

Abstract
The identification of activating mutations in NOTCH1 in over 50% of T-cell acute lymphoblastic leukemias (T-ALL) has generated major interest in the elucidation of the mechanisms of transformation downstream of oncogenic NOTCH and in the targeting of the NOTCH signaling pathway in this disease. Small molecule γ-secretase inhibitors (GSIs) block NOTCH1 signaling in T-ALL lymphoblasts, yet, the clinical development of GSIs has been held back by the development of gastrointestinal toxicity and their weak antileukemic effects against human T-ALL. However, new therapeutic strategies aiming to optimize the use of anti-NOTCH1 therapies for T-ALL, including combination therapies with molecularly targeted drugs and glucocorticoids, have started to emerge as result of improved understanding of the molecular mechanisms that mediate the effects of GSIs in leukemic cells and the intestinal epithelium. This review focuses on the molecular basis of NOTCH1-induced transformation, the mechanisms of action of oncogenic NOTCH1 and clinical significance of NOTCH1 mutations in T-ALL.
T-cell acute lymphoblastic leukemia (T-ALL) is characterized by infiltration of the bone marrow with immature lymphoblasts expressing T-cell immunophenotypic markers. T-ALL accounts for 10% to 15% of pediatric and 25% of adult acute lymphoblastic leukemia cases. T-ALL patients frequently show a large tumor burden with hyperleukocytosis, large mediastinal masses and pleural effusions. In addition, T-ALL patients have a higher risk of central nervous system infiltration at diagnosis. These aggressive features of T-ALL were originally associated with a very poor prognosis. Thus, in the early days of combination chemotherapy, when treatment of B-precursor ALL started to make significant progress in overall survival, results in T-ALL patients lagged behind with cure rates under 10%, due to a high incidence of relapse. Since then, therapy intensification has improved the outcome of T-ALL and current 5-year relapse-free survival rates are now over 75% in children and over 50% in adults. However, T-ALL remains a significant clinical problem. Thus the prognosis for those T-ALL patients who present with resistant disease or relapse after the induction of complete remission remains particularly poor, highlighting the need to develop more specific and highly effective antileukemic drugs. Although the specific biological and molecular mechanisms that account for the aggressiveness and poor therapy response of T-ALL remain to be elucidated, cytogenetic, molecular and genomic studies have shown that T-ALL is a disease primarily caused by aberrant activation of the NOTCH1 signaling pathway 1.
The NOTCH1 signaling pathway
The NOTCH1 receptor functions as a ligand activated transcription factor that directly transduces extracellular signals in the cell surface into changes in gene expression in the nucleus. The fundamental components of the NOTCH pathway include the Delta and Serrate family of ligands (Delta-like 1, 3 and 4; and Jagged 1 and 2), four distinct NOTCH receptors (NOTCH1-4), and the RBPJ/CSL (CBF1/Su(H)/LAG-1) DNA binding protein 1.
All four NOTCH receptors are class I transmembrane glycoproteins expressed at the cell surface as heterodimers consisting of an N-terminal extracellular (NEC) fragment and a C-terminal transmembrane-intracellular subunit (N™) (Figure 1) generated from a precursor polypeptide (proNOTCH1) by proteolytic processing by a furin protease in the trans-Golgi network. The NEC portion of the receptor consist of a number of epidermal growth factor (EGF)-like repeats which mediate the interaction with Delta-like and Jagged ligands; followed by three LIN-12/NOTCH repeats (LNR) 1. The NEC and N™ subunits remain associated in the resting receptor via the HD or heterodimerization domain, which consists of the C-terminal part of the NEC and the N-terminal part of the N™ subunits. The HD and LNR repeat domains are closely associated and fold together to form a molecular lock that plays a critical role in preventing the spontaneous activation of the receptor in the absence of ligand-receptor interaction. The N™ subunit contains a single-pass transmembrane domain followed by a RAM domain and a series of ankyrin repeats, a transactivation domain and several nuclear localization signals, which together function as a ligand activated transcription factor 1. Finally, the C-terminal part of the receptor contains a PEST [proline (P), glutamic acid (E), serine (S) and threonine (T) rich] sequence, which limits the intensity and duration of NOTCH activation via targeting of the activated receptor for proteasomal degradation in the nucleus.
Figure 1. The NOTCH1 signaling pathway.
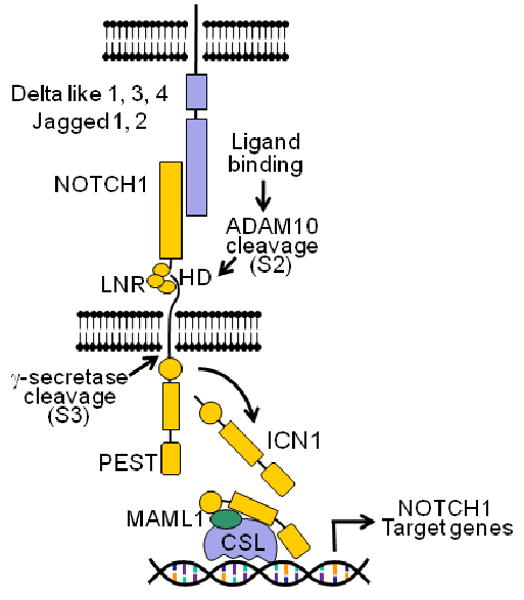
Interaction of the NOTCH1 receptor with Delta-like and Jagged ligands expressed on the surface of a neighbor cells triggers the proteolytic cleavage of the receptor, first by an ADAM metalloprotease (S2 cleavage) and subsequently by the γ-secretase complex (S3 cleavage), which releases the intracellular domains of NOTCH1 (ICN1) from the membrane. ICN1 translocates to the nucleus and interacts with DNA via the RBPJ/CSL DNA binding protein and recruits the MAML1 coactivator to activate the expression of NOTCH1 target genes.
Activation of NOTCH receptors typically occurs via cell-cell contact and interaction of a NOTCH protein with a Delta-like or Jagged ligand expressed on the surface of a neighboring cell. Current models of NOTCH activation support that ligand-receptor interaction induces a conformational change in the LNR repeats which results in: (i) dissociation of the HD subunits, (ii) exposure of an otherwise protected metalloprotease cleavage site in the C-terminal of the HD domain and (iii) release of the intracellular domain of NOTCH1 from the membrane via a double proteolytic processing of the receptor (Figure 1).
Two proteolytic systems mediate NOTCH activation. First, an extracellular metalloprotease typically ADAM10 or ADAM17, cleaves the receptor in the C-terminal portion of the HD domain (S2 cleavage), this generates a truncated protein with a short extracellular stub which is recognized and subsequently processed (S3 cleavage) by the γ-secretase complex, an aspartyl protease responsible for the endomembrane cleavage of NOTCH1 and the release of the intracellular domains of NOTCH in the cytosol. Once released from the membrane, the intracellular domains of NOTCH (ICN) rapidly translocate to the nucleus where they form a transcriptional complex with the RBPJ/CSL DNA binding protein and activate gene expression via recruitment of coactivators of the MAML family. Notably, transcriptional activation of NOTCH target genes is coupled with an active mechanism that ensures the rapid termination of NOTCH signaling. Thus, the recruitment of the RNA polymerase II holoenzyme to the ICN-MAML-RBPJ/CSL complex triggers phosphorylation of the PEST domain of the receptor, and its proteasomal degradation via the FBXW7-SCF ubiquitin ligase complex 1. In this context, cyclin-dependent kinase 8 has been proposed as one of the enzymes that may mediate phosphorylation of the NOTCH PEST domain. Thus, the NOTCH pathway could be considered a “quantum” signaling mechanism in which one receptor is activated by interaction with one ligand and is irreversibly processed to transduce this signal to the cell nucleus without the involvement of secondary messenger intermediates.
NOTCH1 in T-cell development
NOTCH receptors play a critical role in cell fate specification during development and participate in multiple biological processes. In the hematopoietic system NOTCH1 activation plays a critical role at multiple stages of T-cell development 2. Ablation of NOTCH1 function in hematopoietic progenitors results in a complete block at the earliest stages of T-cell lymphopoiesis due to a failure in the specification of the T-cell lineage 2. Thus, mice harboring a conditional deletion of NOTCH1 fail to develop T-cells, and show ectopic B-cell development in the thymus 2. Conversely, immunodeficient mice expressing a constitutively active form of Notch1 show ectopic T-cell development in the bone marrow and fail to produce B lymphocytes 2. In addition to this early role in T-cell lineage specification, NOTCH1 signaling is continuously required at different stages of T-cell development and plays a role in progression through the early DN1, DN2 and DN3 stages of thymocyte maturation; participates in the regulation of TCRB gene rearrangement; and regulates lineage decisions between αβ vs. γδ lineages and, at least in some systems, between CD4 vs. CD8 lineages 2.
Mechanisms of aberrant NOTCH1 activation in T-ALL
Constitutive activation of NOTCH1 in T-ALL was first demonstrated in rare leukemia cases harboring the t(7;9)(q34;q34.3) translocation, which juxtaposes a truncated NOTCH1 gene –missing the EGF-like, LNR and HD domains– next to the TCRB locus 3,4. Depending on the exact location of the translocation breakpoint and the translation initiation site these truncated NOTCH1 alleles encode membrane bound receptors that are readily processed by the γ-secretase complex due to the lack of HD-LNR repeats, or intracellular proteins devoid of transmembrane domains, which do not require γ-secretase processing for activation 3,4. The strong oncogenic activity of constitutively active NOTCH1 was demonstrated by the rapid development of T-ALL in mice transplanted with hematopoietic progenitors infected with retroviruses driving the expression of ICN1 1. Similarly, transgenic mice expressing ICN1 in hematopoietic progenitor cells or in immature thymocytes developed T-cell tumors 1. Moreover NOTCH1 activation is a common event in T-cell neoplasms induced by retroviral mutagenesis in mice 1. However, the significance of these findings for the pathogenesis of T-ALL remained elusive as aberrant activation of NOTCH1 in human tumors seemed to be restricted to only a handful of cases harboring the t(7;9) translocation.
This perception was radically changed with the identification of activating mutations in NOTCH1 leading to high levels of NOTCH1 signaling in over 60% of human T-ALLs 5. NOTCH1 mutations found in T-ALL affect critical domains responsible for preventing the spontaneous activation of the receptor in the absence of ligand or for terminating NOTCH1 signaling in the nucleus. The most frequent hotspots for mutations in NOTCH1 are exons 26 and 27, which encode the N-terminal and C-terminal components of the heterodimerization domain, respectively (Figures 2) 5. These so called HD mutations are found in approximately 40% of human T-ALLs (Figure 3).
Figure 2. Oncogenic forms of NOTCH1 in T-ALL.
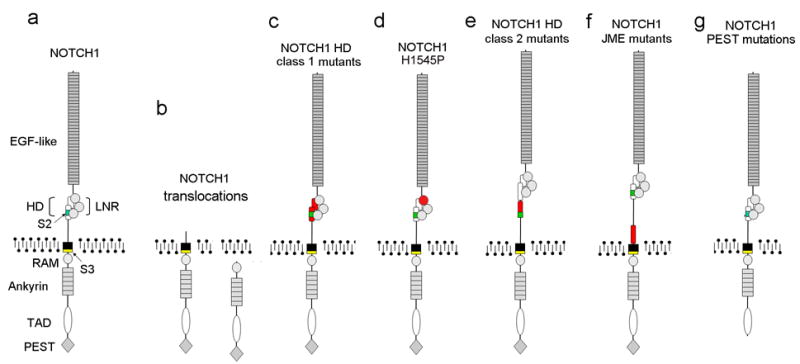
Aberrant activation of NOTCH signaling can be triggered by mutations in the NOTCH1 gene. (a) Structure of the wild type NOTCH1 receptor. Functional domains of NOTCH1 are annotated. EGF-like: EGF-like repeats. HD: heterodimerization domain. LNR: LNR repeats. RAM: RAM domain. Ankyrin: ankyrin repeats. TAD: transactivation domain. PEST: PEST domain. S2: metalloprotease cleavage site (green). S3: γ-secretase cleavage site (yellow). (b) Translocations of NOTCH1 to the TCR loci induce the expression of truncated forms of NOTCH1. (c) NOTCH1 HD class1 mutations destabilize the structure of the HD-LNR repeats responsible for maintaining the receptor in resting configuration. (d) The NOTCH1 H1545P mutation impairs the protection of the S2 cleavage site by the HD-LNR repeat complex. (e) NOTCH1 HD class2 mutations displace the S2 metalloprotease cleavage site outside the HD-LNR complex. (f) NOTCH1 JME alleles increase the separation of the HD-LNR repeat complex from the membrane. (g) NOTCH1 ΔPEST mutations delete the C-terminal part of the receptor and impairing the degradation of activated NOTCH1 in the nucleus. Sequences altered by the different NOTCH1 mutations are highlighted in red.
Figure 3. Prevalence of activating mutations in NOTCH1 in T-ALL.
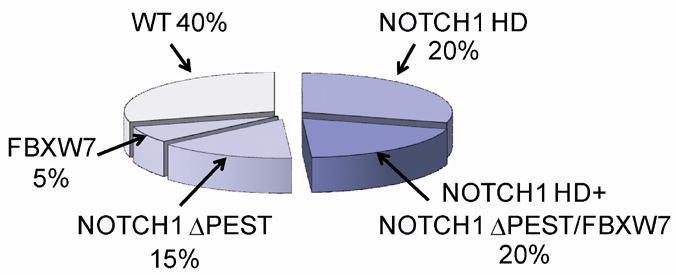
NOTCH1 HD and ΔPEST mutations account for the majority of activating mutations in NOTCH1 in T-ALL. An additional 1% of patients harbor translocations involving NOTCH1 and the TCR loci. NOTCH1 JME alleles are found in 3% of all T-ALLs. In total about 15% of T-ALL cases harbor deletions or mutations in FBXW7, which impair the degradation of activated NOTCH1 in the nucleus and are functionally related to NOTCH1 ΔPEST mutant alleles.
The resolution of the crystal structure of NOTCH1 and the LNR-HD complex explains the mechanism of action for NOTCH1 HD mutant alleles. In this model the N-terminal and C-terminal subunits of the HD domain are closely associated. The LNR repeats fold over the two HD subunits and hold them together 6. In addition, the third LNR domain of the receptor directly protects the metalloprotease S2 cleavage site located in the C-terminal HD subunit 6. Most NOTCH1 HD mutations (class 1, HD1) are typically single amino acid substitutions and small in-frame deletions and insertions (Figure 2). These mutations result in ligand-hypersensitivity or ligand-independent NOTCH1 activation by reducing the stability of the LNR-HD complex 7. A second, group of HD mutations (class 2, HD2) are longer insertions located at the distal part of the HD domain encoded in exon 27. These class 2 HD mutations result in high levels of ligand independent NOTCH1 activation by relocating the S2 metalloprotease cleavage site out of reach of the protective LNR-HD complex (Figure 2) 7. Recently, a novel mutation (NOTCH1 H1545P), located in the LNR repeats, has been shown to trigger ligand-independent increases in NOTCH1 signaling despite inducing only mild destabilization of the LNR-HD complex, suggesting that it may activate NOTCH1 cleavage by selectively releasing the inhibitory effect of the LNR-repeats on the ADAM cleavage site located in the C-terminal HD domain 6 (Figure 2). A fourth group of mutations resulting in aberrant NOTCH1 activation at the membrane are Juxtamembrane Expansion Mutants or JME NOTCH1 alleles 8. These mutations consist of internal tandem duplications in the 3′ end of intron 27 and/or in the proximal region of exon 28 which displace the LNR-HD complex away from the membrane without altering the primary structure of any of its components (Figure 2). Although the exact mechanism mediating the activation of JME NOTCH1 mutants is not fully understood yet, activation of JME NOTCH1 alleles requires ADAM and γ-secretase processing and results in very high levels of NOTCH1 signaling.
NOTCH1 ΔPEST mutations, present in 20-25% of T-ALLs, are typically frameshift or nonsense nucleotide substitutions generating premature stop codons in the C-terminal portion of the receptor (Figure 3). The loss of the PEST domain results in increased levels of activated NOTCH1 due to impaired degradation of the activated receptor by the proteasome 5,9. Similarly, 15% of T-ALL cases harbor mutations or deletions in FBXW7 9-11. FBXW7 mutations found in T-ALL cluster in three critical arginine residues involved in the interaction of this F-box protein with NOTCH1 (Figure 3). Proteins encoded by the FBXW7 mutants have impaired substrate recognition function and promote increased stability of activated NOTCH1, mimicking the effect of NOTCH1 ΔPEST mutations 9,10. However, FBXW7 also mediates the proteasomal degradation of additional oncoproteins such as c-MYC, JUN, Cyclin E and mTOR 12. Thus, the biological consequences of FBXW7 inactivation in T-ALL lymphoblasts are broader than those derived from deletion of the NOTCH1 PEST domain and may include increased cell metabolism, cell growth and cell cycle progression.
Importantly, about 15% of T-ALLs present NOTCH1 HD and ΔPEST mutations in cis, this is in the same NOTCH1 transcript (Figure 3) 5. Similarly, about 5% of T-ALLs harbor a FBXW7 mutations and a concomitant NOTCH1 HD mutation (Figure 3) 9,10. The presence of a NOTCH1 HD allele and a PEST deletion or FBXW7 mutation induces ligand independent activation at the membrane and impaired degradation of the activated receptor in the nucleus resulting in very high levels of NOTCH1 signaling 5,9.
The presence of multiple hits promoting NOTCH1 signaling in the same leukemic clone shows a lasting pressure to acquire ever higher levels of NOTCH1 signaling during T-cell transformation and is consistent with experimental data showing that different NOTCH mutant alleles have marked differences in their potential to induce T-cell transformation in mice 13. Expression of NOTCH1 mutations inducing high levels of NOTCH1 activation such as NOTCH1 HD1-ΔPEST and NOTCH1 HD2 alleles in hematopoietic progenitors promoted T-cell transformation 13. However, more common, weaker alleles, such as NOTCH1 HD1 or ΔPEST mutations forced the differentiation of hematopoietic progenitors to the T-cell lineage but were not sufficient to induce the development of overt leukemia 13. Thus, not all NOTCH1 mutations seem to be equal and differences in the level of NOTCH signaling may be strongly associated with different roles in T-ALL. Strong NOTCH1 alleles may work as drivers of T-cell transformation while weaker alleles may have a more limited role in promoting T-cell lineage specification and further growth and proliferation in preleukemic cells harboring other oncogenic lesions. Notably, weak NOTCH1 alleles which failed to induce T-ALL by themselves accelerated T-cell transformation in hematopoietic progenitors expressing the k-ras oncogene 13. Most notably, despite the presumably secondary role of the NOTCH1 alleles in T-cell transformation in NOTCH1 plus k-ras tumors, these leukemias were still dependent on continuous NOTCH signaling for proliferation and survival 13. Determining whether NOTCH1 mutations in human T-ALL show a similar functional heterogeneity may have important implications for the design of anti-NOTCH1 therapies in T-ALL.
The identification of a NOTCH1 HD mutation in neonatal blood spots of a pediatric patient developing a T-ALL at 6 years of age show that NOTCH1 mutations may occur as initiating events in preleukemic clones of prenatal origin 14. However, subclonal NOTCH1 mutations are frequently detected in the analysis of T-ALL samples and a switch from NOTCH1 mutated leukemia at diagnosis to a NOTCH1 wild type tumor or to a different NOTCH1 mutated clone at relapse is not rare 15. Therefore, NOTCH1 mutations may work as initiating events or as secondary events during disease progression in the pathogenesis of T-ALL.
NOTCH1 mutations as prognostic markers in T-ALL
Analysis of the prognostic significance of NOTCH activation in T-ALL has shown that NOTCH1 mutations are not associated with unfavorable outcome and that in some series they may confer better prognosis. Breit et al. analyzed the prognostic significance of NOTCH1 mutations in 157 pediatric T-ALL patients from the pediatric ALL-Berlin-Frankfurt-Munster (BFM) 2000 study and found that NOTCH1 mutations were associated with favorable minimal residual disease and improved relapsed free survival 16. Similarly, combined analysis of NOTCH1 mutations and FBXW7 mutations in 55 T-ALL and 14 T-cell lymphoblastic lymphoma patients treated in the Japan Association of Childhood Leukemia Study (JACLS) protocols ALL-97 and NHL-98 showed an improved outcome for patients harboring NOTCH1 and/or FBXW7 mutations 17. In contrast, analysis of a series of 72 pediatric T-ALL patients, treated in the Dutch Childhood Oncology Group (DCOG) protocols ALL-7, ALL-8 or ALL-9 failed to demonstrate a different prognosis for patients whose leukemias harbored activating mutations in NOTCH1, suggesting that differences in therapy may influence the effect of NOTCH1 mutations on prognosis 18.
In the case of adult T-ALL, analysis of 141 samples from patients treated the Lymphoblastic Acute Leukemia in Adults (LALA)-94 (n = 87) and the GRAALL-2003 (n = 54) trials found a positive association of NOTCH1 and/or FBXW7 mutations with favorable prognosis 11. However, a recent analysis of a series of 88 adult T-ALL patients treated according to the MRC UKALLXII/ECOGE2993 protocol failed to detect significant differences in outcome associated with the presence of NOTCH1 and or FBXW7 mutations 19. Thus, as in the case of children, the prognostic impact of NOTCH1 mutations in adults with T-ALL may be influenced by differences in therapy.
NOTCH1 as therapeutic target in T-ALL
The fundamental importance of NOTCH1 mutations in T-ALL is highlighted by the potential role of NOTCH1 as a molecular therapeutic target for the treatment of this disease. Given the strict requirement of γ-secretase cleavage for NOTCH1 activation, pharmacologic inhibition of this proteolytic complex effectively abrogates oncogenic NOTCH1 signaling in T-ALL cells. Importantly, the presenilin γ-secretase complex plays a central role in Alzheimer's disease. Thus, pathologic processing of the APP amyloid precursor protein by γ-secretase generates amyloidogenic Aβ peptides whose accumulation in neurodegenerative plaques constitutes one of the hallmark events contributing to the pathogenesis of Alzheimer's disease.
Small molecule γ-secretase inhibitors (GSIs), block the generation of amyloidogenic Aβ peptides and may contribute to slow down the progression of human dementia. Notably, these inhibitors, originally designed to inhibit the processing of APP in Alzheimer's disease, can also effectively block NOTCH1 signaling in T-ALL. Thus GSI treatment of T-ALL cell lines harboring activating mutations in NOTCH1 induced rapid clearance of intracellular activated NOTCH1 protein and transcriptional downregulation of NOTCH1 target genes 4,5,20. Moreover, these biochemical and transcriptional changes were associated with the induction of G1 cell cycle arrest and a decrease in cell size in some T-ALL cell lines 4,5,20-23.
These results, stimulated the initiation of the Dana-Farber Cancer Institute 04-390 trial, an open label, non-randomized, phase I clinical trial testing the activity of MK-0752, an oral GSI developed by Merck for the treatment of relapsed T-ALL patients 24. This study initially enrolled 7 T-ALL patients, 4 of whom showed mutations in NOTCH1. However, despite some correlative data showing downregulation of NOTCH1 target genes in T-cell lymphoblasts, there were no objective clinical responses. In addition a significant number of patients showed marked fatigue and dose limiting gastrointestinal toxicity, most probably derived from inhibition of NOTCH signaling in the gut 24. A modification in the protocol seeking to reduce gut toxicity using a 3 days on-4 days off dosing schedule was introduced. However, the study was closed in October of 2006 having achieved no objective responses.
The dismal results of the DFCI 04-390 trial highlighted several major problems in the clinical development of GSIs as anti-NOTCH1 therapies for the treatment of T-ALL. First, GSIs seem to exert primarily a cytostatic effect against human T-ALLs with minimal or no apoptosis 4,5. This limited antileukemic effect of GSIs against T-ALL was most probably responsible for the lack of effective clinical responses in this trial. However it should also be noted that, most human T-ALL cell lines harboring activating mutations in NOTCH1 do not require continuous NOTCH1 signaling for proliferation and survival and show primary resistant to GSIs 5,22. Should this also be the case of primary T-ALL lymphoblasts, this could an additional problem contributing to the lack of response to anti-NOTCH1 therapies. Finally, the gastrointestinal toxicity observed in T-ALL patients treated with MK-0752 is most probably an on target side effect as a number of experimental models have shown that systemic inhibition of NOTCH signaling induces cell cycle arrest and accumulation of mucus-secreting goblet cells in the intestinal epithelium 25. Overcoming these difficulties will require improved understanding of the oncogenic mechanisms controlled by NOTCH1 in T-ALL and a better appreciation of the genes and pathways that mediate the effects of GSIs in both T-cell lymphoblasts and intestinal progenitor cells.
What are the genes and pathways controlled by NOTCH1 in T-cell transformation?
Despite the prominent role of NOTCH1 in T-cell development and T-ALL, only a handful of NOTCH1 target genes had been identified at the time that GSIs entered clinical trials for the treatment of T-ALL. Since then, a number of studies have addressed the identification of genetic programs and direct target genes downstream of NOTCH1 in T-ALL. These analyses have shown that the genetic program controlled by oncogenic NOTCH1 in T-ALL is dominated by genes involved in anabolic pathways. Thus inhibition of NOTCH signaling with a GSI in T-ALL induces the transcriptional downregulation of ribosome biosynthesis, protein translation, and nucleotide and amino acid metabolism genes 20,21. Strikingly, ChIP-on-chip analysis of NOTCH1 targets showed that many of these genes are directly controlled by oncogenic NOTCH1 20. However the role of NOTCH1 in promoting cell growth and metabolism goes beyond the direct transcriptional upregulation of these direct target genes. Thus NOTCH1 directly promotes the expression of the cMYC oncogene, 20,21,26 which in turn, further enhances multiple processes involved in anabolic cell growth 20,27. Indeed, many of the anabolic genes directly controlled by NOTCH1 are also direct targets of c-MYC 20. This NOTCH1-MYC feed forward-loop transcriptional regulatory network highlights the central role of anabolic pathways controlled by NOTCH1 in promoting cell growth and T-cell transformation.
A prominent role of NOTCH1 as a regulator of cell growth is further supported by evidence indicating that activation of the PI3K-AKT-mTOR pathway is an important effector downstream of NOTCH1 activation in T-cell progenitors and in T-ALL. The PI3K-AKT signal transduction pathway mediates multiple cellular responses triggered by the engagement of growth factor receptors, including increased cell growth, proliferation and survival and its function is antagonized by the PTEN tumor suppressor gene 22. In a seminal paper on the role of NOTCH signaling T-cell precursors, Ciofani and coworkers showed that Notch signals regulate cell growth (cell size, glucose uptake and glycolysis) and promote cell survival via activation of the PI3K-Akt signaling pathway 28. In addition, phosphoproteomic analysis of T-ALL cell lines treated with NOTCH1 inhibitors showed a marked decrease in the phosphorylation of mTOR targets 23. NOTCH1 signaling seems to promote increased PI3K-AKT-mTOR signaling by multiple mechanisms. Early work by Sade and coworkers showed that NOTCH1 can activate AKT via the p56lck tyrosine kinase in T-cells 29. In addition, NOTCH1 can enhance the activity of the PI3K signaling pathway in T-cell progenitors and T-ALL lymphoblasts via transcriptional downregulation of the PTEN tumor suppressor gene by HES1, a transcriptional repressor directly downstream of NOTCH1 signaling 22.
In addition to promoting cell anabolism, oncogenic NOTCH1 signaling has a direct proliferative effect on T-cell precursors and promotes G1/S cell cycle progression in T-ALL 30-32. NOTCH1 activation upregulates the expression of CCND3, CDK4, and CDK6 in T-ALL and CCND3 is a direct NOTCH1 target gene 30. In addition, Notch1 induces transcription of the S phase kinase-associated protein 2 (SKP2), and the proteasome-mediated degradation of CDKN1B (p27/Kip1) and CDKN1A (p21/Cip1), promoting premature entry into S phase in hematopoietic progenitors 31. Moreover, inhibition of NOTCH signaling in T-ALL cell lines induced upregulation of the cyclin-dependent kinase inhibitors CDKN2D (p19/INK4d) and CDKN1B (p27/Kip1), leading to derepression of RB and subsequent exit from the cell cycle 32.
Finally, oncogenic NOTCH1 interacts with important signaling pathways controlling the growth, proliferation and survival of T-ALL cells. Thus, NOTCH1 signaling enhances NFKB activity by increasing NFKB expression 9, promoting NFKB nuclear retention 35 and via activation of IκB kinase 36. In addition, NOTCH1 directly controls the expression of the interleukin 7 receptor alpha chain in T-cell progenitors and T-ALL 33 and may also regulate the activity of p53 34.
Overall, accumulating evidence supports a central role of NOTCH1 signaling in promoting cell metabolism, growth and proliferation and in enhancing the activity of signaling pathways that reinforce these functions and also promote cell survival. An intriguing finding with the potential of having a major impact in the clinic is the observation that ex vivo inhibition of NOTCH signaling in human primary T-ALL cells can block the engraftment of these tumors in immunodeficient mice. These results suggest that blocking NOTCH1 signaling may reduce the self renewal capacity of T-ALL lymphoblasts and/or selectively affect the leukemia initiating cell population 37.
Modulators of clinical response to GSI in T-ALL
Despite the limited clinical experience with the use of anti-NOTCH therapies in T-ALL, the results of the DFCI 04-390 protocol seem to indicate that at least some T-ALLs treated with maximum tolerated doses of the MK-0752 GSI developed gastrointestinal toxicity associated with inhibition of NOTCH signaling in the gut but failed to show a clinical response. These results suggest that inhibition of NOTCH signaling has at best a modest antileukemic effect against human primary T-ALL lymphoblasts. In fact, human T-ALL cell lines harboring activating mutations in NOTCH1 show only a delayed and modest cytostatic response with minimal or no apoptosis upon treatment with GSIs 4,5, and many of them are completely resistant to NOTCH1 inhibition 5. Notably, this is in contrasts with mouse T-cell leukemias induced by constitutive activation of NOTCH1 in which inhibition of NOTCH signaling with a GSI induces a robust antileukemic effect and a clear apoptotic response 13,38.
The identification of cell growth and metabolism as critical downstream effectors of NOTCH signaling suggests that additional mutations in oncogenic pathways promoting cell growth may ameliorate the effects of inhibition of NOTCH signaling in T-ALL lymphoblasts. Analysis of GSI sensitive and resistant T-ALL cell lines has shown that mutational loss of PTEN and consequent constitutive activation of the PI3K-AKT-mTOR pathway can induce GSI resistance by bypassing the requirement for NOTCH1 signaling for continuous growth and metabolism 22. In addition, forced expression of c-MYC can overcome the cell cycle arrest induced by NOTCH1 inhibition in some T-ALL cell lines 23. Finally, mutations in FBXW7 are more prevalent in GSI resistant T-ALL cell lines suggesting that increased c-MYC, JUN, cyclin E and mTOR protein stability may lead to reduced sensitivity to NOTCH1 inactivation in these tumors 9,10. These studies in T-ALL cell lines warrant the prospective evaluation of the role of PTEN loss, MYC overexpression and FBXW7 mutations in the resistance to NOTCH1 inhibition in the clinic.
Intestinal toxicity due to inhibition of NOTCH signaling
NOTCH signaling plays a critical role in lineage specification decisions by which equivalent multipotential precursor cells become committed to specific cell lineages in multiple tissues. Intestinal epithelium cells express Notch1 and Notch2 receptors and NOTCH signaling plays an important role in homeostasis of intestinal progenitors 39. Inactivating Notch signaling by conditional targeting of RBPJ/CSL or via conditional knockout of Notch1 and Notch2 in the gut results in increased numbers of secretory cells at the expense of the absorptive enterocytes and induces a marked block in cell proliferation in the intestinal crypt. Similarly, inhibition of NOTCH signaling with a GSI induces secretory metaplasia in the gut characterized primarily by the accumulation of mucus-secreting goblet cells and the loss of the proliferative compartment 25. In addition, deletion of Hes1, a transcriptional repressor directly controlled by Notch resulted in the generation of excessive numbers of secretory cells 25. Notably, Klf4, a transcription factor inhibitor of cell cycle progression and a critical factor required for the generation of intestinal goblet cells, is related to the two main histological features associated with GSI-induced gut toxicity, namely, accumulation of secretory goblet cells and a block in cell proliferation 25. A role of Klf4 in GSI induced gut toxicity is supported by the demonstration that Klf4 is a HES1 direct target gene whose expression is downregulated by NOTCH signaling in the gut 25. Consequently, inhibition of NOTCH signaling induces Klf4 upregulation in the intestine of mice treated with a GSI 25. Given the pleiotrophic effects of NOTCH signaling, multiple factors will probably contribute to the pathogenesis of GSI-induced gut toxicity, however, the identification of Klf4 as an indirect target downregulated by NOTCH1 signaling strongly points to Klf4 as a critical mediator of GSI-induced gut toxicity 25.
Combination therapies with GSIs in the treatment of T-ALL
It is clear that the development of successful anti-NOTCH therapies for T-ALL will require strategies to increase the antileukemic effects of these drugs and to overcome the obstacle of GSI induced gut toxicity. In this regard, the combination of GSIs with CDK inhibitors 32, drugs targeting NFKB signaling 9, or small molecule inhibitors of the PI3K-AKT-mTOR pathway 22,23,38 may increase the antileukemic effects of these panNOTCH inhibitors. In addition, a number of approaches including intermittent treatment schedules and parenteral formulations of GSIs may facilitate increased tolerance to GSI therapy. However, the most straight forward strategy to introduce GSIs in the forefront of T-ALL therapy would probably consist of combination therapies with the core drugs used in the treatment of T-ALL. The observation that constitutive activation of NOTCH1 signaling can protect developing thymocytes against glucocorticoid-induced apoptosis suggested that GSIs and glucocorticoids could have a synergistic antileukemic effect in T-ALL25. Additionally, T-ALL cell lines treated for two weeks with a GSI showed increased responses to glucocorticoids treatment 40 and inhibition of NOTCH signaling with a GSI sensitized glucocorticoid-resistant T-ALL cell lines to glucocorticoid induced apoptosis 25. Most notably, in vivo testing of GSIs and glucocorticoids in combination in a mouse model of glucocorticoid resistant T-ALL showed that glucocorticoid treatment has a direct protective effect against GSI-induced intestinal toxicity in mice 25. Overall these results strongly suggest that glucocorticoid treatment may enhance the antileukemic effects of GSIs and ameliorate the intestinal toxicity typically associated with systemic inhibition of NOTCH signaling.
Concluding remarks and future directions
Recent progress in the characterization of the mechanisms that mediate the oncogenic activity of NOTCH signaling have opened new possibilities for the development of anti-NOTCH therapies in the treatment of T-ALL. In addition, numerous recent reports have described a role for aberrant NOTCH signaling in the pathogenesis of solid tumors and tumor angiogenesis, suggesting a broader role for anti-NOTCH therapies in the treatment of human cancer. Overall these results have created a renewed interest in the NOTCH signaling pathway as a therapeutic target and generated momentum for the design of new clinical trials testing the efficacy and safety of anti-NOTCH therapies in the clinic.
Acknowledgments
Supported by the NIH (grants R01CA120196 and R01CA129382) and the Leukemia and Lymphoma Society (grants 1287-08 and 6237-08) (AF). Adolfo Ferrando is a Leukemia & Lymphoma Society Scholar.
References
- 1.Aster JC, Pear WS, Blacklow SC. Notch Signaling in Leukemia. Annu Rev Pathol. 2008;3:587–613. doi: 10.1146/annurev.pathmechdis.3.121806.154300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tanigaki K, Honjo T. Regulation of lymphocyte development by Notch signaling. Nat Immunol. 2007;8:451–456. doi: 10.1038/ni1453. [DOI] [PubMed] [Google Scholar]
- 3.Ellisen LW, Bird J, West DC, et al. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991;66:649–661. doi: 10.1016/0092-8674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- 4.Palomero T, Barnes KC, Real PJ, et al. CUTLL1, a novel human T-cell lymphoma cell line with t(7;9) rearrangement, aberrant NOTCH1 activation and high sensitivity to gamma-secretase inhibitors. Leukemia. 2006;20:1279–1287. doi: 10.1038/sj.leu.2404258. [DOI] [PubMed] [Google Scholar]
- 5.Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 6.Gordon WR, Roy M, Vardar-Ulu D, et al. Structure of the Notch1-negative regulatory region: implications for normal activation and pathogenic signaling in T-ALL. Blood. 2009;113:4381–4390. doi: 10.1182/blood-2008-08-174748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malecki MJ, Sanchez-Irizarry C, Mitchell JL, et al. Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol Cell Biol. 2006;26:4642–4651. doi: 10.1128/MCB.01655-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sulis ML, Williams O, Palomero T, et al. NOTCH1 extracellular juxtamembrane expansion mutations in T-ALL. Blood. 2008;112:733–740. doi: 10.1182/blood-2007-12-130096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson BJ, Buonamici S, Sulis ML, et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med. 2007;204:1825–1835. doi: 10.1084/jem.20070872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Neil J, Grim J, Strack P, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204:1813–1824. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asnafi V, Buzyn A, Le Noir S, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood. 2009;113:3918–3924. doi: 10.1182/blood-2008-10-184069. [DOI] [PubMed] [Google Scholar]
- 12.Minella AC, Clurman BE. Mechanisms of tumor suppression by the SCF(Fbw7) Cell Cycle. 2005;4:1356–1359. doi: 10.4161/cc.4.10.2058. [DOI] [PubMed] [Google Scholar]
- 13.Chiang MY, Xu L, Shestova O, et al. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J Clin Invest. 2008;118:3181–3194. doi: 10.1172/JCI35090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eguchi-Ishimae M, Eguchi M, Kempski H, Greaves M. NOTCH1 mutation can be an early, prenatal genetic event in T-ALL. Blood. 2008;111:376–378. doi: 10.1182/blood-2007-02-074690. [DOI] [PubMed] [Google Scholar]
- 15.Mansour MR, Duke V, Foroni L, et al. Notch-1 mutations are secondary events in some patients with T-cell acute lymphoblastic leukemia. Clin Cancer Res. 2007;13:6964–6969. doi: 10.1158/1078-0432.CCR-07-1474. [DOI] [PubMed] [Google Scholar]
- 16.Breit S, Stanulla M, Flohr T, et al. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood. 2006;108:1151–1157. doi: 10.1182/blood-2005-12-4956. [DOI] [PubMed] [Google Scholar]
- 17.Park MJ, Taki T, Oda M, et al. FBXW7 and NOTCH1 mutations in childhood T cell acute lymphoblastic leukaemia and T cell non-Hodgkin lymphoma. Br J Haematol. 2009 doi: 10.1111/j.1365-2141.2009.07607.x. [DOI] [PubMed] [Google Scholar]
- 18.van Grotel M, Meijerink JP, Beverloo HB, et al. The outcome of molecular-cytogenetic subgroups in pediatric T-cell acute lymphoblastic leukemia: a retrospective study of patients treated according to DCOG or COALL protocols. Haematologica. 2006;91:1212–1221. [PubMed] [Google Scholar]
- 19.Mansour MR, S M, Duke V, Foroni L, Jenkinson S, Koo K, Allen CG, Gale RE, Buck G, Richards S, Paietta E, Rowe JM, Tallman MS, Goldstone AH, Ferrando AA, Linch DC. Relationship and prognostic implications of NOTCH1 and FBXW7 mutations in adult patients with T-cell acute lymphoblastic leukaemia treated on the UKALLXII/ECOG2993 protocol. Journal of Clinical Oncology. 2009 doi: 10.1200/JCO.2009.22.0996. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palomero T, Lim WK, Odom DT, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103:18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weng AP, Millholland JM, Yashiro-Ohtani Y, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20:2096–2109. doi: 10.1101/gad.1450406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palomero T, Sulis ML, Cortina M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13:1203–1210. doi: 10.1038/nm1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan SM, Weng AP, Tibshirani R, Aster JC, Utz PJ. Notch signals positively regulate activity of the mTOR pathway in T-cell acute lymphoblastic leukemia. Blood. 2007;110:278–286. doi: 10.1182/blood-2006-08-039883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deangelo D, Stone R, Silverman L, et al. A phase I clinical trial of the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. Journal of Clinical Oncology, 2006 ASCO Annual Meeting Proceedings Part I. 2006;24:6585. [Google Scholar]
- 25.Real PJ, Tosello V, Palomero T, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med. 2009;15:50–58. doi: 10.1038/nm.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma VM, Calvo JA, Draheim KM, et al. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol Cell Biol. 2006;26:8022–8031. doi: 10.1128/MCB.01091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Margolin AA, Palomero T, Sumazin P, Califano A, Ferrando AA, Stolovitzky G. ChIP-on-chip significance analysis reveals large-scale binding and regulation by human transcription factor oncogenes. Proc Natl Acad Sci U S A. 2009;106:244–249. doi: 10.1073/pnas.0806445106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciofani M, Zuniga-Pflucker JC. Notch promotes survival of pre-T cells at the beta-selection checkpoint by regulating cellular metabolism. Nat Immunol. 2005;6:881–888. doi: 10.1038/ni1234. [DOI] [PubMed] [Google Scholar]
- 29.Sade H, Krishna S, Sarin A. The anti-apoptotic effect of Notch-1 requires p56lck-dependent, Akt/PKB-mediated signaling in T cells. J Biol Chem. 2004;279:2937–2944. doi: 10.1074/jbc.M309924200. [DOI] [PubMed] [Google Scholar]
- 30.Joshi I, Minter LM, Telfer J, et al. Notch signaling mediates G1/S cell-cycle progression in T cells via cyclin D3 and its dependent kinases. Blood. 2009;113:1689–1698. doi: 10.1182/blood-2008-03-147967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dohda T, Maljukova A, Liu L, et al. Notch signaling induces SKP2 expression and promotes reduction of p27Kip1 in T-cell acute lymphoblastic leukemia cell lines. Exp Cell Res. 2007;313:3141–3152. doi: 10.1016/j.yexcr.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 32.Rao SS, O'Neil J, Liberator CD, et al. Inhibition of NOTCH signaling by gamma secretase inhibitor engages the RB pathway and elicits cell cycle exit in T-cell acute lymphoblastic leukemia cells. Cancer Res. 2009;69:3060–3068. doi: 10.1158/0008-5472.CAN-08-4295. [DOI] [PubMed] [Google Scholar]
- 33.González-García Sara, GP M, Martín-Gayo Enrique, Ballestar Esteban, Esteller Manel, Bornstein Rafael, Luis de la Pompa José, Ferrando Adolfo A, Toribio María L. CSL/MALM-dependent Notch1 signaling controls T lineage-specific IL-7Ra gene expression in early human thymopoiesis and leukemia. J Exp Med. 2009 doi: 10.1084/jem.20081922. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beverly LJ, Felsher DW, Capobianco AJ. Suppression of p53 by Notch in lymphomagenesis: implications for initiation and regression. Cancer Res. 2005;65:7159–7168. doi: 10.1158/0008-5472.CAN-05-1664. [DOI] [PubMed] [Google Scholar]
- 35.Shin HM, Minter LM, Cho OH, et al. Notch1 augments NF-kappaB activity by facilitating its nuclear retention. EMBO J. 2006;25:129–138. doi: 10.1038/sj.emboj.7600902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song LL, Peng Y, Yun J, et al. Notch-1 associates with IKKalpha and regulates IKK activity in cervical cancer cells. Oncogene. 2008;27:5833–5844. doi: 10.1038/onc.2008.190. [DOI] [PubMed] [Google Scholar]
- 37.Armstrong F, de la Grange PB, Gerby B, et al. NOTCH is a key regulator of human T-cell acute leukemia initiating cell activity. Blood. 2009;113:1730–1740. doi: 10.1182/blood-2008-02-138172. [DOI] [PubMed] [Google Scholar]
- 38.Cullion K, Draheim KM, Hermance N, et al. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood. 2009 doi: 10.1182/blood-2008-02-136762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riccio O, van Gijn ME, Bezdek AC, et al. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27(Kip1) and p57(Kip2) EMBO Rep. 2008;9:377–383. doi: 10.1038/embor.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Keersmaecker K, Lahortiga I, Mentens N, et al. In vitro validation of gamma-secretase inhibitors alone or in combination with other anti-cancer drugs for the treatment of T-cell acute lymphoblastic leukemia. Haematologica. 2008;93:533–542. doi: 10.3324/haematol.11894. [DOI] [PubMed] [Google Scholar]