

Abstract
Phagocytic clearance of apoptotic cells by macrophages is an essential part in the resolution of inflammation. It coincides with activation of repair mechanisms, including accumulation of extracellular matrix. A possible link between clearance of apoptotic debris and accumulation of extracellular matrix has not been investigated. Production of collagen was measured in primary fibroblasts co-cultured with macrophages. Ingestion of apoptotic cells by monocyte-derived macrophages led to upregulation of collagen. Direct contact between macrophages and fibroblasts was not required for collagen upregulation. Macrophages produced TGF- β following ingestion of apoptotic cells, but the levels of this cytokine were lower than those required for a significant upregulation of collagen. Simultaneously, the levels of TGF-β-induced (TGFBI), or keratoepithelin/BIGH3, mRNA and protein were increased. In contrast, primary alveolar macrophages stimulated collagen production without exposure to apoptotic cells; there was no further increase in the levels of TGFBI, mRNA or protein, or collagen after ingestion of apoptotic cells. Stimulation of fibroblasts with TGFBI downregulated MMP14 levels, decreased DNA binding by p53, increased DNA binding by PU.1, and upregulated collagen protein but not mRNA levels. Overexpression of MMP14 or p53, or siRNA-mediated inhibition of PU.1 led to an increase in MMP14 and a decline in collagen levels, whereas siRNA-mediated inhibition of MMP14 led to elevation of collagen levels. In conclusion, monocyte-derived but not alveolar macrophages produce TGFBI following ingestion of apoptotic cells, leading to downregulation of MMP14 levels in fibroblasts through a mechanism involving p53 and PU.1, and to subsequent accumulation of collagen.
Keywords: cytokines, fibroblasts, matrix metalloproteinases, macrophages, apoptosis, collagen, fibrosis
INTRODUCTION
Macrophages are important functional contributors to normal wound healing/repair process; they participate in the clearance of apoptotic debris that accumulates as a result of primary injury and subsequent inflammatory response. Clearance of apoptotic debris is a major non-phlogistic function of macrophages; ingestion of apoptotic debris causes dramatic phenotypic changes in these cells. Of particular importance, the production of TGF-β by macrophages is accelerated following phagocytosis of apoptotic debris, suggesting potential anti-inflammatory (1–4) and pro-fibrotic (see 5 for a review) effects of this phenomenon. Based on these observations, it is reasonable to hypothesize that tissue fibrosis may be a consequence of disturbed clearance mechanisms of apoptotic debris by macrophages. Fibrosis, often viewed as an exaggerated repair process, is a major debilitating factor and a cause of death in patients with various diseases (5). Although macrophages may not be absolutely necessary for wound healing in macrophageless (PU.1 null) mice (apoptotic debris in the wounds is cleared by “stand-in” fibroblast phagocytes in such animals), the wounds heal with significantly less inflammation, lower levels of TGF-β, and less fibrosis in the absence of macrophages (6). Macrophages appear to be intimately involved in the regulation of tissue fibrosis in the lung (7–9), kidney (10, 11), and liver (12); the apoptotic mechanisms are often involved in the mechanism of tissue fibrosis in the lung (13), kidney (14), and liver (15).
Little is known about pro- and anti-fibrotic regulation by macrophages in relation to phagocytotic clearance of apoptotic debris. It is unclear whether TGF-β production following the uptake of apoptotic debris by macrophages is sufficient to drive tissue fibrosis, or more complex mechanisms, such as so-called alternative macrophage activation (8,16) or yet unknown novel mechanisms are necessary. Of note, the levels of TGF-β production by macrophages following phagocytosis of apoptotic debris are relatively low (within 100 pg/ml, see ref. 4), and thus may be insufficient for direct activation of collagen production in fibroblasts (17, 18).
In this study, we sought to investigate the effects of phagocytosis of apoptotic or necrotic cells by macrophages on the rate of collagen production by primary fibroblasts in cell culture, and to begin addressing the molecular mechanisms of such effects. We report that phagocytosis of apoptotic but not necrotic debris by monocyte-derived but not alveolar macrophages stimulates collagen production in co-cultures with primary fibroblasts. This regulation is mediated by transforming growth factor beta induced (TGFBI) protein also called keratoepithelin, or BIGH3. We report that the production of TGFBI by macrophages leads to upregulation of collagen protein but not mRNA in primary fibroblasts. This effect of TGFBI is mediated by a decrease in the levels of MMP14 mRNA and protein in a p53-dependent and PU.1-dependent fashion. Thus, we describe a novel mechanism by which macrophages that ingest apoptotic cells may regulate normal wound healing and, if exaggerated, possibly fibrosis. This mechanism may be a novel target for future therapies aimed at facilitating repair or preventing and treating fibrosis.
MATERIALS AND METHODS
Macrophage and fibroblast cell culture
Macrophages were derived from human peripheral blood monocytes (monocyte-derived macrophages [MDM]), bronchoalveolar lavage fluids (alveolar macrophages [AM]), or a human monocytic cell line THP-1 (THP-1-derived macrophages [TDM]). To produce MDM, peripheral blood mononuclear cells (PBMC) were isolated from freshly drawn peripheral blood by density gradient centrifugation using Ficoll-Paque (Amersham Biosciences, Piscataway, NJ) and resuspended in RPMI 1640 medium supplemented with 20% human serum, 10 mM HEPES, pH 7.4, 2 mM L-glutamine, 1 mM sodium pyruvate, 0.1 mM non-essential amino acid mix, 5 X 10−5 M 2-ME, and 5 μg/ml gentamicin sulfate. The cells were cultured overnight in 6-well plates (Becton Dickinson, Franklin Lakes, NJ), in a 5% CO2 humidified air atmosphere at 37°C. The non-adherent cells were removed and the adherent cells were cultured for an additional five days, and were termed monocyte-derived macrophages. Alveolar macrophages were obtained from broncho-alveolar lavage fluids derived from two adult healthy individuals or from eight patients with interstitial lung disease associated with systemic sclerosis (19–21). The protocols for drawing blood and for broncho-alveolar lavage procedures were approved by the University of Maryland Institutional Review Board. Human monocytic line THP-1 was obtained from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in the same medium, except that 10% FBS was used instead of human serum. The THP-1-derived macrophages were obtained by stimulating these cells with 200 nM/ml phorbol-12-myristate-13-acetate (PMA) purchased from Cell Signaling (Danvers, MA).
Four primary pulmonary fibroblast cultures (PF1-PF4) derived from different adult healthy donors were purchased from Cambrex (Walkersville, MD) and each tested separately in independent experiments. Fibroblast cultures were maintained in T75 culture flasks as previously described (22–24). In all experiments fibroblast cell lines were tested in passages three to seven.
Apoptotic and necrotic cellular debris
Jurkat cells (human T cell line) were purchased from ATCC and maintained according to the supplier’s recommendations. Apoptosis of Jurkat cells was induced by incubation with 0.5 μg/ml staurosporine (Sigma Aldrich, St. Louis, MO) at 37°C for 6–8 h or, alternatively, by exposure to UV irradiation at 90 mJ/cm2 followed by culture for 3–4 h. The percentage of apoptotic cells was quantified by flow-cytometry analysis by using Annexin V and propidium iodide staining (Sigma Aldrich, St Louis, MO) and was within 70–80%. Necrotic debris was generated by three cycles of freezing-thawing involving freezing the cells in liquid nitrogen and then thawing them at 37°C.
Phagocytosis assays
Jurkat cells were labeled with the dye TAMRA (5− [and 6−]-carboxy tetramethyl rhodamine succinimidyl ester) (Molecular Probes, Eugene, OR). TAMRA-labeled cells were added to cultured macrophages at a ratio of 5:1 and incubated for 2 h at 37°C. For confocal microscopy experiments, vital staining of macrophages was performed with calcein AM (Molecular Probes) immediately before assays. At the end of the incubation period, the monolayer was vigorously washed with ice-cold phosphate-buffered saline to remove unbound and bound but unengulfed apoptotic cells. The complete removal of non-ingested apoptotic debris after washing was confirmed by co-localization of TAMRA-stained apoptotic material and calcein AM-stained macrophages via confocal microscopy at ×400 magnification using Zeiss LSM 510 laser scanning confocal microscope. The phagocytosis was assessed by fluorescent or confocal microscopy. The percentage of macrophages that ingested TAMRA-labeled apoptotic cells was determined as the percent phagocytosis (number of macrophages, per 100, that ingested at least one apoptotic particle) in three different wells. Also, the conditioned supernatant media from these cell cultures were collected and used for stimulation of fibroblast cultures as described below.
Macrophage-fibroblast co-cultures and conditioned medium experiments
Fibroblasts were seeded in 6 well-tissue culture plates (Becton Dickinson, Franklin Lakes, NJ) at a sub-confluent density of 150,000 cells/well and grown for 24 h in the same conditions as described above, except that low-serum 1640 RPMI medium supplemented with 50μM ascorbic acid, and 50 μM BAPN (β-aminopropionitrile) was used. Then, macrophages were added to each well at a concentration of 250,000 cells/well for additional 24 h, followed by adding 1.5 mln apoptotic Jurkat cells for 2 h. After 2 h, the adherent cells were washed to remove non-ingested apoptotic cells and fresh medium was added for additional 24 h before analyzing these cultures for collagen or cytokine production. In separate experiments, fibroblasts were stimulated with the conditioned media collected from macrophage cultures following phagocytosis assays, without or with neutralizing anti-TGF-β antibody 1D11 or isotype control Ig (both from R&D Systems, Minneapolis, MN). Fibroblast proliferation was tested as described in (24). Briefly, after 5 to 7 days of co-culture, macrophages were removed and fibroblast proliferation tested using CellTiter AQueous 96 Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI) per manufacturer’s recommendations.
Transwell assay
To determine whether cell-to-cell contacts are necessary to mediate the effects of macrophages on fibroblasts, or whether soluble factors are sufficient for the interactions between these two cell types, Transwell assays were performed. In these assays, macrophages were separated from fibroblast monolayers by a membrane with 3.0 μm pore size in the 6 well-Transwell plates (Corning Costar Corp., Cambridge, MA). Primary lung fibroblasts were seeded in the lower chamber whereas macrophages that engulfed apoptotic cells were placed in the upper chamber, using the same cell culture medium as described above. These co-cultures were incubated for 24 hours before analyzing the levels of collagen or cytokines.
Western blotting
Preparation of cell lysates, immunoprecipitation of BIGH3 protein, normalization of protein concentration in the samples with BioRad assays, electrophoretic separation and Western blotting were performed as previously described (22). Goat antibodies for BIGH3 were purchased from R&D Systems. Goat antibodies for MMP14 were purchased from Santa-Cruz Biotechnology (Santa Cruz, CA). Western blotting assays for collagen were performed using rabbit affinity purified anti-collagen type I antibody (Rockland, Gilbertsville, PA).
Nucleofection of primary fibroblast cultures
Nucleofection with collagen promoter-chloramphenicol acetyltransferase (CAT) reporter constructs (25), MMP14− or p53-encoding constructs (under control of CMV promoter, OriGene Technologies, Rockville, MD), siRNA directed against collagen α2(I) or MMP14 or PU.1, or non-targeting control siRNA (all from Santa-Cruz Biotechnology, Santa Cruz, CA) was performed using Basic Nucleofector kit reagents from Amaxa (Gaithersburg, MD), following manufacturer’s recommendations. Transfected fibroblasts were cultured for 48 h before treatment with 300 ng/ml TGFBI (R&D Systems, Minneapolis, MN). The efficiency of target depletion was assessed by measuring the levels of corresponding mRNA by Q-PCR.
Collagen production assays
Production of collagen was measured in cell cultures utilizing the metabolic labeling of collagen with 14C-proline as described in (25, 26). Briefly, fibroblast monolayers were pulsed with L-[U-14C]-proline (Amersham Biosciences, Piscataway, NJ) at 1μCi/ml for the final 12 hours of incubation. Purified bacterial collagenase type III was purchased from Sigma Aldrich (St. Louis, MO). Fibroblasts were then ruptured by repeated freeze-thawing, and part of each sample digested with collagenase type III. The samples were pelletted with 20% TCA containing 0.1% L-proline, and then the pellets were resuspended and washed twice with 5% TCA and 95% ice-cold ethanol. The samples were assayed in a liquid scintillation counter in order to determine the amount of collagenase-digestible and non-digestible 14C-labeled protein. Alternatively, collagen protein levels in cell culture supernatants were measured in Western blotting assays as described above.
The activity of the collagen α2(I) promoter was measured in primary fibroblasts transfected with collagen promoter-chloramphenicol acetyltransferase (CAT) reporter constructs as described (25).
ELISA
ELISA kits for TGF-β1, IL-4, and IL-13, were purchased from R&D Systems (Minneapolis, MN) and assays performed following the manufacturer’s recommendations. Fibroblast culture supernatants and whole cell lysates were activated by acidification prior to the assay to quantify the levels of IL-4, IL-13, and total (active and latent) TGF-β1. All samples were assessed in duplicates. Low-serum cell culture medium containing 0.5% dialyzed FBS had no detectable TGF-β1, IL-4, or IL-13 and was used as a negative control in these assays.
Profiling of gene expression with DNA arrays
Expression of 367 genes for cytokines and cytokine receptors in macrophages was profiled with cDNA macroarrays (SuperArray, Frederick, MD) at 0, 2, 6, 12, 24 h of exposure to apoptotic debris. Developed membranes were scanned and hybridization intensities for each spot were measured using Image Quant software (Molecular Dynamics, Sunnyvale, CA) and background subtracted. Numeric spot density data were exported into a spreadsheet software for data analyses. Results were confirmed by real-time PCR assays for selected genes as indicated in the Results section.
Analyses of DNA binding by transcription factors
Nuclear extracts from TGFBI-activated and control fibroblast cultures were prepared using nuclear extraction kit from Active Motif (Carlsbad, CA) and adjusted for total protein content using Bio-Rad assays. DNA binding by 345 different transcription factors was evaluated using protein/DNA TranSignal™ system (Panomics, Redwood City, CA), following the manufacturer’s recommendations. To validate selected results of the protein-DNA array experiments, electromobility shift assay kit (Active Motiff), including p53-specific consensus sequence probe and corresponding mutant probe, was used as described (23).
Real-time PCR quantification of mRNA levels
Total RNA purification, reverse transcription, and real-time PCR were performed using LightCycler (Roche, Indianapolis, IN), as previously described (22). Quantification of internal control 18S ribosomal RNA was performed as reported previously (23). The PCR reaction mixture included the recommended components of the FastStart DNA Master Hybridization Probes Hot Start Reaction Mix (Roche, Indianapolis, IN).The fold difference in gene expression relative to 18S ribosomal RNA between treated and untreated cultures was calculated using the 2−ΔΔCt method (26). The primers and the hybridization probes for collagen α2(I) mRNA were designed and prepared by TIB Molbiol (Adelphia, NJ). The primers for collagen α2(I) mRNA were: forward, 5′ – GAT GGT GAA GAT GGT CCC ACA GG – 3′ and reverse, 5′ – GGT CGT CCG GGT TTT CCA GGG T – 3′. The hybridization probes were labeled with fluorescein at the 3′-terminus (3FL) of one probe and with LightCycler Red at the 5′-terminus (5LC) of the other probe. The probes were 3FL 5′ – TTC CAA GGA CCT GCT GGT GAG CCT – 3′ and 5LC 5′ – TGA ACC TGG TCA AAC TGG TCC TGC AG – 3′. TGFBI-specific primers and PU.1-specific primers were designed and tested for specificity by SuperArray (Frederick, MD), and their specificity has been additionally confirmed in our preliminary experiments. Real-time PCR assays (RT2 Profiler™, SuperArray, Frederick, MD) were utilized to measure expression of 84 genes related to extracellular matrix in fibroblasts, following the manufacturer’s recommendations.
RESULTS
Collagen production but not proliferation of fibroblasts is upregulated in co-cultures with macrophages following ingestion of apoptotic cells
TAMRA-labeled apoptotic cells were co-cultured with either THP-1 derived macrophages (TDM), monocyte-derived macrophages (MDM), or alveolar macrophages (AM) for 2 h. The subsequent washing completely removed the non-ingested apoptotic material (Figure 1). On average, 31 ± 6% of macrophages have engulfed at least one apoptotic particle. There was no significant difference in the percent phagocytosis between the three types of macrophages (p > 0.05, one-way ANOVA). Since fibroblast proliferation and collagen turnover jointly define fibrosis, we tested whether ingestion of apoptotic debris affects these two processes in the macrophage-fibroblast co-cultures. Phagocytosis of TDM, MDM, or AM did not affect proliferation rates in the macrophages-fibroblast co-cultures (p > 0.05, two-tailed Student’s t-test comparing co-cultures of primary pulmonary fibroblasts with macrophages that engulfed or did not engulf apoptotic cells).
Figure 1.

Confocal microscopy of calcein AM-stained THP-1 macrophages (green) after 2 h co-culture with TAMRA-labeled apoptotic cells (red) followed by washing. Four serial sections of a macrophage are shown in A1–A4, top to bottom; and two sections of a different macrophage are shown in B1–B2, top to bottom. Notice that ingested apoptotic material remains inside macrophages whereas non-ingested debris is removed by washing.
The initial experiments were performed with TDM. The results suggested that these non-activated macrophages or those exposed to necrotic Jurkat cells failed to affect collagen production in co-cultures with pulmonary fibroblasts (Figure 2). However, ingestion of the apoptotic cells by TDM caused a significant increase in collagen production in co-cultures, as judged by 14C-proline incorporation and Western blotting assays (Figure 2). These experiments were repeated on thirteen independent occasions, in duplicates or triplicates, using 14C-proline incorporation, and on eight independent occasions using Western blotting for collagen in primary fibroblast cultures from four different unrelated donors, with consistent results. These observations suggested that ingestion of apoptotic debris by macrophages may have a profibrotic effect. Further experiments included monocyte-derived and alveolar macrophages.
Figure 2.

Regulation of collagen levels in macrophage-fibroblast co-cultures. Primary fibroblasts (Fib) were cultured with or without THP-1-derived macrophages (TDM). The macrophages were or were not exposed to apoptotic (Apopt) or necrotic (Necr) Jurkat cells. Panel A shows fold change in total (closed bars) or collagenase-sensitive (open bars) levels of metabolic incorporation of 14C-proline (mean CPM ± SD of quadruplicate cultures). Significant increases in 14C-proline incorporation (p < 0.05) are indicated with asterisks. Panel B shows representative Western blotting results for collagen type I in these co-cultures.
Co-culturing primary fibroblasts with MDM did not significantly influence collagen production in fibroblasts when compared with fibroblasts cultured alone (p > 0.05, Figure 3). Co-culturing MDM that have ingested apoptotic cells with fibroblasts caused a significant increase in collagen production (Figure 3). These results were consistently observed in two independent experiments using primary fibroblast cultures from four different unrelated donors. A different pattern of modulation of collagen production was observed when primary fibroblasts were co-cultured with AM from two healthy volunteers or eight patients with interstitial lung disease. Collagen production was increased when primary fibroblasts were co-cultured with non-stimulated AM (Figure 3). There was no further increase in collagen production in fibroblast co-cultures with AM that have ingested apoptotic cells (Figure 3). No statistically significant difference was observed between AM macrophages from healthy donors and from patients with interstitial lung disease in these relatively small subsets of volunteers (not shown, p > 0.05). These observations suggested that macrophages derived from monocytes (TDM or MDM) respond to apoptotic cells by increasing their profibrotic potential, whereas alveolar macrophages appear to be pre-activated by the pulmonary milieu in their ability to induce accumulation of collagen, and fail to further upregulate collagen levels following exposure to apoptotic cells.
Figure 3.

Regulation of collagen levels in co-cultures of monocytes-derived macrophages (MDM) or alveolar macrophages (AM) and fibroblasts. The bars represent fold change in total (closed bars) or collagenase-sensitive (open bars) metabolic incorporation of 14C-proline (mean CPM ± SD of quadruplicate cultures). The collagen production was upregulated when fibroblasts were co-cultured with MDM or AM alone and the levels of collagen increased further significantly when MDM have ingested apoptotic cells (Apopt). The levels of collagen did not change in co-cultures of fibroblasts with AM that have ingested apoptotic cells. Significant increases in 14C-proline incorporation (p < 0.05) are indicated with asterisks.
The profibrotic effect of macrophages on fibroblasts is mediated by soluble factors
We then sought to determine whether direct cell-to-cell contacts between macrophages and fibroblasts are necessary to stimulate collagen production, or secretion of soluble profibrotic factors by macrophages may be sufficient to mediate such an effect. To address this question, two types of experiments were performed. Macrophages were either co-cultured with fibroblasts with or without separation with a semipermeable membrane (Transwell assays) (Figure 4A), or, separately, the conditioned supernatant from the macrophage cultures were transferred into fibroblast cultures and the effect on collagen production was evaluated (Figure 4B). Macrophages that have ingested apoptotic debris stimulated collagen production in fibroblast monolayers even when separated by a Transwell membrane (Figure 4A). Also, the conditioned medium from macrophages that have ingested apoptotic debris stimulated collagen production in fibroblast monolayers (Figure 4B). These observations suggested that soluble factors produced by macrophages following ingestion of apoptotic debris drive the increase in collagen levels. To further investigate this mechanism, we considered a well-known upregulation in production of TGF-β, a potent profibrotic cytokine, by macrophages following ingestion of apoptotic debris (4). A potent neutralizing anti-TGFβ antibody 1D11 clone added to the conditioned medium from macrophages that have ingested apoptotic debris, had a statistically significant, yet relatively modest attenuating effect on the upregulation of collagen levels (Figure 4B). ELISA assays revealed that indeed the levels of total TGF-β1 were increased in macrophage cultures 2 h and 24 h of exposure to apoptotic debris (Figure 5). Of important notice, the levels of TGF-β1 production are consistent with the previous observation (4), and may not be sufficient to directly drive collagen production in fibroblasts (17, 18, 25), particularly because no active TGF-β was detected in the co-cultures by ELISA assays. Also, TGF-β is known to increase collagen production transcriptionally, but our real-time PCR experiments for collagen α2(I) mRNA revealed no increase in collagen mRNA in the macrophage-fibroblast co-cultures (not shown). Therefore, a possibility was considered that factors other than TGF-β might contribute to upregulation of collagen levels by the macrophages following ingestion of apoptotic debris.
Figure 4.

Effect of macrophage-derived soluble factors on 14C-proline incorporation by primary fibroblasts. The bars represent fold change in total (closed bars) or collagenase-sensitive (open bars) metabolic incorporation of 14C-proline (mean CPM ± SD of quadruplicate cultures). In Panel A, macrophages that have ingested apoptotic debris were separated from fibroblast monolayers with Transwell membranes. In Panel B, the medium conditioned by macrophages that have ingested apoptotic cells [(TDM+Apopt) sup] was transferred into fibroblast cultures, without or with added anti-TGF-β antibody or isotype control immunoglobulin. Single and double asterisks indicate significant differences (p < 0.05) from fibroblasts cultured alone, whereas double asterisks indicate the significant difference between co-cultures treated with isotype control and anti-TGF-β antibody. Direct contact between macrophages and fibroblasts is not necessary to upregulate the collagen levels; the effect is mediated, at least in part, by soluble factors produced by macrophages.
Figure 5.

Production of total TGF-β1 in cultures of monocyte-derived macrophages (MDM) after 2 h or 24 h of exposure to apoptotic cells (Apopt) measured by ELISA, mean pg/ml values ± SD of triplicate cultures. Levels of active TGF-β were below detection level in these assays (not shown)
Transforming growth factor beta induced (TGFBI) is generated by macrophages following exposure to apoptotic cells
Transcriptomic profiling of macrophages using cDNA arrays revealed that out of nearly 400 cytokine, chemokine, and their receptor genes represented on the array, only one, transforming growth factor β-induced (TGFBI), also known as keratoepithelin, or beta-ig-h3 (BIGH3), was consistently increased shortly (2 h) following ingestion of apoptotic debris (not shown). Reverse transcriptase-real-time PCR assays confirmed that the steady-state levels of TGFBI mRNA increased significantly when THP-1 derived (TDM), and monocyte-derived macrophages (MDM) engulfed apoptotic cells compared to non-stimulated macrophages (Figure 6A). MDM were tested on two independent occasions, and TDM and AM were tested on four independent occasions, in duplicate cultures, with consistent results. Further experiments defined the dynamics of increase in levels of TGFBI mRNA in macrophage cultures following ingestion of apoptotic debris (Figure 6B). The latter experiments were repeated on two different occasions with consistent results (p < 0.05, one-way ANOVA). In the THP-1 derived macrophages, engulfment of apoptotic Jurkat T cells stimulated an increase in TGFBI protein levels, as judged by the density of the bands in Western blotting analyses (Figure 6C). No differences in steady-state levels of TGFBI mRNA and TGFBI protein were observed between activated and control cultures of alveolar macrophages (AM) (Figure 6A,D). The Western blotting experiments were repeated on two different occasions in each of these macrophage types.
Figure 6.

TGFBI protein and mRNA production by macrophage cultures. Panel A shows that the steady-state levels of TGFBI mRNA increased significantly in THP-1 derived (TDM) and monocyte-derived macrophages (MDM) cultures following ingestion of apoptotic debris for 2h, but not in alveolar macrophages (AM) cultures. Significant changes (p < 0.05) are indicated with asterisks. Panel B shows fold changes in the steady-state levels of TGFBI mRNA (measured by reverse transcriptase – real time PCR) in THP-1 derived macrophage cultures following exposure to apoptotic debris for indicated times. Significant changes (p < 0.05) are indicated with asterisks. Panels C and D show Western blotting for TGFBI protein in THP-1 derived (C) and alveolar (D) macrophage cultures.
Others have previously reported that so-called alternatively activated macrophages express elevated levels of TGFBI (27). Another group also reported that such macrophages stimulate collagen production in co-cultures with fibroblasts (28). Therefore, a possibility was considered that phagocytosis of apoptotic debris may convert macrophages toward an alternative phenotype that is characterized by the cell surface expression of CD163 and CD206 (29). We conducted flow-cytometric analyses of TDM and MDM macrophages before and after phagocytosis of apoptotic debris and observed no increase in the cell surface expression of CD163 and CD 206 at 24 h post incubation, as judged by lack of change in the mean fluorescence intensity (data not shown). Levels of IL-4 and IL-13, cytokines known to promote alternative activation of macrophages, were below detection thresholds of 0.25 pg/ml (IL-4) and 62.5 pg/ml (IL-13) in all macrophage culture supernatants; these concentrations are significantly lower than those necessary for alternative activation of macrophages (28) or for upregulation of collagen production in fibroblasts (5,24). Therefore, ingestion of apoptotic cells causes a selective increase in the expression levels of TGFBI without alternative activation of macrophages.
TGFBI upregulates collagen levels in primary fibroblasts without activation of collagen production
The finding of increased TGFBI production by macrophages following ingestion of the apoptotic debris suggested that this cytokine may directly regulate collagen accumulation in fibroblast cultures. To test this possibility, fibroblast cultures were incubated for various times with various doses of rhTGFBI, and the levels of collagen accumulation were measured. The experiments showed that TGFBI directly regulated collagen levels in cultured primary fibroblasts in a dose- (Figure 7A,B) and time- (Figure 7C,D) dependent fashion. These experiments were repeated on at least two occasions, in duplicates, for each of the four primary fibroblast cultures, with consistent results.
Figure 7.

Time and dose-dependent effect of rhTGFBI on collagen protein levels in primary lung fibroblast cultures. Collagen levels were measured by metabolic incorporation of 14C-proline (Panels A,C), or using Western blotting technique (Panels B,D,E). Significant increases in 14C-proline incorporation (p < 0.05) are indicated with asterisks. In Panels A and B, fibroblasts were activated for 48 h with increasing concentrations of rhTGFBI as indicated. In Panels C, fibroblast cultures were incubated for indicated times without or with 100 ng/ml rhTGFBI. In Panel D, fibroblasts were cultured for 72 h, with 100 ng/ml rhTGFBI added to the cultures for the final 24, 48, or 72 h as indicated. In Panel E, fibroblasts were transfected with collagen α2(I) siRNA or control siRNA as indicated, and the effect of rhTGFBI on collagen protein levels was assessed. Notice the decline in basal collagen production but persistent upregulation in collagen level in response to TGFBI stimulation (repeated on two independent occasions with consistent results).
The experiments were performed to determine whether the increase in collagen levels stimulated by TGFBI was due to transcriptional upregulation of the collagen gene expression. To test whether collagen gene expression was transcriptionally upregulated by TGFBI, collagen α2(I) mRNA steady state levels were measured by Q-PCR and showed no differences in steady-state levels of collagen α2(I) mRNA between TGFBI-treated and control cultures (not shown). The response of the collagen α2(I) promoter-CAT reporter constructs was evaluated and no differences were observed (not shown). Finally, activation of DNA binding by transcription factors known to regulate the activity of collagen gene promoter (Smad3/4, Sp1, AP1, Ets) was tested using transcription factor array approach (TranSignal™ Protein/DNA system, Panomics, Fremont, CA), and again, no differences were detected (not shown). These results suggested that the observed upregulation of collagen protein levels by TGFBI (see Figure 7) was not due to transcriptional regulation at the level of the collagen gene promoter. To further confirm this conclusion, siRNA-mediated inhibition of collagen α2(I) was performed (Figure 7E). Despite the decrease in the total collagen level, the responsiveness to stimulation with TGFBI was not affected, further supporting the notion that collagen level regulation by TGFBI does not occur at the level of production.
Stimulation with TGFBI inhibits expression of MMP14
A possibility was considered that TGFBI upregulates collagen levels in fibroblast cultures by attenuating collagen turnover. To address such a possibility, the experiments were performed in which primary fibroblasts were incubated for 6 h with or without 300 ng/ml TGFBI. The expression levels of 84 genes related to connective tissue biology were analyzed using reverse transcriptase – real time PCR approach (SuperArray, Frederick, MD). Expression of matrix metalloproteinase (MMP) 14 was consistently decreased following stimulation with TGFBI (Figure 8A). Western blotting analyses confirmed that MMP14 protein levels decreased significantly in fibroblast cultures activated with TGFBI for 24 h or 48 h, compared to non-activated fibroblasts (Figure 8B). These experiments were repeated on three independent occasions with primary fibroblast cultures from different donors, with consistent results. MMP14 is a critical factor for collagen turnover by fibroblasts (30). This decrease in MMP14 levels (Figure 8) is consistent with the increase in collagen levels (Figure 7), as collagen turnover is likely to be downregulated due to lower levels of MMP14. To further confirm the inverse link between the levels of MMP14 and collagen, primary fibroblasts were transfected with either an MMP14-encoding plasmid construct or the corresponding “blank” plasmid (Figure 9B,C). As expected, the levels of MMP14 following the transfection were increased (Figure 9B) and the levels of collagen reciprocally decreased (Figure 9C). Reciprocally, siRNA-mediated inhibition of MMP14 expression in fibroblasts led to an increase in collagen levels compared to control siRNA-transfected fibroblasts (Figure 9C).
Figure 8.

MMP14 mRNA (A) and protein (B) levels in primary fibroblasts. In Panel A, mRNA was purified from control or TGFBI-stimulated fibroblasts and reverse transcribed into cDNA; real-time PCR amplification was performed using primers for a housekeeping gene GAPDH and for MMP14 gene. The amplification curves for GAPDH corresponding to control and TGFBI-stimulated fibroblasts closely overlapped, whereas amplification of MMP14 template in cDNA from TGFBI-stimulated fibroblasts occurred two cycles later, suggesting approximately 22 = 4 fold lower levels of MMP14 mRNA. In Panel B, primary fibroblasts were cultured for indicated times without or with 300 ng/ml of rhTGFBI. Lysates were normalized for total protein, and Western blotting assays conducted using anti-MMP14 antibody.
Figure 9.

DNA binding by p53 (A) and effects of MMP14 or p53 overexpression, or MMP14 or PU.1 siRNA inhibition on the expression levels of MMP14 and collagen (B,C). In Panel A, electromobility shift assays were used, on two independent occasions, to validate the results of the protein-DNA array experiments. The p53-specific 32P-labeled probe was incubated with nuclear lysates from non-stimulated control or TGFBI-activated fibroblasts, as indicated, or in the presence of 10-fold excess of the mutant (scrambled) or unlabeled (cold) specific probe, as indicated. The cold but not the scrambled probe inhibited p53 binding (arrows), suggesting that the binding is specific. In Panels B and C, fibroblast cultures were transfected with MMP14-encoding or p53-encoding plasmid, or with MMP14 or PU.1 siRNA as indicated. Western blotting assays were performed using antibodies against MMP14 or collagen. At least three independent experiments were conducted for each panel shown, with consistent results.
Regulation of MMP14 and collagen by TGFBI is dependent on PU1 and p53
Parallel semi-quantitative screening of DNA binding by transcription factors (TranSignal™ Protein/DNA Array, Panomics) revealed a significant upregulation of DNA binding by PU.1 and a downregulation of DNA binding by p53 in response to stimulation of fibrolasts with 300 ng/ml rhTGFBI (not shown). These observations suggested that PU.1 might be a repressor, whereas p53 might be an activator of MMP14 expression. To confirm the validity of these observations in protein/DNA arrays, electromobility shift assays were performed to assess DNA binding by p53 (Figure 9A). To test whether PU.1 is involved in the regulation of MMP14 and collagen levels, PU.1 expression in fibroblasts was inhibited using siRNA transfection technique. Real-time PCR analyses revealed that levels of PU.1 mRNA were decreased three fold in PU.1 siRNA-transfected fibroblasts compared to control siRNA-transfected fibroblasts (not shown). Simultaneously, expression levels of MMP14 increased (Figure 9B) and levels of collagen decreased in PU.1 siRNA-transected fibroblasts (Figure 9C). Transient transfection of fibroblast cultures with p53-encoding plasmid construct was performed to determine whether overexpression of p53 leads to increase in MMP14 and a decrease in collagen protein levels. As expected, MMP14 protein levels increased (Figure 9B) and collagen levels decreased (Figure 9C) in fibroblasts transfected with p53-encoding plasmid. These observations suggest that PU.1 and p53 are involved in the regulation of MMP14 and collagen levels in fibroblasts.
DISCUSSION
This study addressed the issue of macrophages’ involvement in the regulation of extracellular matrix following ingestion of apoptotic cells. Macrophages derived from a monocytic cell line THP-1 (see Figure 2), as well as macrophages derived from primary monocytes (see Figure 3), stimulated upregulation of collagen protein levels in co-cultures with fibroblasts, following ingestion of apoptotic cells. In contrast, primary alveolar macrophages stimulated upregulation of collagen levels even without exposure to apoptotic cells, and this effect was not further increased following ingestion of apoptotic debris (see Figure 3). The apparent “preactivation” of alveolar macrophages may be due to the specialized TGF-β-rich pulmonary environment (35) that predisposes alveolar macrophages to a more profibrotic basal phenotype. The effect of ingestion of apoptotic cells by macrophages on collagen production by fibroblasts in co-cultures was preserved if the macrophages and fibroblasts were separated by a Transwell membrane (see Figure 4A), suggesting that soluble factors produced by macrophages mediated the effect on collagen levels. If macrophages were exposed to apoptotic cells separately, and the conditioned medium was then transferred into the cultures of the primary fibroblast monolayers, the effect was still preserved (see Figure 4B), further supporting the notion that soluble factor(s) were produced by macrophages following ingestion of apoptotic cells, and that those soluble factors regulated accumulation of collagen.
An obvious candidate for such a soluble factor would be TGF-β, a factor whose expression in macrophages does increase following ingestion of apoptotic debris (4), and that is a known potent profibrotic factor (5). Indeed, ELISA assays revealed that total TGF-β was elevated in the macrophage culture supernatants following ingestion of apoptotic debris (see Figure 5). However, such an increase in TGF-β is unlikely to explain the observed increase in collagen for the following reasons. First, a concentration of at least 500 pg/ml of active TGF-β is required for upregulation of collagen production (17, 18, 25), whereas in these assays, total TGF-β did not exceed 325 pg/ml, and active TGF-β was not detectable. Second, TGF-β is known to upregulate collagen production transcriptionally, leading to elevated steady-state levels of collagen mRNA, but no such increase in collagen α2(I) mRNA was observed by real-time PCR in fibroblast-macrophage co-cultures following ingestion of apoptotic cells. Finally, the increase in collagen levels was somewhat attenuated but still significant in the presence of neutralizing anti-TGF-β antibody (see Figure 4B). Together, these observations suggest that an additional soluble factor may promote upregulation of collagen protein levels.
To address this possibility, profiling of gene expression for cytokines and related factors has been performed in fibroblasts from co-cultures with macrophages. In three independent experiments, expression of mRNA for transforming growth factor beta-induced (TGFBI), also termed Beta-IG-H3 (BIGH3), or keratoepithelin, was increased. Real-time PCR experiments confirmed this observation in THP-1-derived and primary monocyte-derived, but not in alveolar macrophages (see Figure 6A), in a time-dependent fashion (see Figure 6B). Western blotting analyses for TGFBI confirmed that expression of this protein increased following ingestion of apoptotic but not necrotic cells (see Figure 6C). This effect on TGFBI expression was observed in THP-1-derived macrophages (see Figure 6C) but not in alveolar macrophages (see Figure 6D). Thus, expression of TGFBI in macrophages and the effect on collagen accumulation in co-cultures with fibroblasts are both upregulated following ingestion of apoptotic cells in THP-1-derived and primary monocyte-derived, but not in alveolar macrophages (compare Figure 6 with Figures 2,3). The observed non-responsiveness of alveolar macrophages may be due to their “preactivation” in the TGF-β-rich pulmonary environment (35). These observations suggest that TGFBI may be an important factor mediating the profibrotic effect of macrophages that ingested apoptotic cells. While producing TGFBI (27), and acting profibrotically (28) similarly to alternatively activated macrophages, the macrophages that ingested apoptotic cells did not express other markers of alternative activation such as CD163 or CD206 (29). There was no increase in the levels of cytokines IL-4 or IL-13 that are known to facilitate alternative macrophage activation or upregulation of collagen in fibroblasts.
To investigate the possible profibrotic involvement of TGFBI, recombinant human (rh) TGFBI was added to cultured primary fibroblasts and collagen levels assessed (see Figure 7). Increases in collagen protein levels were observed in response to stimulation with rhTGFBI in a dose- and time-dependent fashion (see Figure 7A-D). However, TGFBI did not stimulate an increase in collagen mRNA according to real-time PCR data (not shown). Also, a collagen promoter-CAT reporter construct that has been previously described to respond to TGF-β activation (25), was transfected into fibroblast and was responsive to TGF-β stimulation but non-responsive to TGFBI stimulation (not shown). Profiling of DNA binding by transcription factor arrays showed no increase in the activity of Smad3/4, Sp1, AP1, and Ets (factors known to regulate activity of the collagen gene promoter). Finally, siRNA-mediated inhibition of collagen production (see Figure 7E) did not affect the TGFBI-stimulated increase in collagen levels. Together, these observations suggest that TGFBI regulates collagen levels through a mechanism that is different from upregulation of collagen production.
A possibility was considered that TGFBI may regulate collagen turnover, e.g. by inhibiting the levels of matrix metalloproteinases in fibroblasts. To address such a possibility, real-time PCR experiments were conducted in fibroblasts that were or were not activated with rhTGFBI, to compare steady-state levels of mRNA for genes related to extracellular matrix. Real-time PCR-based system (RT2 Profiler™, SuperArray) was utilized to simultaneously compare expression of 84 extracellular matrix relevant genes. The differences between TGFBI-activated and control cells were observed for MMP14 (also termed MT1-MMP), whose mRNA levels decreased four fold following activation with TGFBI (see Figure 8A). Western blotting assays revealed a similar decrease in MMP14 protein following stimulation of fibroblasts with TGFBI (see Figure 8B).
MMP14 is able to directly degrade various extracellular matrix components, including type I, II, and III collagens, gelatin, fibronectin, vitronectin, tenascin, entactin, and laminin-1, as well as to directly activate pro-MMP-2(31, 32). The loss of MMP14 leads to significant disturbances of connective tissue metabolism. Fibroblasts from MMP14-deficient mice completely lose the ability to degrade collagen fibrils, resulting in severe and progressive fibrosis in many tissues of MMP14-deficient mice (33). In our study, overexpression of MMP14 in cultured fibroblasts caused downregulation of collagen levels, whereas siRNA-mediated inhibition of MMP14 expression led to increased collagen levels (Figure 9C), confirming previous observations of MMP14 involvement in collagen turnover by fibroblasts (30).
Since the levels of MMP14 decreased at both mRNA and protein levels, the possibility of transcriptional regulation of MMP14 by TGFBI was considered. To assess possible transcription factors that might be involved in downregulation of MMP14, profiling of DNA binding activity by various transcription factors was assessed in TGFBI-stimulated in comparison with control fibroblasts. Experiments with transcription factor arrays revealed that out of 345 transcription factors analyzed, two factors have significantly changed their DNA binding activity in fibroblasts in response to activation with TGFBI. Activity of PU.1 was increased and activity of p53 was decreased following fibroblast activation with TGFBI. Inhibition of PU.1 expression with siRNA in cultured fibroblasts led to an increase in MMP14 expression levels and a decrease in collagen levels (see Figure 9B,C). This observation is consistent with the notion that PU.1 is a repressor of MMP14 expression, as stimulation with TGFBI activates DNA binding by PU.1 and simultaneously causes a decrease in MMP14 expression (see Figure 8). Overexpression of p53, a possible activator of MMP14 expression, led to a similar increase in MMP14 and decrease in collagen expression (see Figure 9B,C). The later observation is consistent with the previous report (34) that suggests an inhibitory effect of p53 on TGF-β-stimulated collagen expression.
Based on the observations summarized above, a sequence of event connecting ingestion of apoptotic cells by macrophages with collagen accumulation in fibroblasts appears to be as presented in Figure 10. Physiologically, the studied mechanisms are likely to be relevant to inflammation resolution and healing, but, if exaggerated, may possibly contribute to tissue fibrosis. Pharmacological targeting of this pathway may be useful in developing future therapies for delayed wound healing or for tissue fibrosis.
Figure 10.
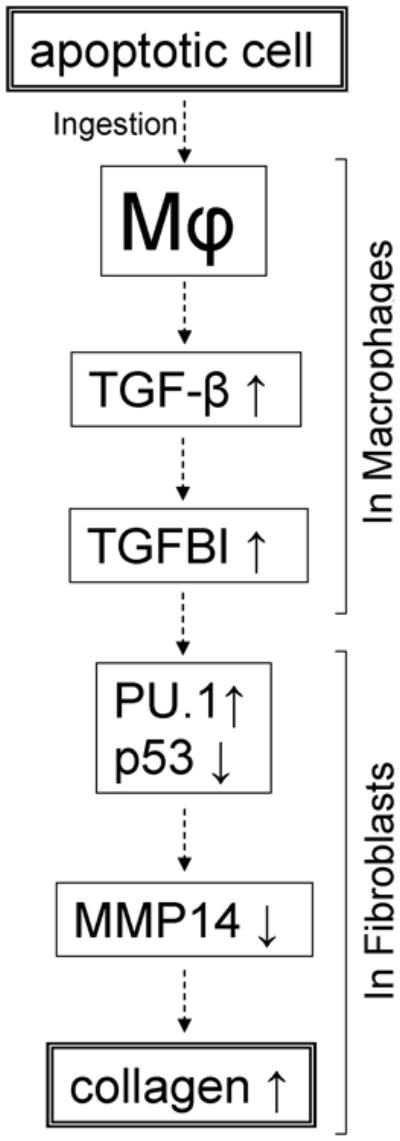
Proposed outline of the sequential regulation of collagen expression in fibroblasts by macrophages following exposure to apoptotic cells.
Acknowledgments
This work was supported by a National Institutes of Health Grant 1 R01 HL074067 (to S.P.A.), a Veterans Administration Merit Review grant (to S.P.A.), and Maryland Chapter of Arthritis Foundation Research Awards (to I.G.L. and S.P.A.).
We thank Drs. Barry S. Handwerger, Jeffrey D. Hasday, and Simeon E. Goldblum for critical discussions of this work and Ms. Jung Choi for technical help.
References
- 1.Huynh ML, V, Fadok A, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xiao YQ, Malcolm K, Worthen GS, Gardai S, Schiemann WP, Fadok VA, Bratton DL, Henson PM. Cross-talk between ERK and p38 MAPK mediates selective suppression of pro-inflammatory cytokines by transforming growth factor-beta. J Biol Chem. 2002;277:14884–14893. doi: 10.1074/jbc.M111718200. [DOI] [PubMed] [Google Scholar]
- 3.McDonald PP, V, Fadok A, Bratton D, Henson PM. Transcriptional and translational regulation of inflammatory mediator production by endogenous TGF-beta in macrophages that have ingested apoptotic cells. J Immunol. 1999;163:6164–6172. [PubMed] [Google Scholar]
- 4.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atamas SP, White B. Cytokine regulation of pulmonary fibrosis in scleroderma. Cytokine Growth Factor Rev. 2003;14:537–550. doi: 10.1016/s1359-6101(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 6.Martin P, D’Souza D, Martin J, Grose R, Cooper L, Maki R, McKercher SR. Wound healing in the PU.1 null mice--tissue repair is not dependent on inflammatory cells. Curr Biol. 2003;13:1122–1128. doi: 10.1016/s0960-9822(03)00396-8. [DOI] [PubMed] [Google Scholar]
- 7.Reynolds HY. Lung inflammation and fibrosis: an alveolar macrophage-centered perspective from the 1970s to 1980s. Am J Respir Crit Care Med. 2005;171:98–102. doi: 10.1164/rccm.200406-788PP. [DOI] [PubMed] [Google Scholar]
- 8.Prasse A, Pechkovsky DV, Toews GB, Jungraithmayr W, Kollert F, Goldmann T, Vollmer E, Muller-Quernheim J, Zissel G. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med. 2006;173:781–792. doi: 10.1164/rccm.200509-1518OC. [DOI] [PubMed] [Google Scholar]
- 9.Zhang-Hoover J, Sutton A, van Rooijen N, Stein-Streilein J. A critical role for alveolar macrophages in elicitation of pulmonary immune fibrosis. Immunology. 2000;101:501–511. doi: 10.1046/j.1365-2567.2000.00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamate J, Sato K, Ide M, Nakanishi M, Kuwamura M, Sakuma S, Nakatsuji S. Participation of different macrophage populations and myofibroblastic cells in chronically developed renal interstitial fibrosis after cisplatin-induced renal injury in rats. Vet Pathol. 2002;39:322–333. doi: 10.1354/vp.39-3-322. [DOI] [PubMed] [Google Scholar]
- 11.Chow FY, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in streptozotocin-induced diabetic nephropathy: potential role in renal fibrosis. Nephrol Dial Transplant. 2004;19:2987–2996. doi: 10.1093/ndt/gfh441. [DOI] [PubMed] [Google Scholar]
- 12.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, Scabilloni JF, Antonini JM, Rojanasakul Y, Castranova V, Mercer RR. Induction of secondary apoptosis, inflammation, and lung fibrosis after intratracheal instillation of apoptotic cells in rats. Am J Physiol Lung Cell Mol Physiol. 2006;290:L695–L702. doi: 10.1152/ajplung.00245.2005. [DOI] [PubMed] [Google Scholar]
- 14.Docherty NG, O’Sullivan OE, Healy DA, Fitzpatrick JM, Watson RW. Evidence that inhibition of tubular cell apoptosis protects against renal damage and development of fibrosis following ureteric obstruction. Am J Physiol Renal Physiol. 2006;290:F4–F13. doi: 10.1152/ajprenal.00045.2005. [DOI] [PubMed] [Google Scholar]
- 15.Canbay A, Higuchi H, Bronk SF, Taniai M, Sebo TJ, Gores GJ. Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology. 2002;123:1323–1330. doi: 10.1053/gast.2002.35953. [DOI] [PubMed] [Google Scholar]
- 16.Mora AL, Torres-Gonzalez E, Corredor C, Ritzenthaler J, Xu J, Roman J, Brigham K, Stecenko A. Activation of alveolar macrophages via the alternative pathway in herpesvirus-induced lung fibrosis. Am J Respir Cell Mol Biol. 2006;35:466–473. doi: 10.1165/rcmb.2006-0121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kahari VM, Chen YQ, Su MW, Ramirez F, Uitto J. Tumor necrosis factor-alpha and interferon-gamma suppress the activation of human type I collagen gene expression by transforming growth factor-beta 1. Evidence for two distinct mechanisms of inhibition at the transcriptional and posttranscriptional levels. J Clin Invest. 1990;86:1489–1495. doi: 10.1172/JCI114866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fine A, Goldstein RH. The effect of transforming growth factor-beta on cell proliferation and collagen formation by lung fibroblasts. J Biol Chem. 1987;262:3897–3902. [PubMed] [Google Scholar]
- 19.Atamas SP, Yurovsky VV, Wise R, Wigley FM, Goter Robinson CJ, Henry P, Alms WJ, White B. Production of type 2 cytokines by CD8+ lung cells is associated with greater decline in pulmonary function in patients with systemic sclerosis. Arthritis Rheum. 1999;42:1168–1178. doi: 10.1002/1529-0131(199906)42:6<1168::AID-ANR13>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 20.Luzina IG, Atamas SP, Wise R, Wigley FM, Xiao HQ, White B. Gene expression in bronchoalveolar lavage cells from scleroderma patients. Am J Respir Cell Mol Biol. 2002;26:549–557. doi: 10.1165/ajrcmb.26.5.4683. [DOI] [PubMed] [Google Scholar]
- 21.Luzina IG, Atamas SP, Wise R, Wigley FM, Choi J, Xiao HQ, White B. Occurrence of an activated, profibrotic pattern of gene expression in lung CD8+ T cells from scleroderma patients. Arthritis Rheum. 2003;48:2262–2274. doi: 10.1002/art.11080. [DOI] [PubMed] [Google Scholar]
- 22.Atamas SP, I, Luzina G, Choi J, Tsymbalyuk N, Carbonetti NH, Singh IS, Trojanowska M, Jimenez SA, White B. Pulmonary and activation-regulated chemokine stimulates collagen production in lung fibroblasts. Am J Respir Cell Mol Biol. 2003;29:743–749. doi: 10.1165/rcmb.2003-0078OC. [DOI] [PubMed] [Google Scholar]
- 23.Luzina IG, Tsymbalyuk N, Choi J, Hasday JD, Atamas SP. CCL18-stimulated upregulation of collagen production in lung fibroblasts requires Sp1 signaling and basal Smad3 activity. J Cell Physiol. 2006;206:221–228. doi: 10.1002/jcp.20452. [DOI] [PubMed] [Google Scholar]
- 24.Atamas SP, I, Luzina G, Dai H, Wilt SG, White B. Synergy between CD40 ligation and IL-4 on fibroblast proliferation involves IL-4 receptor signaling. J Immunol. 2002;168:1139–1145. doi: 10.4049/jimmunol.168.3.1139. [DOI] [PubMed] [Google Scholar]
- 25.Luzina IG, Highsmith K, Pochetuhen K, Nacu N, Rao JN, Atamas SP. PKCalpha mediates CCL18-stimulated collagen production in pulmonary fibroblasts. Am J Respir Cell Mol Biol. 2006;35:298–305. doi: 10.1165/rcmb.2006-0033OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luzina IG, Papadimitriou JC, Anderson R, Pochetuhen K, Atamas SP. Induction of prolonged infiltration of T lymphocytes and transient T lymphocyte -dependent collagen deposition in mouse lungs following adenoviral gene transfer of CCL18. Arthritis Rheum. 2006;54:2643–2655. doi: 10.1002/art.21950. [DOI] [PubMed] [Google Scholar]
- 27.Gratchev A, Guillot P, Hakiy N, Politz O, Orfanos CE, Schledzewski K, Goerdt S. Alternatively activated macrophages differentially express fibronectin and its splice variants and the extracellular matrix protein betaIG-H3. Scand J Immunol. 2001;53:386–392. doi: 10.1046/j.1365-3083.2001.00885.x. [DOI] [PubMed] [Google Scholar]
- 28.Song E, Quyang N, Horbelt M, Antus B, Wang M, Exton MS. Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol. 2000;204:19–28. doi: 10.1006/cimm.2000.1687. [DOI] [PubMed] [Google Scholar]
- 29.Porcheray V, Viaud S, Rimaniol AC, Leone C, Samah B, Dereuddre-Bosquet N, Dormont D, Gras G. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol. 2005;142:481–489. doi: 10.1111/j.1365-2249.2005.02934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee H, Overall CM, McCulloch CA, Sodek J. A critical role for the membrane-type 1 matrix metalloproteinase in collagen phagocytosis. Mol Biol Cell. 2006;17:4812–4826. doi: 10.1091/mbc.E06-06-0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, Seiki M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370:61–65. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 32.Okada A, Tomasetto C, Lutz Y, Bellocq JP, Rio MC, Basset P. Expression of matrix metalloproteinases during rat skin wound healing: evidence that membrane type-1 matrix metalloproteinase is a stromal activator of pro-gellatinase A. J Cell Biol. 1997;137:67–77. doi: 10.1083/jcb.137.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, Mankani M, Robey PG, Poole AR, Pidoux I, Ward JM, Birkedal-Hansen H. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99:81–92. doi: 10.1016/s0092-8674(00)80064-1. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh AK, Bhattacharyya S, Varga J. The tumor supressor p53 abrogates Smad-dependent collagen gene induction in mesenchymal cells. J Biol Chem. 2004;279:47455–47463. doi: 10.1074/jbc.M403477200. [DOI] [PubMed] [Google Scholar]
- 35.Takabayshi K, Corr M, Hayashi T, Redecke V, Beck L, Guiney D, Sheppard D, Raz E. Induction of a homeostatic circuit in lung tissue by microbial compounds. Immunity. 2006;24:475–487. doi: 10.1016/j.immuni.2006.02.008. [DOI] [PubMed] [Google Scholar]