

Abstract
Tumor suppressor genes on the X chromosome may skew the gender distribution of specific types of cancer1,2. T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematological malignancy with an increased incidence in males3. In this study, we report the identification of inactivating mutations and deletions in the X-linked plant homeodomain finger 6 (PHF6) gene in 16% of pediatric and 38% of adult primary T-ALL samples. Notably, PHF6 mutations are almost exclusively found in T-ALL samples from male subjects. Mutational loss of PHF6 is significantly associated with leukemias driven by aberrant expression of the homeobox transcription factor oncogenes TLX1 and TLX3. Overall, these results identify PHF6 as a new X-linked tumor suppressor in T-ALL and point to a strong genetic interaction between PHF6 loss and aberrant expression of TLX transcription factors in the pathogenesis of this disease.
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive malignancy in which multiple genetic defects collaborate in the transformation of T-cell progenitors4,5. Notably, T-ALL has a 3-fold higher incidence in males3, while other immature hematologic tumors such as precursor B-lineage ALL (BCP-B-ALL) are equally frequent in males and females3.
To identify a possible X-linked tumor suppressor in T-ALL, we performed an X chromosome targeted mutation analysis in tumor DNA samples from 12 male T-ALL cases. For each sample, we performed in-solution DNA capture of 7,674 regions encompassing 3,045,708 nucleotides corresponding to 5,215 X chromosome exons using the Agilent Sure Select oligonucleotide capture system6. DNA samples enriched for X chromosome exons were then analyzed by next generation sequencing using the SOLiD 3 platform. This analysis identified 66 candidate novel non-synonymous single nucleotide variants and 7 positions with high confidence calls for containing complex variants such as insertions or deletions (Fig. 1a). Dideoxynucleotide DNA sequencing of PCR products encompassing affected exons confirmed the presence of 61/66 (92%) of these single nucleotide variants and 4/7 (57%) of the more complex variants, including 2 insertions and 2 deletions (Supplementary Tables 1 and 2). Sequence analysis of paired DNA samples obtained at the time of clinical remission showed that most of these variants corresponded to previously unreported germline polymorphisms. However, and most notably, we also identified three somatically acquired changes corresponding to two non-synonymous single nucleotide substitutions (c.902A>G, p.T300A and c.990A>G, p.H330R) and a frameshift-creating insertion of 5 nucleotides (c.124_125insAGGCA, p.H43fs) in the PHF6 (plant homeodomain finger 6) gene (Fig. 1a).
Figure 1.

Next generation sequencing and array CGH analysis of the X chromosome identifies PHF6 mutations in human T-ALL. (a) Overview of mutation screening approach of the human X chromosome exome in a panel of tumor DNA samples from 12 male T-ALL cases using oligonucleotide sequence capture and next generation sequencing with SOLiD3. After filtering and confirmation of high throughput sequencing data, analysis of corresponding remission DNA samples led to the identification of three somatically acquired changes in the PHF6 gene. (b) Schematic overview of the recurrent genomic deletions involving chromosomal band Xq26.3 in 8 human T-ALL samples. Specific genes located in Xq26.3 are shown. (c) Detailed view of a representative oligo array-CGH plot of leukemia DNA/control DNA ratios (blue tracing) versus the dye-swap experiment (red tracing) in a patient harboring an Xq26.3 deletion. (d) DNA quantitative PCR analysis of PHF6 copy number dose in female and male reference genomic DNAs and 2 primary samples from male T-ALL cases harboring Xq26.3 deletions.
In a complementary approach, we analyzed X chromosome array comparative genome hybridization (array-CGH) data from 246 primary T-ALL samples (179 male and 67 female) in a multi-centre setting. These analyses revealed the presence of recurrent deletions in chromosomal band Xq26 in 8 out of 246 (∼3%) T-ALL samples (Table 1). For 3 del(X)(q26) positive T-ALL cases, we performed array-CGH analysis against the corresponding remission material, which showed that these Xq26 deletions are somatically acquired leukemia-associated genetic events (Table 1). Re-analysis of all 8 del(X)(q26) positive T-ALL cases on a custom high-resolution chromosome X oligonucleotide array (Fig. 1b,c) narrowed down the common minimally deleted region to an area of 80 kb containing the PHF6 gene. Consistently, quantitative PCR analysis confirmed loss of the PHF6 locus in the del(X)(q26) positive cases (Fig. 1d). The convergent findings of our X chromosome exon mutation analysis and analysis of copy number alterations by array-CGH thus identified the PHF6 gene as a new tumor suppressor mutated and deleted in T-ALL.
Table 1. Characteristics of 38 primary T-ALL samples showing PHF6 inactivation.
| PHF6 lesion | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | Sex | Age | WBC (×109/l) | Immuno-phenotype | Genetic subtype | NOTCH1 | Type of alteration | Predicted protein | Germline/somatic |
| PHF6 deletions | |||||||||
| 1 | M | Ped | 77 | Cortical | TLX3 | mut | Deletion (0.55 Mb) | NA | |
| 2 | M | Ped | 46 | Pre-T | TLX3 | wt | Deletion (0.23 Mb) | NA | |
| 3 | M | Ped | 31 | Pre-T | TLX3 | NA | Deletion (1.50 Mb) | NA | |
| 4 | M | Ped | 2 | Pre-T | unknown | NA | Deletion (0.27 Mb) | NA | |
| 5 | M | Ped | NA | Cortical | HOXA | NA | Deletion (1.90 Mb) | Somatic | |
| 6 | M | Ped | NA | Cortical | unknown | NA | Deletion (0.20 Mb) | Somatic | |
| 7 | M | Ped | NA | Cortical | TLX1 | NA | Deletion (0.08 Mb) | Somatic | |
| 8 | M | Adult | NA | Pre-T | Unkn | NA | Deletion (0.11 Mb) | NA | |
| PHF6 mutations | |||||||||
| 9 | M | Ped | 185 | Cortical | TLX3 | wt | Nonsense | p.G122X | NA |
| 10 | M | Ped | 417 | Pre-T | TLX3 | mut | Nonsense | p.R116X | NA |
| 11 | F | Ped | 280 | Pre-T | TLX1 | mut | Frameshift | p.F172fs | NA |
| 12 | M | Ped | 405 | Pre-T | TLX3 | mut | Frameshift | p.Y303fs | NA |
| 13 | M | Ped | 159 | Pre-T | TLX1 | mut | Nonsense | p.K235X | NA |
| 14 | M | Ped | 500 | Pre-T | TLX3 | mut | Frameshift | p.A41fs | NA |
| 15 | M | Ped | 347 | Cortical | HOXA | mut | Nonsense | p.K274X | NA |
| 16 | M | Ped | 129 | Cortical | unknown | mut | Frameshift | p.D333fs | NA |
| 17 | M | Ped | 174 | Cortical | TLX3 | mut | Nonsense | p.R225X | NA |
| 18 | M | Ped | 27 | Cortical | TLX1 | wt | Nonsense | p.R116X | NA |
| 19 | M | Ped | 310 | Cortical | TAL1 | mut | Missense | p.C283R | NA |
| 20 | M | Ped | 189 | Cortical | TAL1 | mut | Frameshift | p.C28fs | NA |
| 21 | M | Adult | 170 | Cortical | TLX3 | wt | Frameshift | p.H44fs | NA |
| 22 | M | Adult | 21 | Cortical | TLX1 | mut | Frameshift | p.H43fs | Somatic |
| 22 | M | Adult | 21 | Cortical | TLX1 | mut | Frameshift | p.H43fs | Somatic |
| 23 | M | Adult | NA | Pre-T | TLX3 | mut | Frameshift | p.T98fs | Somatic |
| 24 | M | Adult | 14 | Pre-T | TLX3 | wt | Frameshift | p.Y105fs | NA |
| 25 | M | Adult | 28 | Mature | TLX3 | wt | Frameshift | p.S158fs | NA |
| 26 | M | Adult | NA | Cortical | TLX3 | mut | Missense | p.C215Y | NA |
| 27 | M | Adult | NA | Pre-T | Unkn | mut | Missense | p.C215F | NA |
| 28 | M | Adult | 31 | Cortical | Unkn | mut | Missense | p.C215Y | NA |
| 29 | M | Adult | NA | Mature | TLX3 | mut | Missense | p.T300A | Somatic |
| 30 | M | Adult | 21 | Cortical | TLX1 | wt | Missense | p.A311P | NA |
| 31 | M | Adult | 30 | Mature | Unkn | wt | Missense | p.C280Y | NA |
| 32 | M | Adult | 23 | Cortical | Unkn | wt | Missense | p.H329R | Somatic |
| 33 | M | Ped | NA | NA | TAL1 | NA | Nonsense | p.R257X | Somatic |
| 34 | M | Ped | NA | NA | TLX1 | NA | Frameshift | p.S191fs | Somatic |
| 35 | M | Adult | NA | NA | TLX1 | NA | Missense | p.C215F | Somatic |
| 36 | M | Adult | NA | NA | TLX1 | NA | Nonsense | p.Y303X | NA |
| 37 | M | Adult | NA | NA | Unkn | NA | Nonsense | p.R274X | NA |
| 38 | M | Adult | NA | NA | Unkn | NA | Frameshift | p.H135fs | NA |
Ped, pediatric; NA, not available; mut, mutated; wt, wild-type.
PHF6 encodes a plant homeodomain (PHD) factor containing four nuclear localization signals and two imperfect PHD zinc finger domains7 with a proposed role in the control of gene expression7. Notably, inactivating mutations in PHF6 cause the Börjeson-Forssman-Lehmann syndrome (BFLS; MIM#301900), a relatively uncommon type of X-linked familial syndromic mental retardation which has not been associated with increased incidence of T-ALL7-9. Quantitative RT-PCR analysis demonstrated ubiquitous expression of PHF6 transcripts in human tissues, with the highest levels of expression in thymus, ovary and thyroid and moderate levels of expression in spleen, testes and adipose tissue (Supplementary Fig. 1). Consistent with these results, PHF6 was readily detected by immunohistochemistry in mouse thymus (Supplementary Fig. 1). Finally, quantitative RT-PCR analysis of human thymocyte populations at different stages of development showed variable levels of PHF6 expression, with marked upregulation of PHF6 transcripts in CD4/CD8 double positive cells (Supplementary Fig. 1).
Mutation analysis of PHF6 in an extended panel of pediatric and adult T-ALL primary samples identified truncating or missense mutations in PHF6 in 38% (16/42) of adult and ∼16% (14/89) of pediatric T-ALL samples (Fig. 2a and Table 1). In all available cases (7/30), analysis of matched buccal and/or bone marrow remission genomic DNA confirmed the somatic origin of PHF6 mutations (4/21 frameshift mutations and 3/9 missense mutations) (Fig. 2b and Table 1). Finally, no mutations in PHF6 were identified in DNA samples from BCP-B-ALLs (n = 62), suggesting that mutational loss of PHF6 in lymphoid tumors could be restricted to T-ALL.
Figure 2.
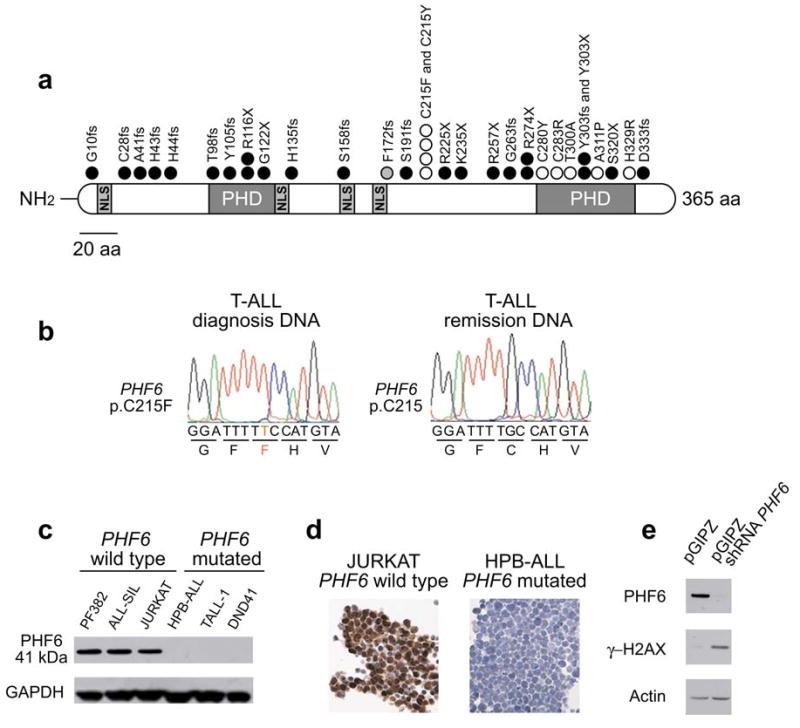
PHF6 mutations and expression in T-ALL lymphoblasts. (a) Structure of the PHF6 protein including four nuclear localization signals and two imperfect PHD zinc finger domains. Overview of all PHF6 mutations identified in primary T-ALL samples and T-ALL cell lines. Filled circles represent nonsense and frameshift mutations, whereas missense mutations are depicted as open circles. Circles filled in gray indicate mutations identified in female T-ALL cases. (b) Representative DNA sequencing chromatograms of paired diagnosis and remission genomic T-ALL DNA samples showing a somatic mutation in exon 7 of PHF6. (c) Western blot analysis of T-ALL cell lines revealed complete loss of PHF6 protein expression in the PHF6 mutated T-ALL cell lines. (d) PHF6 immunostaining in the Jurkat and HPB-ALL, wild-type and mutant T-ALL cell lines, respectively. (e) Western blot analysis of PHF6 and gamma-H2AX expression in HEK293T cells upon PHF6 shRNA knockdown. Actin levels are shown as loading control.
Nonsense and frame-shift mutations accounted for 70% (21/30) of all PHF6 mutations identified in our series and were evenly distributed throughout the gene. Missense mutations accounted for the remaining 30% (9/30) of PHF6 lesions and recurrently involved codon C215 and the second zinc finger domain of the protein (Fig. 2a). DNA sequence analysis of PHF6 in a panel of 15 well characterized T-ALL cell lines (Supplementary Table 3) showed the presence of truncating mutations in the PHF6 gene in the DND41, HPB-ALL and T-ALL1 cell lines. Western blot analysis and immunohistochemical staining of PHF6 demonstrated robust expression and nuclear localization of PHF6 in PHF6-wild-type tumors and complete loss of PHF6 protein in T-ALL cell lines harboring mutations in PHF6 (Fig. 2c,d).
PHD finger-containing proteins have been implicated in numerous cellular functions, including transcriptional regulation and in some instances as specialized reader modules that recognize the methylation status of histone lysine residues10. In addition, PHF6 has been reported to be phosphorylated during mitosis11 and by the ATM and ATR kinases upon DNA damage12, which suggests a dynamic regulation of PHF6 during cell cycle and DNA repair. Consistent with this notion, shRNA knockdown of PHF6 resulted in increased levels of phosphorylated H2AX (gamma-H2AX), a posttranslational modification associated with the presence of DNA double strand breaks13 (Fig. 2e).
Sex determination in humans is controlled by differential representation of the X and Y chromosomes, with presence of an XY pair in the male genome and two copies of chromosome X in females. The presence of numerous genes in the non-autosomal region of the X chromosome could result in a genetic imbalance between male and female cells, which is compensated by random chromosomal inactivation of one copy of chromosome X in female cells14. However, allelic expression analysis has shown that some genes can escape X chromosome inactivation in certain tissues1,2,15. To test the possibility that PHF6 could escape X-inactivation in T-ALL cells, we performed allelic expression analysis of a silent SNP (rs17317724) located in the 3′ untranslated region of PHF6 in lymphoblasts from 3 informative female T-ALL cases. In each of these samples, PHF6 was monoallelilically expressed, suggesting that biallelic expression of PHF6 is not commonly found in T-ALL (Fig. 3a). Most notably, we found that PHF6 mutations are almost exclusively found in male T-ALL cases. PHF6 mutations were present in 29/92 (32%) males and only in 1/39 (∼2.5%) females (P < 0.001; Fig. 3b and Supplementary Table 4). Moreover, all 8 PHF6 deletions identified by array-CGH analysis were found in male T-ALL cases, and each of the three cell lines with mutations in PHF6 were derived from male T-ALL cases.
Figure 3.
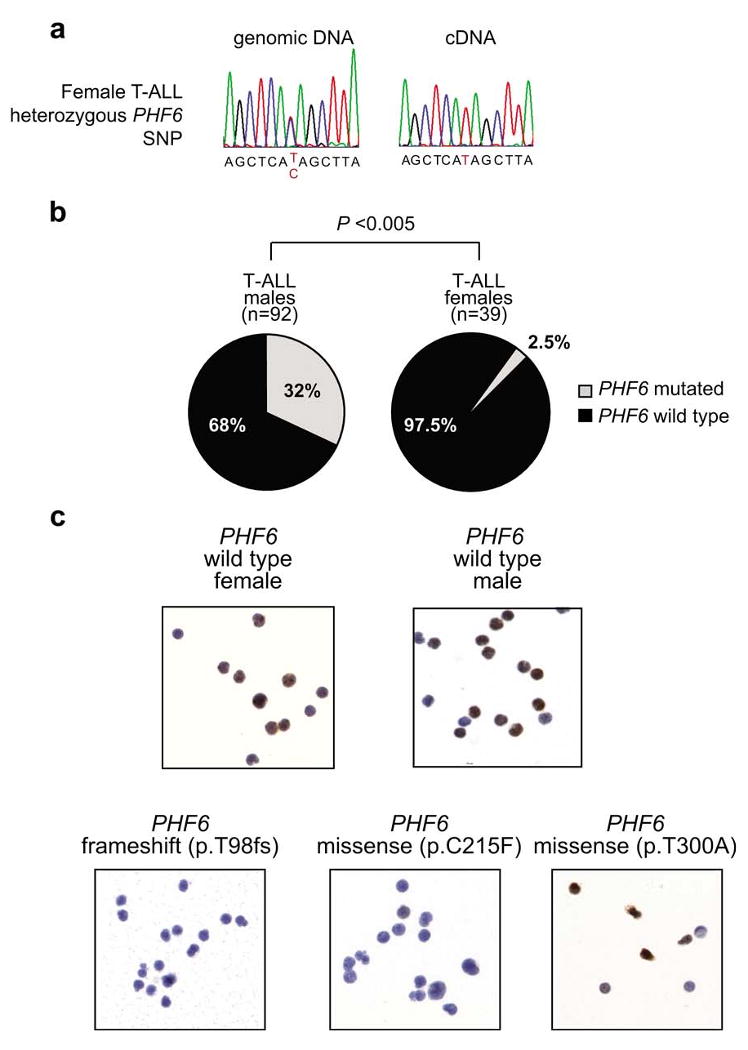
PHF6 expression in T-ALL lymphoblasts. (a) Sequence analysis of paired genomic DNA and cDNA samples shows monoallelic expression of PHF6 SNP rs17317724 in lymphoblasts from a wild-type PHF6 female T-ALL case. (b) Differential distribution of PHF6 mutations in T-ALL samples from male and female cases. (c) Immunohistochemical analysis of PHF6 expression in wild type and mutant T-ALL lymphoblasts.
Immunohistochemical analysis of PHF6 expression in wild-type primary T-ALL samples showed positive PHF6 immunostaining (n = 5; 3 males and 2 females), while cases with PHF6 truncating mutations (n = 4) (Fig. 3c) or a point mutation in C215 (p.C215F) were negative for PHF6 protein expression (Fig. 3c). In contrast, primary T-ALL cells harboring a PHF6 point mutation in the PHD2 domain (p.T300A) were positive for PHF6 protein expression (Fig. 3c). Overall, these results suggest that truncating mutations and point mutations in C215 impair PHF6 expression, while amino acid substitutions in the PHD2 domain of PHF6 may selectively impair the tumor suppressor function of this protein.
Leukemic transformation of immature thymocytes is the result of a multistep process involving numerous genetic abnormalities, which can be associated with different clinical features, including age and prognosis. Notably, PHF6 mutations were significantly more prevalent in adult (16/42; 38%) than in pediatric T-ALLs (14/89; 16%) (P = 0.005; Fig. 4a). Detailed genetic information was available for T-ALL cases treated in DCOG clinical trials (n = 65) (Supplementary Table 5). In this cohort, PHF6 mutations were significantly associated with the aberrant expression of TLX1 and TLX3 (P < 0.005; Fig. 4b and Supplementary Table 5), two related oncogenes activated by chromosomal translocations in T-ALL16-18. No significant associations were observed between PHF6 mutations and mutations in NOTCH1, FBXW7 or PTEN in either pediatric (n = 65) or adult (n = 34) T-ALL cohorts (Supplementary Tables 5 and 6). Overall survival in PHF6 wild-type pediatric T-ALL cases treated on DCOG protocols19 was 65% (33/51) vs. 71% (10/14) for PHF6-mutated cases (log-rank P = 0.71) (Fig. 4c). Overall survival in PHF6 wild-type adult T-ALL leukemias treated in the ECOG2993 clinical trial was 36% (7/12) vs. 58% (8/22) for PHF6-mutated samples (log-rank P = 0.24) (Fig. 4d).
Figure 4.

Clinical and biological characteristics associated with PHF6 mutations in T-ALL. (a) Frequencies of PHF6 mutations in pediatric and adult T-ALL samples. (b) Differential distribution of PHF6 mutations in TLX1/TLX3 positive and negative T-ALL samples. (c) Kaplan-Meier curve of overall survival in pediatric T-ALL patients from DCOG trials ALL7, ALL8 and ALL9 with and without PHF6 mutations. (d) Kaplan-Meier survival curve in adult T-ALL patients with and without mutations in PHF6 treated in ECOG clinical trial ECOG2993.
Overall, these results identify PHF6 as a new X-linked tumor suppressor gene and strongly suggest a specific interaction between the oncogenic programs activated by aberrant expression of TLX transcription factors and the mutational loss of PHF6 in the pathogenesis of T-ALL.
Supplementary Material
Acknowledgments
This study was supported by the Fund for Scientific Research (FWO) Flanders (postdoctoral grants to P.V.V. and T.T., PhD grant to J.V.M., senior clinical investigator award to B.P. and project grants G.0198.08 and G.0869.10N to F.S.); the GOA-UGent (grant no. 12051203); the IWT-Vlaanderen (SBO grant no. 060848); the Children Cancer Fund Ghent (F.S.); Leukemia Research UK (C.H.); the Stichting Kinderen Kankervrij (KiKa; Grant no. KiKa 2007-012 to L.Z.); the Belgian Program of Interuniversity Poles of Attraction; the Belgian Foundation Against Cancer; the Austrian Ministry of Science and Research (GEN-AU Child, GZ 200.136/1-VI/1/2005 to S.S.), the National Library of Medicine (1R01LM010140-01 to R.R. and H.K.); the ECOG and DCOG tumor banks; grants from Plan Nacional (BFU 2007-60990 and PlanE2009-0110 to M.L.T.), Comunidad de Madrid (S-SAL0304-2006 to M.L.T), Fundación MM (M.L.T), Instituto de Salud Carlos III (RECAVA RD06/0014/1012 to M.L.T), an Institutional Grant from the Fundación Ramón Areces (M.L.T.), the Alex's Lemonade Stand Foundation Young Investigator Award (T.P.); a Northeast Biodefence Center ARRA award (U54-AI057158 to R.R.); the National Institutes of Health (R01CA120196 and R01CA129382 to A.F.); the Rally Across America Foundation (A.F); the Swim Across America Foundation (A.F.) and the Golfers Against Cancer Foundation (A.F.). A.F. is a Leukemia & Lymphoma Society Scholar. We thank the Pediatric Cardiosurgery Units from Centro Especial Ramón y Cajal and Ciudad Sanitaria La Paz (Madrid, Spain) for thymus samples.
Footnotes
AUTHOR CONTRIBUTIONS: P.V.V. performed array CGH and mutation analysis of PHF6 and wrote the manuscript. T.P. performed exon capture and next-generation sequencing of T-ALL samples and wrote the manuscript. H.K. analyzed next-generation sequencing data. J.V.d.M. performed additional array-CGH analysis and PHF6 mutation screening in T-ALL and BCP-ALL samples. T.T., N.V.R. and A.W.L. performed experiments. M.C. and C.C.-C. performed and analyzed histological and immunohistochemical staining. J.P collaborated on PHF6 mutation screening in BCP-ALL samples. C.J.H. and C.S. collaborated on additional screening for genomic PHF6 deletions in T-ALL. Y.B., B.D.M and B.C. collaborated on the PHF6 mutation screening. R.P. M.P., S.S. and J.S. collaborated on the multi-center array-CGH study. S.G.-G. and M.L.T. performed the isolation of T-cell progenitor cells for expression analysis of PHF6. X.Y. performed survival analysis of ECOG T-ALL patients. J.G. provided critical reagents and discussion. E.S. provided samples and correlative clinical data from DCOG. E.P., J.M.R. and P.H.W. provided samples and correlative clinical data from ECOG. J.M. and L.Z. collaborated on the multi-center array-CGH study and PHF6 mutation analysis, provided molecular data on the characterization of T-ALL and performed survival analysis of PHF6 mutations in the DCOG series. R.R. designed and directed the analysis of next generation sequencing results. F.S. and B.P. designed the studies and directed research. A.F. designed the studies, directed research and wrote the manuscript.
References
- Carrel L, Cottle AA, Goglin KC, Willard HF. A first-generation X-inactivation profile of the human X chromosome. Proc Natl Acad Sci USA. 1999;96:14440–14444. doi: 10.1073/pnas.96.25.14440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- Goldberg JM, et al. Childhood T-cell acute lymphoblastic leukemia: the Dana-Farber Cancer Institute acute lymphoblastic leukemia consortium experience. J Clin Oncol. 2003;21:3616–3622. doi: 10.1200/JCO.2003.10.116. [DOI] [PubMed] [Google Scholar]
- Aifantis I, Raetz E, Buonamici S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol. 2008;8:380–390. doi: 10.1038/nri2304. [DOI] [PubMed] [Google Scholar]
- Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- Gnirke A, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lower KM, et al. Mutations in PHF6 are associated with Borjeson-Forssman-Lehmann syndrome. Nat Genet. 2002;32:661–665. doi: 10.1038/ng1040. [DOI] [PubMed] [Google Scholar]
- Borjeson M, Forssman H, Lehmann O. An X-linked, recessively inherited syndrome characterized by grave mental deficiency, epilepsy, and endocrine disorder. Acta Med Scand. 1962;171:13–21. doi: 10.1111/j.0954-6820.1962.tb04162.x. [DOI] [PubMed] [Google Scholar]
- Turner G, et al. The clinical picture of the Borjeson-Forssman-Lehmann syndrome in males and heterozygous females with PHF6 mutations. Clin Genet. 2004;65:226–232. doi: 10.1111/j.0009-9163.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- Baker LA, Allis CD, Wang GG. PHD fingers in human diseases: disorders arising from misinterpreting epigenetic marks. Mutat Res. 2008;647:3–12. doi: 10.1016/j.mrfmmm.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, et al. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci USA. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- Lowndes NF, Toh GW. DNA repair: the importance of phosphorylating histone H2AX. Curr Biol. 2005;15:R99–R102. doi: 10.1016/j.cub.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Payer B, Lee JT. X chromosome dosage compensation: how mammals keep the balance. Annu Rev Genet. 2008;42:733–772. doi: 10.1146/annurev.genet.42.110807.091711. [DOI] [PubMed] [Google Scholar]
- Carrel L, Willard HF. Heterogeneous gene expression from the inactive X chromosome: an X-linked gene that escapes X inactivation in some human cell lines but is inactivated in others. Proc Natl Acad Sci USA. 1999;96:7364–7369. doi: 10.1073/pnas.96.13.7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrando AA, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1:75–87. doi: 10.1016/s1535-6108(02)00018-1. [DOI] [PubMed] [Google Scholar]
- Soulier J, et al. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL) Blood. 2005;106:274–286. doi: 10.1182/blood-2004-10-3900. [DOI] [PubMed] [Google Scholar]
- Van Vlierberghe P, et al. The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood. 2008;111:4668–4680. doi: 10.1182/blood-2007-09-111872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Grotel M, et al. The outcome of molecular-cytogenetic subgroups in pediatric T-cell acute lymphoblastic leukemia: a retrospective study of patients treated according to DCOG or COALL protocols. Haematologica. 2006;91:1212–1221. [PubMed] [Google Scholar]
- Marks DI, et al. T-cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993) Blood. 2009;114:5136–5145. doi: 10.1182/blood-2009-08-231217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumble SM, et al. SHRiMP: accurate mapping of short color-space reads. PLoS Comput Biol. 2009;5:e1000386. doi: 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khiabanian H, VanVlierberghe P, Palomero T, Ferrando AA, Rabadan R. ParMap, an algorithm for the identification of complex genomic variations in nextgen sequencing data. Nature Precedings. posted online 12 January 2010 (hdl:10101/npre201041451) [Google Scholar]
- Clappier E, et al. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood. 2007;110:1251–1261. doi: 10.1182/blood-2006-12-064683. [DOI] [PubMed] [Google Scholar]
- Erdogan F, et al. Impact of low copy repeats on the generation of balanced and unbalanced chromosomal aberrations in mental retardation. Cytogenet Genome Res. 2006;115:247–253. doi: 10.1159/000095921. [DOI] [PubMed] [Google Scholar]
- Lahortiga I, et al. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat Genet. 2007;39:593–595. doi: 10.1038/ng2025. [DOI] [PubMed] [Google Scholar]
- Voss AK, et al. Protein and gene expression analysis of Phf6, the gene mutated in the Borjeson-Forssman-Lehmann Syndrome of intellectual disability and obesity. Gene Expr Patterns. 2007;7:858–871. doi: 10.1016/j.modgep.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garcia S, et al. CSL-MAML-dependent Notch1 signaling controls T lineage-specific IL-7Rα gene expression in early human thymopoiesis and leukemia. J Exp Med. 2009;206:779–791. doi: 10.1084/jem.20081922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.