line and equation are the best linear curve fit to the data. The cyst values calculated from the two methods are significantly correlated (p<0.001).
We also examined the correlation between qPCR and the primulin-staining method at the two different sample depths (the top 1 cm of each core, and the next 1–3 cm). There is a significant relationship (p<0.01) between qPCR and primulin counts at the two depths (Fig. 5), but at the 1–3 cm depth there is much more scatter in the data, and the underestimation of the qPCR counts is much more pronounced (slope = 0.46±0.15). The slope of the relationship for the top 1 cm is 0.82±0.16, indicating that the qPCR counts are still slightly lower than the estimates from primulin-staining. The differences observed between the two depths may be due to the condition of the cysts in the different layers. The top 1 cm of sediment contains cysts that were deposited more recently; in the Gulf of Maine it corresponds to roughly the last decade of cyst and sediment deposition (Keafer et al., 1992). Cysts in the lower 1–3 cm have been in the sediments much longer, and there may be a higher proportion of cysts where the nucleic acids have degraded over time, even if the cysts themselves are not empty.
Fig. 5.

Comparison of PCR and microscope methods for samples collected at different sediment depths. The samples shown in Figure 4 are compared separately based upon the sediment depth at which the sample was collected. (A) 70 samples were analyzed from the top 1cm of sediment and (B) 59 samples were derived from the 1–3cm depth. The solid lines and equations are the best linear curve fit to the data. The cyst values calculated from the two methods are significantly correlated (p<0.005).
We examined the ratio between the cyst densities calculated by qPCR and primulin-staining, to better understand the variation between the two methods. The qPCR and primulin values are within a two-fold range of each other for a majority (56.6%) of the samples analyzed. An additional 31% of the samples show less than a five-fold difference between the two methods; a little more than half of these have higher primulin values relative to qPCR. Another 9.3% of samples are different by 5 to 10-fold, meaning that the estimates for virtually all of the samples (96.9%) are within an order of magnitude of each other. There are several factors that could contribute to the observed differences between the two methods. As was mentioned previously, the two methods quantify different targets – DNA copies for qPCR and cyst capsules for primulin-staining. Degraded cysts could be counted with the primulin-staining method but not by qPCR, leading to overestimation by primulin-staining. There is also some variability inherent in the sediment processing, which is compounded by variability in the sample preparation and counting methods. The samples for primulin-staining and qPCR are replicate subsamples of the same single core sample. Because sample processing and counting is so time-consuming, replicate cores are generally not analyzed. Thus, we do not have a good idea of the core-to-core variability within a station, or even within subsamples from a single station counted by one method. Because of this, we are unable to determine whether the numbers obtained at individual stations are significantly different from one another, although we can still assess the overall performance of the qPCR method, as we have done here.
The number and type of processing steps used for the two methods is also different, which could contribute to the observed variability. After the sediment is sonicated and sieved, half of the resuspended cyst preparation is removed and collected on a filter for qPCR, and the remainder is preserved in the seawater for primulin staining. The primulin staining protocol requires 3 sequential centrifugation steps, after which the cyst pellet is resuspended in different solutions; each centrifugation step could lead to the loss of cysts. For qPCR, the cyst lysate is made in the original sample tube and then transferred to the DNA purification column. The primary loss comes from DNA extraction efficiency - cyst lysis and DNA purification. We tried to mitigate any effect of extraction efficiency by using the same DNA purification methods for our copy number calibration and unknown samples. Thus, our estimate of ribosomal copies per cyst reflects the number of extracted ribosomal copies per cyst, and as such is an underestimate of the total number of ribosomal copies per cyst. Despite being subjected to the same lysis and DNA purification methods, however, the natural samples do contain sediment matrix, which could reduce the extraction efficiency of the natural samples relative to gradient-purified cysts. Percy et al. (this volume) show that this is in fact the case; cysts that are first purified using sodium polytungstate gradients show lower Ct values than cysts extracted with the accompanying sediment.
3.6 Choice of PCR or primulin staining methods for cyst studies
The qPCR method described here represents a promising alternative to primulin-staining and microscopy for the enumeration of cysts from marine sediments. The two methods have often complementary strengths and weaknesses. Both methods require the same sediment collection and processing methods, including core collection, sonication, and sieving. The qPCR method has a higher throughput for sample analysis, enabling the extraction and assay of 24 samples in 6–7 hours. About 8–10 samples can be counted by primulin staining in the same time. Both methods require some operator experience – taxonomic expertise in the case of primulin-staining and careful pipetting and sterile technique in the case of qPCR. The qPCR method uses fewer person-hours per sample compared to primulin-staining, but the reagent costs are significantly higher. The qPCR method might be more desirable for large-scale cyst mapping, where large numbers of samples are generated and a higher sample analysis rate is necessary. In many cases, however, the decision may be most influenced by the practical issue of the relative cost and availability of person-hours and materials, as qPCR requires much more of the latter and primulin-staining relies heavily on the former.
5. Conclusions
The quantitative real-time PCR (qPCR) method described here represents a promising alternative to primulin-staining and microscopy for the enumeration of Alexandrium cysts of the toxic North American/Group I ribotype. Analysis of manually isolated Alexandrium cysts from the northeastern U.S. shows that cysts contain ~28,000 ribosomal DNA copies per cell, which is much less than the number of ribosomal copies found in vegetative cells. A comparison of cyst densities determined by qPCR and primulin-staining shows a significant correlation between the two methods, although the qPCR counts tend to be lower than the primulin values. This underestimation is less pronounced in those samples collected from the top 1 cm of sediment, and more pronounced in those derived from the next 1–3 cm of the core. While the qPCR and primulin-staining methods generate similar data, the ultimate choice of cyst counting technique will be influenced by the different relative costs of labor and materials between the two methods.
Fig. 6.

Comparison of the cyst counts obtained by the qPCR and primulin-staining/microscopy methods. The extent of variation between the methods was assessed by calculating the ratio of cysts/cc determined by primulin-staining to the cysts/cc calculated by the qPCR method. For example, a ratio of 0.5–2.0 corresponds to ≤ 2-fold difference in values between the two methods. The data are shown as the percent of total samples within the given ratio range.
Acknowledgments
We thank the scientists and crew of the R/V Cape Hatteras for their assistance with sample collection, particularly C. Pilskaln, J. Brown, K. Norton, and J. Lawrence and colleagues. We also thank E. Harrison and L. McCauley for processing and counting numerous cyst samples. This work was supported by NSF Grant OCE-0402707 (DLE, DMA), NOAA ECOHAB Grant NA04NOS4780274 (DMA, BAK), NSF/NIEHS Centers for Oceans and Human Health NSF Grant OCE-0430724 and NIEHS Grant 1P50-ES01274201 (DMA, DLE) and EU SEED grant GOCE-CT-2005-003875 (JL, LP).
Footnotes
Only A. tamarense has been documented in W. Europe, whereas both A. tamarense and A. fundyense occur in the northeastern U.S. (Anderson et al. 1994). We consider A. tamarense and A. fundyense to be varieties of the same species (Scholin et al. 1995) as antibody probes cannot distinguish between them, and they share the same ribotype. Only detailed analysis of thecal plates on individual cells can provide this resolution. For the purpose of this study, the name A. tamarense is used to refer to both forms.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson DM. Toxic algal blooms and red tides: a global perspective. In: Okaichi T, Anderson DM, Nemoto T, editors. Red tides: biology, environmental science, and toxicology. Elsevier Science Publishing Co., Inc; New York: 1989. pp. 11–16. [Google Scholar]
- Anderson DM, Aubrey DG, Tyler MA, Coats DW. Vertical and horizontal distributions of dinoflagellate cysts in sediments. Limnology and Oceanography. 1982;27 (4):757–765. [Google Scholar]
- Anderson DM, Fukuyo Y, Matsuoka K. Cyst methodologies. In: Hallegraeff GM, Anderson DM, Cembella AD, editors. Manual on Harmful Marine Microalgae, Monographs on Oceanographic Methodology. UNESCO; 2003. [Google Scholar]
- Anderson DM, Glibert PM, Burkholder JM. Harmful algal blooms and eutrophication: Nutrient sources, composition and consequences. Estuaries. 2002;25 (4b):704–726. [Google Scholar]
- Anderson DM, Kulis DM, Binder BJ. Sexuality and cyst formation in the dinoflagellate Gonyaulax tamarensis: Cyst yield in batch cultures. Journal of Phycology. 1984;20 (3):418–425. [Google Scholar]
- Anderson DM, Stock CA, Keafer BA, Bronzino Nelson A, Thompson B, McGillicuddy JDJ, Keller MD, Matrai PA, Martin JL. Alexandrium fundyense cyst dynamics in the Gulf of Maine. Deep Sea Research II. 2005a;52 (19–21):2522–2542. [Google Scholar]
- Anderson DM, Townsend DW, McGillicuddy DJ, Jr, Turner JT. The ecology and oceanography of toxic Alexandrium fundyense Blooms in the Gulf of Maine. Deep-Sea Research Part II: Topical Studies in Oceanography. 2005b;52 (19–21):2365–2368. [Google Scholar]
- Balech E. The genus Alexandrium Halim (Dinoflagellata) Sherkin Island Marine Station; Cork, Ireland: 1995. [Google Scholar]
- Bolch CJS. The use of sodium polytungstate for the separation and concentration of living dinoflagellate cysts from marine sediments. Phycologia. 1997;36 (6):472–478. [Google Scholar]
- Brosnahan M, Kulis DM, Solow AR, Erdner D, Percy L, Lewis J, Anderson DM. Outbreeding lethality between toxic, Group I and nontoxic, Group III Alexandrium tamarense spp. isolates: predominance of heterotypic encystment and implications for mating interactions and biogeography. Deep-Sea Resarch II. 2010 doi: 10.1016/j.dsr2.2009.09.005. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craib JS. A sampler for taking short undisturbed cores. Journal du Conseil International pour l’Exploration de la Mer. 1965;30 (1):34–39. [Google Scholar]
- Dale B. Dinoflagellate resting cysts: benthic plankton. In: Fryxell GA, editor. Survival Strategies of the Algae. Cambridge University Press; Cambridge: 1983. pp. 69–136. [Google Scholar]
- Dyhrman ST, Erdner D, Du JL, Galac M, Anderson DM. Molecular quantification of toxic Alexandrium fundyense in the Gulf of Maine using real-time PCR. Harmful Algae. 2006;5 (3):242–250. doi: 10.2307/1543269. [DOI] [PubMed] [Google Scholar]
- Franks PJS, Anderson DM. Alongshore transport of a toxic phytoplankton bloom in a buoyancy current: Alexandrium tamarense in the Gulf of Maine. Marine Biology. 1992;116 (1):153–164. [Google Scholar]
- Galluzzi L, Penna A, Bertozzini E, Vila M, Garcés E, Magnani M. Development of a Real-Time PCR Assay for Rapid Detection and Quantification of Alexandrium minutum (a Dinoflagellate) Applied and Environmental Microbiology. 2004;70 (2):1199–1206. doi: 10.1128/AEM.70.2.1199-1206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallegraeff GM. A review of harmful algal blooms and their apparent global increase. Phycologia. 1993;32:79–99. [Google Scholar]
- He R, McGillicuddy DJ, Jr, Anderson DM, Keafer BA. Historic 2005 toxic bloom of Alexandrium fundyense in the western Gulf of Maine: 2. Coupled biophysical numerical modeling. Journal of Geophysical Research - Oceans. 2008;113:C07040. doi: 10.1029/2007JC004602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi-Tanabe S, Nagai S, Sako Y. Species-specific detection and quantification of the toxic dinoflagellate Alexandrium tamarense by Taq Man PCR using 5′-3′ exonuclease activity. Marine Biotechnology. 2004;6:S30–S34. doi: 10.1007/s10126-004-4128-4. [DOI] [PubMed] [Google Scholar]
- Kamikawa R, Nagai S, Hosoi-Tanabe S, Itakura S, Yamaguchi M, Uchida Y, Baba T, Sako Y. Application of real-time PCR assay for detection and quantification of Alexandrium tamarense and Alexandrium catenella cysts from marine sediments. Harmful Algae. 2007;6 (3):413–420. [Google Scholar]
- Keafer BA, Buesseler KO, Anderson DM. Burial of living dinoflagellate cysts in estuarine and nearshore sediments. Marine Micropaleontology. 1992;20 (2):147–161. [Google Scholar]
- LaJeunesse TC, Lambert G, Andersen RA, Coffroth MA, Galbraith DW. Symbiodinium (Pyrrhophyta) genome sizes (DNA content) are smallest among dinoflagellates. Journal of Phycology. 2005;41 (4):880–886. [Google Scholar]
- Lilly EL, Halanych KM, Anderson DM. Species boundaries and global biogeography of the Alexandrium tamarense complex (Dinophyceae) Journal of Phycology. 2007;43 (6):1329–1338. [Google Scholar]
- Lilly EL, Kulis DM, Gentien P, Anderson DM. Paralytic shellfish poisoning toxins in France linked to a human-introduced strain of Alexandrium catenella from the Western Pacific: evidence from DNA and toxin analysis. Journal of Plankton Research. 2002;24 (5):443–452. [Google Scholar]
- McGillicuddy JDJ, Anderson DM, Lynch DR, Townsend DW. Mechanisms regulating the large-scale seasonal fluctuations in Alexandrium fundyense populations in the Gulf of Maine: results froma physical-biological model. Deep Sea Research II. 2005;52:2698–2714. [Google Scholar]
- Moorthi SD, Countway PD, Staufeer BA, Caron DA. Use of quantitative real-time PCR to investigate the dynamics of the red tide dinoflagellate Lingulodinium polyedrum. Microbial Ecology. 2006;52:136–150. doi: 10.1007/s00248-006-9030-3. [DOI] [PubMed] [Google Scholar]
- Nejstgaard JC, Frischer ME, Simonelli P, Troedsson C, Brakel M, Adiyaman F, Sazhin AF, Artigas LF. Quantitative PCR to estimate copepod feeding. Marine Biology. 2008;153:565–577. [Google Scholar]
- Galluzzi L, Bertozzini E, Penna A, Garcés E, Magnani M. Analysis of rRNA gene content in the Mediterranean dinoflagellate Alexandrium catenella and A. taylori: implications for the quantitative real-time PCR-based monitoring methods. Journal of Applied Physiology. 2009 (in press) [Google Scholar]
- Popels LC, Cary SC, Hutchins DA, Forbes R, Pustizzi F, Gobler CJ, Coyne KJ. The use of quantitative polymerase chain reaction for the detection and enumeration of the harmful alga Aureococcus anophagefferens in environmental samples along the United States East Coast. Limnology and Oceanography: Methods. 2003;1:92–102. [Google Scholar]
- Porteous LA, Armstrong JL. Recovery of bulk DNA from soil by a rapid, small-scale extraction method. Current Microbiology. 1991;22 (6):345–348. [Google Scholar]
- Prokopowich CD, Gregory TR, Crease TJ. The correlation between rDNA copy number and genome size in eukaryotes. Genome. 2003;46:48–50. doi: 10.1139/g02-103. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell D. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Press; Cold Spring Harbor: 2001. [Google Scholar]
- Scholin CA, Herzog M, Sogin M, Anderson DM. Identification of group- and strain-specific genetic markers for globally distributed Alexandrium (Dinophyceae). 2. Sequence analysis of a fragment of the LSU rRNA gene. Journal of Phycology. 1994;30 (6):999–1011. [Google Scholar]
- Stock CA, McGillicuddy DJ, Jr, Solow AR, Anderson DM. Evaluating hypotheses for the initiation and development of Alexandrium fundyense blooms in the western Gulf of Maine using a coupled physical-biological model. Deep-Sea Research II. 2005;52:2715–2744. [Google Scholar]
- Turpin DH, Dobell PER, Taylor FJR. Sexuality and cyst formation in Pacific strains of the toxic dinoflagellate Gonyaulax tamarensis. Journal of Phycology. 1978;14 (2):235–238. [Google Scholar]
- Vila M, Garcés E, Maso M, Camp J. Is the distribution of the toxic dinoflagellate Alexandrum catenella expanding along the NW Mediterranean coast? Marine Ecology Progress Series. 2001;222:73–83. [Google Scholar]
- Wang W, Chen KS, Xu C. DNA quantification using EvaGreen and a real-time PCR instrument. Analytical Biochemistry. 2006;356 (2):303–305. doi: 10.1016/j.ab.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Itakura S, Imai I, Ishida Y. A rapid and precise technique for enumeration of resting cysts of Alexandrium spp. (Dinophyceae) in natural sediments. Phycologia. 1995;34:207–214. [Google Scholar]