

Abstract
It is demonstrated that it is possible to coat the individual fibers of wood-based nanocellulose with polypyrrole using in situ chemical polymerization to obtain an electrically conducting continuous high-surface-area composite. The experimental results indicate that the high surface area of the water dispersed material, to a large extent, is maintained upon normal drying without the use of any solvent exchange. Thus, the employed chemical polymerization of polypyrrole on the microfibrillated cellulose (MFC) nanofibers in the hydrogel gives rise to a composite, the structure of which—unlike that of uncoated MFC paper—does not collapse upon drying. The dry composite has a surface area of ∼90 m2/g and a conductivity of ∼1.5 S/cm, is electrochemically active, and exhibits an ion-exchange capacity for chloride ions of 289 C/g corresponding to a specific capacity of 80 mAh/g. The straightforwardness of the fabrication of the present nanocellulose composites should significantly facilitate industrial manufacturing of highly porous, electroactive conductive paper materials for applications including ion-exchange and paper-based energy storage devices.
Introduction
Microfibrillated cellulose (MFC), which was first developed in the early 1980s,1,2 is cellulose produced by high-shear mechanical homogenization (often of enzymatically hydrolyzed cellulose pulp) in water,(3) as opposed to microcrystalline cellulose (MCC) produced by hydrolysis with strong mineral acids to a level-off degree of polymerization (DP). Due to the mild character of the enzymatic hydrolysis, the cellulose fibers in MFC are substantially longer than MCC whiskers,(4) and the fibers are therefore capable of producing very thick hydrogels at very low solid content (>99 wt % water content) upon high-shear mechanical homogenization in the absence of protective hydrocolloids, such as carboxymethylcellulose.(5) By controlling the number of passes during high-shear homogenization, microfibrils with diameters in the range 10−100 nm and lengths on the order of several micrometers can be obtained.3,5 Thus, MFC(6) together with bacterial cellulose3,7–9 and green algae cellulose10,11 are examples of native nanocelluloses. Thanks to its special rheological properties, MFC hydrogels were originally envisaged for use as thickening agents in food and cosmetics products. More recent applications include dried MFC-based materials such as paper of exceptionally high tensile strength,(12) optically transparent paper,(13) and flexible organic light-emitting diode (OLED) displays.(14)
Highly porous nanocellulose materials are generally of high interest in the manufacturing of filtration media as well as for biomedical applications, e.g., in dialysis membranes. Upon removal of water, MFC, which is produced in the form of hydrogels with very high water contents, collapses into a dense opalescent cellulose material of high mechanical strength.(12) The collapse of the structure after removal of water is initially in the drying process due to the capillary forces which water exerts on the hydrophilic cellulose microfibrils and later in the drying process due to the strong cooperative hydrogen bonding between the hydroxyl groups on the nanocellulose surfaces. Thus, upon complete removal of liquid, the neighboring microfibrils are drawn into intimate proximity and bond through numerous hydrogen bonds.(15) If, however, the water in MFC hydrogels is replaced by a number of solvents with progressively decreasing polarity, the latter results in a gradual decrease of the capillary forces between the solvent and the hydrophilic cellulose fibrils.(16) Removal of the nonpolar solvent in the last step by drying (or vacuum treatment) prevents the fibrils from collapsing which results in the formation of a highly porous cellulose material, often referred to as cellulose aerogels.(16) Cellulose aerogels can also be obtained via lyophilization or supercritical CO2 extraction.17,18 However, if the latter MFC aerogels are exposed to high relative humidity and hence allowed to reabsorb water, the structure will subsequently collapse upon drying.
In recent years, composite materials of cellulose and conductive polymers have received significant attention. It has been shown that polypyrrole (PPy) can be uniformly coated on cellulose fibers from commercial filter paper by a chemical polymerization-induced adsorption process.(19) More recently, it was demonstrated that composites of cellulose and PPy, in situ polymerized on the individual cellulose fibers (which were extracted from the Cladophora green algae), can be obtained using a similar approach.(20) The high surface area and good electronic conductivity of the latter material made it highly suitable for use in electrochemically controlled ion-exchange21–23 and ultrafast all polymer-based batteries.(24) It was demonstrated that the large surface area of the composite cellulose material ensured the attainment of a relatively high specific charge capacity, whereas the continuous, 50 nm thick PPy coating on the individual cellulose fibers enabled the PPy layers to undergo oxidation and reduction at high rates. Since the latter results were obtained with cellulose from the Cladophora sp. algae while cellulose from land plants is typically used in contemporary industrial processes, it is very interesting to study if composites with similar properties likewise can be obtained with microfibrillated cellulose from wood. It has been reported that MFC cellulose can be coated with polyaniline (PANI) merely by dipping the MFC aerogel in a PANI toluene solution.(17) A cellulose PANI composite using in situ polymerization(25) and a conductive paper made from wood microfibers, carbon nanotubes, and poly(3,4-ethylenedioxythiophene)−poly(styrenesulfonate) (PEDOT−PSS) using a layer-by-layer approach(26) have also been described.
To obtain the high capacities required for efficient extractions of biologically interesting ions and the fast charging and discharging needed in paper-based energy storage devices, the cellulose fibers in the conducting polymer composites must clearly serve the dual purpose of mechanically reinforcing the brittle conductive polymers and enhancing the specific charge capacity by providing a continuous 3-D scaffold of high porosity. In the present work, we describe a straightforward method of coating MFC microfibrils with PPy to obtain highly electroactive cellulose composites with large surface areas. The described manufacturing process is entirely water-based and does not include any time-consuming solvent-exchange and drying steps to retain the large surface area of MFC upon water removal. It is shown that the so obtained composites have surface areas of ∼90 m2/g and conductivities of ∼1.5 S/cm which render them well-suited for use in applications involving large scale extraction of ions and novel energy storage devices. The present paper is, to our knowledge, the first to report on a composite based on wood-derived MFC and PPy.
Experimental Section
Sample Preparation
MFC Hydrogel
MFC was made from a never-dried bleached sulphite softwood cellulose pulp (Domsjö Eco Bright; Domsjö Fabriker AB) consisting of 40% pine (Pinus sylverstris) and 60% spruce (Picea abies) with a hemicellulose content of 13.8% and a lignin content of less than 1%. This pulp was first refined in an Escher-Wyss refiner to 35° SR and was then treated with a small amount of an enzyme (10 ECU/g fiber of a monocomponent endoglucanase enzyme, Novozym 476, Novozymes A/S, Denmark) at 60 °C at pH 7.0 (using a phosphate buffer) for 2 h after (the enzyme activity was also terminated by a 90 °C heat treatment). After the enzyme treatment, the pulp was again beaten to 90° SR in the Escher-Wyss refiner and finally brought to its Na form, by an acid wash followed by adjusting the pH to 8.0 using NaOH. Finally, MFC was obtained by passing the fibers once through a microfluidizer (type M-110EH, Microfluidics Corp., USA).
MFC Paper
MFC paper was obtained by diluting 240 mg of the thick 2 wt % MFC gel in 100 mL of distilled water after which the cellulose cake was collected on a filter paper in a Buchner funnel employing reduced pressure and allowed to dry in air. When dried, the MFC paper was carefully peeled off from the filter paper by moistening the backside of the latter.
Composite Material
The MFC−PPy composite, hereafter referred to as the composite material, was prepared by first dissolving 240 mg of the 2 wt % MFC hydrogel in 100 mL of distilled water. This solution was then mixed with a solution of 3 mL of pyrrole (purchased from VWR Sweden and used as received) in 100 mL of distilled water, the oxidizing solution, prepared by mixing 3 mL of pyrrole (purchased from VWR Sweden and used as received) with a solution of 8 g of FeCl3 in 100 mL of distilled water, and subsequently 160 μL of 37% HCl was added to the resulting mixture. The reaction between the oxidant solution and the MFC dispersion, giving rise to a layer of polypyrrole on the cellulose fibers, was allowed to proceed for 15 min. The resulting structure was then collected on a filter paper in a Buchner funnel employing reduced pressure and thoroughly washed with distilled water. The filter paper was subsequently removed and the cellulose PPy−MFC cake was dried to constant mass on a Petri dish in air.
PPy Powder
PPy powder, used for the thermogravimetric analyses, was produced by mixing 3 mL of pyrrole with 8 g of FeCl3 dissolved in 100 mL of distilled water. The reaction was then allowed to proceed for 15 min after which the solid product was collected on a filter paper within a Buchner funnel using reduced pressure and then thoroughly washed with distilled water.
Sample Analysis
Thermogravimetric Analysis
Thermogravimetric analyses (TGA) of the composite (7.5 mg), the MFC paper (9.4 mg), and of PPy powder (7.1 mg) was performed using a Mettler Toledo TGA/SDTA851 instrument. All samples were placed in inert ceramic crucibles and were heated from 25 to 600 °C at a heating rate of 10 °C/min in the presence of a 20 mL/min flow of air.
Conductivity Measurements
The bulk resistances of the dried and as prepared composite samples were measured at room temperature using an Agilent 34401A Digital Multimeter. The sample conductivity, σ, was obtained from the measured resistance R according to σ = (1/R)(L/wd), where L is the length, w is the width, and d is the thickness of the sample. The employed sample dimensions were 1 cm × 0.5 cm × 0.15 cm. Silver paint was applied at the ends of the samples to ensure good electrical contacts with the clip probes.
Cyclic Voltammetry
Cyclic voltammetric measurements on aqueous solutions were performed in a standard three-electrode setup utilizing an Autolab/GPES interface (ECO Chemie, The Netherlands) with the composite sample (15 mg) as the working electrode, a Pt wire as the counter electrode, and an Ag/AgCl electrode as the reference electrode. The measurements, which were carried out in 2.0 M solutions of sodium chloride, were recorded in the potential interval between −1.0 and +1.0 V employing a scan rate of 5 mV/s.
Scanning Electron Microscopy
Scanning electron microscopy (SEM) images of the MFC paper as well as of the composite were taken with an environmental SEM (FEI/Philips XL 30, The Netherlands) in the high vacuum mode (3.5 × 10−6 mbar). The samples were mounted on aluminum stubs using double-sided adhesive tape. Prior to imaging, Au/Pt was sputtered on the samples to minimize charging effects.
Gas Adsorption Analysis
Nitrogen (N2) and argon (Ar) gas adsorption isotherms were recorded for the composite sample and the MFC paper, respectively, using an ASAP 2020 (Micromeritics, USA) instrument. Both measurements were performed at liquid nitrogen temperatures (i.e., 77 K), and the specific surface areas of the samples were obtained from the isotherms using the BET method.(27)
Results and Discussion
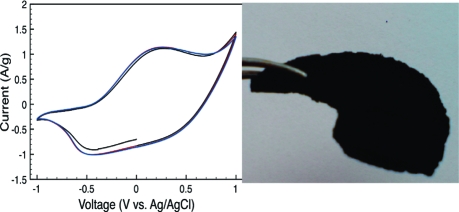
In Figure 1, photographs of the MFC paper and the composite sheets are shown. The MFC paper was a 100 μm thick, highly flexible translucent sheet possessing good mechanical properties. The composite was in the form of a black sheet which had a thickness of roughly 1.5 mm. It was mechanically stable but relatively brittle in comparison both to the MFC paper and to the corresponding previously described composites consisting of algal cellulose and PPy.20–24 To exemplify this, the algal cellulose composite sheets can be bent at an angle of ∼180° without breaking, while corresponding composites of MFC break if they are bent more than ∼5°. The higher brittleness of the composite compared to that for the MFC paper could possibly be due to the fact that the interaction between the PPy coated fibers should involve PPy−PPy bonds, as these are intrinsically weaker than cellulose-to-cellulose bonds. The relatively lower mechanical strength compared to that for the corresponding algae-based composite could be due to the shorter cellulose fiber length and the thinner individual cellulose fibers of the nanocellulose (17 nm5) as opposed to the 30 nm thick fibers of the algal cellulose.(10) Thus, a proper fiber design with tailored dimensions of the fiber thickness and length could most likely make the composite less brittle. Also, the MFC hydrogel is sensitive to low pH and since the iron(III) chloride water solution added to initiate the polymerization has a pH of ∼2, this will cause the hydrogel to partly collapse and also damage the fibers. Both of these effects are expected to negatively influence the mechanical stability of the composite. Thus, polymerizing in a more neutral solution would most likely also help creating a less brittle composite.
Figure 1.

Photographs of MFC paper (a) and a MFC−PPy composite (b).
Figure 2 shows the TGA results of the MFC paper and the composite material compared to that for the PPy powder used as a reference. It is seen that water is continuously evaporating up to a temperature of about 100 °C and that this results in a weight loss of ∼5 wt % for all samples. Polypyrrole is known to degrade in a three-step process, where water leaves the sample first and thereafter a degradation process involving the counterions follows. Finally, the polymer backbone is degraded.28,29 In the first derivative curve (DTGA) of the polypyrrole weight loss, these three processes can be identified as local extremes. The temperature ranges for the loss of the dopant ion (105−315 °C) and for the polymer backbone degradation (315−600 °C) are in agreement with literature values.(29) The result of these processes is a steady degradation of polypyrrole until ∼37 wt % remains at 600 °C. The MFC paper underwent rapid decomposition in the interval between 250 and 350 °C in a process typical for cellulose pyrolysis involving the formation of various anhydro-monosaccharides (including levoglucosenone, levoglucosan, and 1,6-anhydro-β-d-glucopyranose), carbon oxides, and char.(30) This process is also clearly seen in the DTGA curve for the MFC paper which has a monomodal shape with one maximum degradation temperature at 310 °C. At 350 °C, about 61 wt % of the MFC paper was decomposed and a total weight loss of ∼75 wt % was reached at 600 °C. In the region between 230 and 360 °C, the composite lost ∼19 wt %, mainly as a result of the degradation process of the cellulose part in the composite material but also partly caused by the thermal degradation of the polymer backbone in polypyrrole. The process during which the counterion is expelled occurs before the polymer backbone degradation in PPy and is probably responsible for shifting the main composite degradation step to have its maximum degradation temperature at a lower temperature (280 °C) than the maximum degradation temperature of the MFC degradation process (310 °C). It is also seen in the DTGA data that the composite follows the first process of the PPy before the main cellulose degradation and after which it follows the second degradation process of polypyrrole again. This confirms the presence of both MFC and PPy in the composite material. At temperatures above 360 °C, the composite TGA curve followed that of the PPy sample with a small shift in weight percentage corresponding to the residual amount of cellulose degradation products still present in the composite material. This indicates that the majority of the composite is PPy. By considering the extent of decomposition of cellulose and PPy at 400 °C and comparing this to the residual weight of the composite at the same temperature, the mass fraction of PPy in the composite can be estimated to ∼0.7.
Figure 2.

Thermogravimetric analysis showing the weight loss vs temperature for the MFC−PPy composite (solid line), the MFC paper material (dashed line), and PPy (dotted line). In the inset, the derivatives of these curves are shown as a function of temperature.
Conductivity measurements on composite sheets indicated an average electrical conductivity of 1.5 S/cm, which is somewhat higher than that for the algal-cellulose-based composites.(21) This indicates that the ratio of conducting (PPy) to nonconducting (cellulose) material was higher in the wood-based MFC composite, a hypothesis that is in good agreement with the TGA results (cf. above and ref (24)). The electrical conductivity of the MFC paper sheet was, incidentally, nearly 106 times lower than that for the composite. As is seen in Figure 3, depicting a cyclic voltammogram for the composite in 2.0 M NaCl electrolyte, the composite material also showed a high degree of electroactivity. The characteristic redox behavior of PPy(31) is clearly seen, with an oxidation peak at +0.27 V and a reduction peak at −0.45 V vs Ag/AgCl. The onset of an overoxidation peak can also be observed at 0.75 V. To estimate the ion-exchange capacity of the composite, the charge capacity was calculated by integrating the current vs time curve from the anodic scan in the voltammogram. This resulted in a charge capacity of 289 C/g (or 80 mAh/g) for the composite, which is of the same order of magnitude as that previously found for the algal-cellulose-based composite electrodes used in an ultrafast polymer battery.(24) This clearly shows that the present MFC-based composite material could be used as an alternative material to the composite based on cellulose from the Cladophora sp. algae.
Figure 3.

The three first cycles of a cyclic voltammogram for a MFC−PPy composite recorded in a 2.0 M sodium chloride solution employing a scan rate of 5 mV/s.
The SEM image shown in Figure 4a implies a dense, nonporous structure of the MFC paper. In contrast, the SEM image for the composite material (see Figure 4b) indicates the presence of an open, porous structure of intertwined fibers. At certain locations, excess nodular agglomerates are observed for the composite material; these features are typical results from in situ aqueous polymerization of PPy on a hydrophilic surface.(32) Gas adsorption analyses confirmed the low-porosity structure of the MFC paper, since the specific surface area of the MFC paper was too low to be determined using N2 adsorption analyses. Ar adsorption was therefore also employed, since Ar has similar dimensions as N2 but provides a 4-fold gain in measurement sensitivity due to its lower saturation pressure.33,34 On the basis of Ar BET adsorption analyses, a specific surface area of merely 1.5 m2/g was obtained for the MFC paper. The latter results suggest that the MFC nanofibers collapsed into a dense, compact structure upon drying of the MFC hydrogels, in accordance with the previous results.(16) The N2 BET adsorption analyses of the composite material, on the other hand, resulted in a specific surface area of 89 m2/g which is somewhat higher than the values previously obtained for algal-based composites.20–22,24 The present results thus indicate that direct chemical polymerization of pyrrole monomers on MFC nanofibers in the hydrogel gives rise to a composite, the structure of which (unlike that of MFC paper itself) does not collapse upon drying. It can thus be concluded that the inherent large specific surface area of the composite material could be maintained without the use of any special drying method, such as solvent-exchange drying, supercritical CO2 drying, or lyophilization. By covering the individual cellulose fibers with a continuous layer of PPy, the contact angle between water and the composite fibers increases which results in weaker capillary forces during drying and second the PPy blocks hydrogen bonding between the individual fibers during drying. By varying the PPy content of the composite, it was also found that the degree of loss of surface area upon drying decreased with increasing PPy content in the composite. The fact that the straightforward coating of the cellulose fibers with PPy helps maintain the porous, large-surface-area structure of the MFC significantly simplifies large-scale manufacturing of highly porous, electroactive conductive paper composites for applications including ion-exchange separations, dialysis membranes, and electrode materials for paper-based energy storage devices.
Figure 4.

SEM micrograph of the MFC paper (a) and MFC−PPy composite (b). The scale bar in both images corresponds to 1 μm.
Summary and Conclusion
It has been shown that it is possible to manufacture an electronically conductive high-surface area composite material composed of microfibrillated cellulose (MFC) and polypyrrole by direct chemical polymerization of pyrrole on wood-derived nanofibers in hydrogels without the need for sophisticated and time-consuming drying techniques such as solvent-exchange drying or lyophilization. Although brittle, the air-dried composite sheets exhibited a conductivity of 1.5 S/cm and a specific surface area of 89 m2/g. Since the material was found to be electroactive with an ion-exchange capacity for Cl− of 289 C/g (i.e., a specific charge of 80 mAh/g), it is clear that the present material has similar capacities as the corresponding composites based on cellulose from the Cladophora sp. algae recently used in a novel type of rapid-charging paper-based batteries. The present findings consequently give rise to new exciting possibilities regarding large-scale production of inexpensive paper-based materials for energy storage as well as electrochemically controlled extraction and separation of biologically interesting compounds. Work along these lines and on the improvement of the mechanical properties of the composite is currently in progress.
Acknowledgments
The Swedish Foundation for Strategic Research (SSF), the Swedish Research Council (VR), and the Knut and Alice Wallenberg Foundation (KAW) are gratefully acknowledged for financial support of this work.
References
- Herrick F. W.; Casebier R. L.; Hamilton J. K.; Sandberg K. R. J. Appl. Polym. Sci.: Appl. Polym. Symp. 1983, 37, 797–813. [Google Scholar]
- Turbak A.; Snyder F.; Sandberg K. J. Appl. Polym. Sci.: Appl. Polym. Symp. 1983, 37, 815–827. [Google Scholar]
- Nakagaito A. N.; Yano H. Appl. Phys. A 2005, 80, 155–159. [Google Scholar]
- Henriksson M.; Henriksson G.; Berglund L. A.; Lindström T. Eur. Polym. J. 2007, 43, 3434–3441. [Google Scholar]
- Pääkkö M.; Ankerfors M.; Kosonen H.; Nykanen A.; Ahola S.; Österberg M.; Ruokolainen J.; Laine J.; Larsson P. T.; Ikkala O.; Lindström T. Biomacromolecules 2007, 8, 1934–1941. [DOI] [PubMed] [Google Scholar]
- Klemm D.; Schumann D.; Kramer F.; Hessler N.; Koth D.; Sultanova B. Macromol. Symp. 2009, 280, 60–71. [Google Scholar]
- Nishi Y.; Uryu M.; Yamanaka S.; Watanabe K.; Kitamura N.; Iguchi M.; Mitsuhashi S. J. Mater. Sci. 1990, 25, 2997–3001. [Google Scholar]
- Ciechanska D.; Struszczyk H.; Kazimierczak J.; Guzinska K.; Pawlak M.; Kozlowska E.; Matusiak G.; Dutkiewicz M. Fibres Text. East. Eur. 2002, 10, 27–30. [Google Scholar]
- Gelin K.; Bodin A.; Gatenholm P.; Mihranyan A.; Edwards K.; Strømme M. Polymer 2007, 48, 7623–7631. [Google Scholar]
- Mihranyan A.; Edsman K.; Strømme M. Food Hydrocolloids 2007, 21, 267–272. [Google Scholar]
- Strømme M.; Mihranyan A.; Ek R. Mater. Lett. 2002, 57, 569–572. [Google Scholar]
- Henriksson M.; Berglund L. A.; Isaksson P.; Lindström T.; Nishino T. Biomacromolecules 2008, 9, 1579–1585. [DOI] [PubMed] [Google Scholar]
- Nogi M.; Iwamoto S.; Nakagaito A. N.; Yano H. Adv. Mater. 2009, 21, 1595–1598. [Google Scholar]
- Okahisa Y.; Yoshida A.; Miyaguchi S.; Yano H. Compos. Sci. Technol. 2009, 69, 1958–1961. [Google Scholar]
- Battista O. A.Microcrystal polymer science; McGraw-Hill: New York, 1975. [Google Scholar]
- Jin H.; Nishiyama Y.; Wada M.; Kuga S. Colloids Surf., A 2004, 240, 63–67. [Google Scholar]
- Pääkkö M.; Vapaavuori J.; Silvennoinen R.; Kosonen H.; Ankerfors M.; Lindström T.; Berglund L. A.; Ikkala O. Soft Matter 2008, 4, 2492–2499. [Google Scholar]
- Cai J.; Kimura S.; Wada M.; Kuga S.; Zhang L. ChemSusChem 2008, 1, 149–154. [DOI] [PubMed] [Google Scholar]
- Huang J. G.; Ichinose I.; Kunitake T. Chem. Commun. 2005, 1717–1719. [DOI] [PubMed] [Google Scholar]
- Mihranyan A.; Nyholm L.; Garcia Bennett A. E.; Strømme M. J. Phys. Chem. B 2008, 112, 12249–12255. [DOI] [PubMed] [Google Scholar]
- Razaq A.; Mihranyan A.; Welch K.; Nyholm L.; Strømme M. J. Phys. Chem. B 2009, 113, 426–433. [DOI] [PubMed] [Google Scholar]
- Gelin K.; Mihranyan A.; Razaq A.; Nyholm L.; Strømme M. Electrochim. Acta 2009, 54, 3394–3401. [Google Scholar]
- Strømme M.; Frenning G.; Razaq A.; Gelin K.; Nyholm L.; Mihranyan A. J. Phys. Chem. B 2009, 113, 4582–4589. [DOI] [PubMed] [Google Scholar]
- Nyström G.; Razaq A.; Strømme M.; Nyholm L.; Mihranyan A. Nano Lett. 2009, 9, 3635–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattoso L. H. C.; Medeiros E. S.; Baker D. A.; Avloni J.; Wood D. F.; Orts W. J. J. Nanosci. Nanotechnol. 2009, 9, 2917–2922. [DOI] [PubMed] [Google Scholar]
- Agarwal M.; Xing Q.; Shim B. S.; Kotov N.; Varahramyan K.; Lvov Y. Nanotechnology 2009, 20, 215602. [DOI] [PubMed] [Google Scholar]
- Brunauer S.; Emmett P. H.; Teller E. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar]
- Mohammad F.; Calvert P. D.; Billingham N. C. Bull. Mater. Sci. 1995, 18, 255–261. [Google Scholar]
- Carrasco P. M.; Cortazar M.; Ochoteco E.; Calahorra E.; Pomposo J. A. Surf. Interface Anal. 2007, 39, 26–32. [Google Scholar]
- Lin Y.-C.; Cho J.; Tompsett G. A.; Westmoreland P. R.; Huber G. W. J. Phys. Chem. C 2009, 113, 20097–20107. [Google Scholar]
- Kuwabata S.; Yoneyama H.; Tamura H. Bull. Chem. Soc. Jpn. 1984, 57, 2247–2253. [Google Scholar]
- Wang P. C.; Huang Z.; MacDiarmid A. G. Synth. Met. 1999, 101, 852–853. [Google Scholar]
- Webb P. A.; Orr C.. Analytical Methods in Fine Particle Technology; Micromeritics Instrument Corporation: Norcross, GA, 1997. [Google Scholar]
- Mihranyan A.; Forsgren J.; Strømme M.; Engqvist H. Langmuir 2009, 25, 1292–1295. [DOI] [PubMed] [Google Scholar]