

Summary
Soluble diacylglycerol (DAG) kinases function as regulators of diacylglycerol metabolism in cell signaling and intermediary metabolism. We report the first structure of a DAG kinase, DgkB from Staphylococcus aureus, both as the free enzyme and in complex with ADP. The molecule is a tight homodimer, and each monomer comprises two domains with the catalytic center located within the interdomain cleft. Two distinctive features of DkgB are a structural Mg2+ site and an associated Asp•water•Mg2+ network that extends towards the active site locale. Site-directed mutagenesis revealed that these features have important roles in the catalytic mechanism. The key active site residues and the components of the Asp•water•Mg2+ network are conserved in the catalytic cores of the mammalian signaling DAG kinases, indicating that these enzymes use the same mechanism and have similar structures as DgkB.
Introduction
Diacylglycerol (DAG) kinases are key enzymes in lipid metabolism that phosphorylate diacylglycerol formed by the turnover of membrane phospholipids (Sakane et al., 2007). They have crucial but distinct roles in bacterial and mammalian cells. In bacteria, the headgroup of phosphatidylglycerol (PtdGro) is extensively used in the manufacture of structural constituents of the cell, and removal of the headgroup generates DAG (Goldberg et al., 1981; Neuhaus and Baddiley, 2003; Schulman and Kennedy, 1977). DAG is converted to phosphatidic acid (PtdOH) by a DAG kinase that recycles DAG into the cytidine diphosphate-diacylglycerol pathway for phospholipid synthesis, and prevents the lethal accumulation of DAG in bacterial membranes. In mammals, both the substrate DAG and the product PtdOH act as potent signaling molecules. DAG is a precursor in mammalian phospholipid metabolism, but it also serves as an intracellular lipid signal that activates protein kinase C and is involved in cell-cycle regulation, cellular survival, tumorigenesis and apoptosis (Sakane et al., 2007). DAG kinases consume DAG and therefore provide the means of terminating DAG signaling pathways, but at the same time produce another lipid signal, PtdOH. For these reasons, the biochemistry of mammalian DAG kinases (DGKs) has been widely studied (Van Blitterswijk and Houssa, 2000).
Bacterial DAG kinases belong to one of two protein families, DgkA or DgkB. DgkA is an integral membrane protein with three membrane-spanning domains and is the founding member of Pfam01219 that defines a widely-distributed prokaryotic-type of DAG kinase (Oxenoid et al., 2002; Smith et al., 1994). DgkA is well-characterized in Escherichia coli where PtdGro is degraded to DAG by the transfer of the sn-glycerol-1-P headgroup to membrane-derived oligosaccharides (Goldberg et al., 1981; Schulman and Kennedy, 1977). However, we recently reported that the dgkA genes in Gram-positive bacteria actually encode undecaprenol kinases and not DAG kinases (Jerga et al., 2007). We also demonstrated that Gram-positive bacteria instead produce a second prokaryotic DAG kinase isoform, DgkB, which belongs to Pfam00781, the DAG kinase (DGK) superfamily of soluble proteins that share a common catalytic core signature sequence (Jerga et al., 2007). We identified dgkB genes in 38 Gram-positive bacterial species, including the methicillin resistant Staphylococcus aureus (MRSA) DgkB. S. aureus DgkB phosphorylates DAG generated by the transfer of the sn-1-glycerol-P headgroup from PtdGro to lipoteichoic acid (LTA), a polydisperse macroamphiphile that decorates the external surface of Gram-positive bacteria (Cabacungan and Pieringer, 1981; Koch et al., 1984; Koga et al., 1984; Taron et al., 1983). The synthesis of one LTA molecule requires the successive addition of 14 to 33 glycerol-1-P units, and PtdGro turnover is therefore rapid and persistent in many Gram-positive bacteria (Koch et al., 1984; Taron et al., 1983). Thus, DgkB is an essential protein in these bacteria that reintroduces into the phospholipid biosynthetic pathway the large amount of DAG formed by LTA synthesis, and prevents the lethal accumulation of DAG (Jerga et al., 2007).
The Pfam00781 superfamily includes the mammalian DGKs of which there are ten isoforms in humans (Sakane et al., 2007; Van Blitterswijk and Houssa, 2000). The sequence conservation encompasses a number of highly conserved motifs and individual amino acids whose precise functions have yet to be determined. In this study, we report the crystal structures of S. aureus DgkB to 2.4 Å resolution and the Mg2+•DgkB•ADP complex to 2.3 Å resolution. The enzyme has a two-domain architecture, and the active site is located in the inter-domain cleft. With the aid of mutagenesis experiments, we also correlate the structure of S. aureus DgkB with its catalytic properties to provide insights into the mode of nucleotide binding, the catalytic mechanism, and the precise roles of the highly conserved sequence motifs and specific amino acids shared between bacterial and human DAG kinases. Finally, we have identified a conserved Mg2+ binding site 24 Å away from the active site that is required for catalysis, and suggest that it influences activity via a connecting Asp•water•Mg2+ network. This study defines the structure of DgkB and provides mechanistic insights into the role of the common structural folds and conserved amino acids in the DAG kinase superfamily. In addition, it provides the necessary structural framework for the potential development of small molecule therapeutics to combat Gram-positive organisms and to modulate lipid signaling pathways in humans.
Results
Overall Structure of DgkB
Crystallization trials of selenomethionine-labeled DgkB were conducted for de novo structure determination by single anomalous dispersion (SAD). Crystals in space group P42212 were grown that diffract to 2.4 Å and contain a DgkB monomer in the asymmetric unit. Analysis of the final model using PROCHECK (Laskowski et al., 1993) revealed a high quality structure consistent with final Rwork and Rfree values of 22.9% and 27.2%, respectively (Table 1). A DgkB crystal soaked with ADP plus Mg2+ diffracted to 2.3 Å, and the structure was determined from the ligand-free model using rigid body refinement. The final Rwork and Rfree values are 20.6% and 24.6%, respectively (Table 1). The Mg2+ that is visible in our structure is at a site remote from the active site, and the Mg2+ presumed to be present within the ATP-bound active site was not observed. To distinguish between these metals, we refer to the Mg2+ observed in our structure as Mg1 and the Mg2+ involved in ATP binding as Mg2. The ligand-free and Mg1•DgkB•ADP ternary complex structures are virtually identical with an r.m.s.d. on α-carbons of 0.4 Å. The higher quality and higher resolution Mg1•DgkB•ADP structure resulted in a more complete model assignment of several loops, and is discussed in our analysis unless otherwise noted.
Table 1. Data collection and refinement statistics.
| DgkB | Mg1•DgkB•ADP | |
|---|---|---|
| Data collection | ||
| Space group | P42212 | P42212 |
| Cell dimensions | ||
| a,b,c (Å),α,β,γ = 90° | 124.2, 124.2, 48.5 | 123.8, 123.8, 47.68 |
| Wavelength | 0.9794 (Se Peak) | 1.0 |
| Resolution (Å) | 26-2.4 (2.48-2.40) | 30-2.3 (2.38-2.30) |
| Rmerge | 9.4 (37.6) | 12.8 (43.1) |
| I / σI | 28.1 (8.2) | 24.5 (4.2) |
| Completeness (%) | 99.8 (99.5) | 100 (99.8) |
| Redundancy | 8.8 (6.1) | 12.2 (8.3) |
| Refinement | ||
| Resolution (Å) | 26-2.4 | 30-2.3 |
| No. reflections | 14,042 | 16,069 |
| Rwork / Rfree | 22.9/27.2 | 20.6/24.6 |
| No. atoms | ||
| Protein | 2171 | 2324 |
| ADP/Mg2+ | 27/1 | |
| Water | 116 | 152 |
| B-factors | ||
| Protein | 39.7 | 33.9 |
| ADP/Mg2+ | 38.6/11.6 | |
| Water | 43.5 | 36.7 |
| Rms deviations | ||
| Bond lengths (Å) | 0.01 | 0.008 |
| Bond angles (°) | 1.4 | 1.2 |
| Ramachandran plot | ||
| Most favored (%) | 90.5 | 89 |
| Allowed (%) | 9.5 | 11 |
Values in parentheses are for the highest-resolution shell.
DgkB has a two-domain architecture in which domain 2 is inserted into the fold of domain 1 (Figure 1A). Domain 1 has an α/β structure and comprises the N-terminal 124 amino acids and the C-terminal residues 286 to 315. The fold begins with the substructure β1-α1-β2-α2-β3-α3-β4 in which the β-strands create a parallel four-stranded β-sheet with α1 packed on one side, and α2 and α3 on the opposite side. Helix α4 faces the domain interface adjacent to α2 and α3, and helix α5 lies ‘above’ the β-sheet adjacent to α1 and an offset substructure consisting of the β5-β17 anti-parallel strands and the C-terminal loop. Strand β17 follows the fold of domain 2 and inserts between β4 and β5 to stabilize the two-domain architecture. Domain 2 (residues 125 to 285) is a β-sandwich. The ‘bottom’ sheet contains the anti-parallel β-strands β7, β6, β16, β13, β9, and β10, and the ‘top’ sheet comprises the anti-parallel β-strands β15, β14, β8, β10, β11, and β12. Domain 2 also contains two α-helices and a 310-helix that all pack onto the ‘top’ sheet. Helix α6 follows strand β8 via a partially disordered loop (loop β8-α6). Specifically, residues 145-157 could not be modeled, and residues 144, 162 and 165 were modeled as alanines due to insufficient electron density. Helices α7 and α8 follow strand β11. A structured loop region connects strands β10 and β11 (loop β10-β11), and it contains a structural Mg2+ (Mg1) ion binding site.
Figure 1.
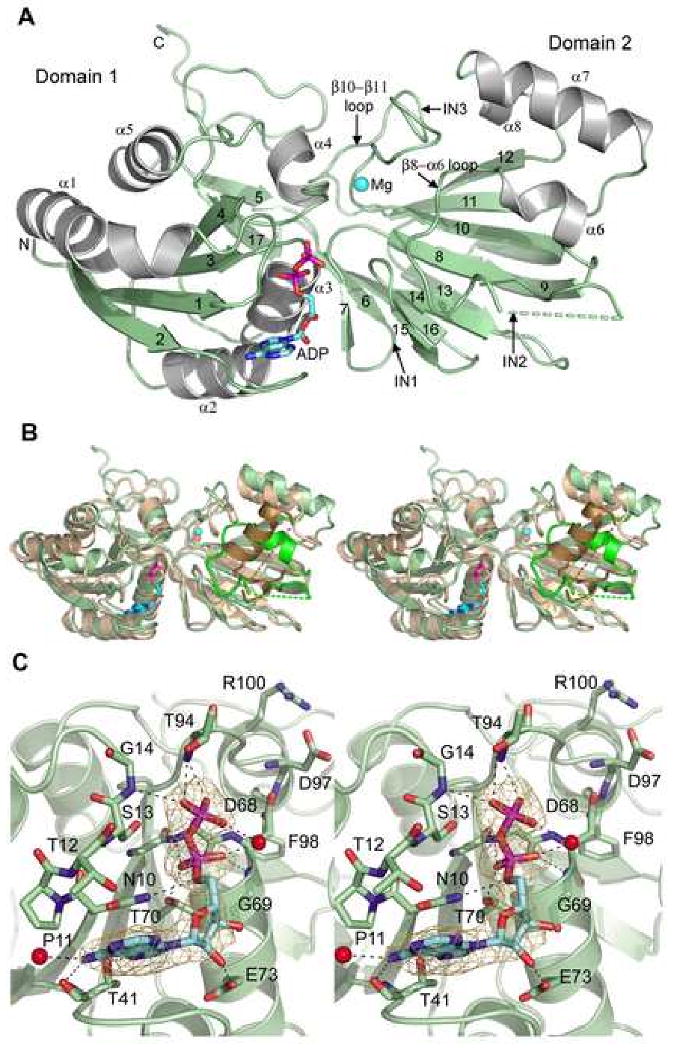
Overall structure of DgkB and a close-up of the nucleotide binding site. (A) Cartoon diagram of the DgkB monomer in the asymmetric unit. Alpha helices are colored grey, β-strands and loops green. ADP carbons and Mg1 (sphere) are colored cyan. Secondary structure elements are labeled. Disordered residues 145-157 are absent in the final model and indicated with a broken line. The predicted locations of the insertions in human DAG kinases (see Fig.2) are labeled IN1-3. (B) Stereo cartoon of the YegS structure (pdb 2BON, chain A) superimposed on DgkB. The orientations of Domains 1 and 2 differ slightly in the two structures, and to highlight their structural similarity each domain of YegS was superimposed independently onto the DgkB structure. DgkB is colored as shown in 1A and YegS is colored tan. Disordered residues are indicated with broken lines. Significant differences reside in the predicted DgkB substrate binding region (β8-α6 loop) highlighted in bright green, and the corresponding YegS region is colored brown. (C) Stereo close-up view of the DgkB nucleotide binding site with omit electron density for ADP contoured to 3σ. Bound waters are shown as red spheres. Hydrogen bonds are indicated by broken lines.
The two-domain architecture of DgkB has similarities to the overall folds described in other proteins. A Dali search (Holm and Sander, 1993) revealed that a similar fold occurs in a Pfam00781 member of unknown substrate specificity referred to as YegS (Bakali et al., 2007; Nichols et al., 2007), and also in the tetrameric NAD kinases (Garavaglia et al., 2002). The most similar structure is E. coli YegS (2BON; Dali Z score = 27.6; r.m.s.d. = 2.2 Å for domain 1 and 1.7 Å for domain 2 excluding structurally unique residues 143-171). YegS, which shares 27% sequence identity with DgkB, is not a bona fide DAG kinase although it is postulated to be a lipid kinase (Bakali et al., 2007; Nichols et al., 2007). For comparison, S. aureus DgkB shares 34-90% sequence homology with other DgkBs and 15-18% sequence identity with human DGKs. YegS phosphorylates the headgroup of phosphatidylglycerol in vitro, but its actual substrate in vivo has not been established (Bakali et al., 2007; Nichols et al., 2007). The overall YegS fold is similar to that of DgkB and it contains the distinctive Mg1 binding site. The Salmonella typhimurium YegS structure has Ca2+ in the cation site, and biophysical studies have shown that the cation stabilizes YegS to heat denaturation (Nichols et al., 2007). A major difference in the DgkB and YegS structures occurs between strands β8 and β9 in domain 2. Starting with residue 141, the proteins take markedly divergent paths until merging again at residue 173. YegS has a 10 residue α-helix in this region that lies perpendicular to the top β-sheet whereas DgkB has the flexible β8-α6 loop followed by the short helix α6 (Figure 1B). This difference is likely to be functionally significant because the helix in YegS and the β8-α6 loop in DgkB lie adjacent to the active site (see below) suggesting a role in substrate recognition.
NAD kinases are also similar to DgkB, and available structures include the inorganic polyphosphate/ATP-NAD kinase family members from Archaeoglobus fulgidus (1Z0S; Dali Z score = 17.2; r.m.s.d. = 2.1 Å for domain 1, 2.9 Å for domain 2), Thermotoga maritima (1YT5; Z score = 16.3; r.m.s.d. = 2.2 Å, 2.1 Å), Mycobacterium tuberculosis (1U0R; Z score = 16.2; r.m.s.d. = 2.4 Å, 2.1 Å), and Salmonella typhimurium (2AN1; Z score = 15.9; r.m.s.d. = 2.5 Å, 2.3 Å). These proteins function as a dimer of dimers with a shared NAD binding pocket between dimers. When compared to DgkB, elements of this dimer interface replace the β8-α6 putative substrate-binding loop. In addition, NAD kinase lacks the helical structures equivalent to α6, α7 and α8, and does not contain the structural Mg1 site in domain 2. Phosphofructokinases (PFKs) (Pfam00365) are distantly related to the above proteins by sequence and structure (Labesse et al., 2002). PFKs are homotetramers with a two-domain architecture in which each domain has a similar αβ topology. The similarity with DgkB is found in domain 1 where PFK contains a core αβ fold equivalent to the β1-α1-β2-α2-β3-α3-β4 substructure of DgkB.
The DgkB Nucleotide Binding Site
The nucleotide is bound to domain 1 with the terminal β-phosphate facing the cleft formed at the domain interface (Figure 1A). The electron density for ADP unambiguously identifies the nucleotide binding site on DgkB and reveals key interactions between the nucleotide and the protein (Figure 1C). The adenine ring is inserted between the β1-α1 turn and the β2-α2 loop, and makes van der Waals contact with residues Asn10 and Pro11, and Glu42 and Ile44, respectively. Thr41 forms two hydrogen bonds to the adenine ring: the carbonyl oxygen is linked to the N6 nitrogen and the OG1 oxygen to the N7 nitrogen. A solvent-exposed water molecule W124 also hydrogen bonds the N6 nitrogen. The ribose ring is not as tightly bound to the protein and forms a single hydrogen bond between the 2′-hydroxyl and the side chain of Glu73 in helix α3. This loose attachment is reflected in the poorer electron density for this portion of the nucleotide. In contrast, the two phosphate groups make tight interactions and are well resolved. The α-phosphate forms three hydrogen bonds, with the backbone amides of Gly67 and Gly69, and the OG1 oxygen of Thr70 at the N-terminus of helix α3. Ser13, located in the turn preceding α1, is in van der Waals contact with the oxygen atom that bridges the α- and β-phosphates, and indirectly supports ADP binding by forming stabilizing hydrogen bonds to Asn10 and Gly67. Specifically, the backbone amide and the OG hydroxyl interact with the carbonyl oxygen and the backbone amide, respectively. Finally, the β-phosphate forms hydrogen bonds with the backbone amide of Gly14, and the backbone amide and OG1 hydroxyl of Thr94 from the β4-α4 loop. Water W26 forms a bridging hydrogen bond between the two phosphates, and the α-phosphate lies at the N-terminus of helix α3 and can benefit from the helix dipole in this region of the binding site (Figure 1C).
The nucleotide binding sequence is conserved and defines members of the soluble DGK superfamily (Pfam00781). Figure 2 shows a sequence alignment of S. aureus DgkB with six other members of the superfamily; three representatives of the most structurally diverse human DGKs, YegS, human sphingosine kinase (SK1) and human ceramide kinase (CERK). The figure also includes the secondary structural elements deduced from our structure, and highlights important features. The alignment reveals three conserved motifs that participate in binding the nucleotide. In DgkB, motif-1 consists of 7ΦΦxNPxSG14 (Φ stands for a hydrophobic residue and x for any residue) and forms one side of the nucleotide binding site. Residues Gly67, Gly69, Asp68, and Glu73 within the second glycine-rich sequence motif-2 63ΦxxGGDGTΦ71 represent the conserved P-loop that is characteristic of the protein family. The P-loop Mg2+ ion (Mg2) is not present in the Mg1•DgkB•ADP structure, but the conserved Asp68 appears to be the ligand for this function based on comparisons with the P-loop structure of PFK (Berger and Evans, 1992; Jiang et al., 2000). Thr94 and Asp97 are central to the motif-3 sequence 89ΦΦPxGTxNDΦxR100 that forms the ‘top’ of the nucleotide binding site. Structural comparison of DgkB with PFK (Berger and Evans, 1992; Jiang et al., 2000) suggests key roles for these residues; Thr94 interacts with the γ-phosphate of ATP and Asp97 coordinates Mg2 via water molecules (equivalent to Thr125 and Asp130 of PFK, respectively).
Figure 2.

Structure-aided sequence alignment of S. aureus DgkB and human DAG kinases. The ten known isoforms of human DAG kinases were included in the similarity calculation, but only the structurally diverse DGKα, DGKζ, and DGKδ (types 1, 4, and 2, respectively) are shown for brevity. Also shown are the sequences of the related superfamily proteins E. coli YegS, human ceramide kinase (CERK) and human sphingosine kinase (SK1). DgkB β-strands and α-helices are shown as green arrows and grey cylinders, respectively. The predicted locations of the insertions in human DAG kinases are indicated by IN1, IN2, and IN3. Motifs I, II, and III refer to important ATP binding regions. Mutational analyses were performed on DgkB residues marked with a black triangle. The most highly conserved residues are in red block, and less conserved residues are indicated by red font. Note: * marks the insertion of an extra domain in type 2 DGKs (DGKδ, DGKη, and DGKκ comprising 296, 286, and 214 amino acids, respectively) and ** marks the insertion of 30 amino acids in CERK.
Although the overall DgkB fold is similar to those of YegS and NAD kinase, our structure is the first to unequivocally identify the nucleotide binding pocket. In the YegS•ADP crystal structure, the adenosine portion was completely disordered, whereas the diphosphate moiety was modeled based on weak electron density. In the A. fulgidus NAD kinase structure (1Z0S) the binding site was deduced from a series of complex structures. Archael NAD kinases also use inorganic polyphosphate in place of ATP (Kawai et al., 2003), and the terminal pyrophosphate binds similarly to the positions of the β and γ phosphates of ATP deduced from the Mg1•DgkB•ADP structure. However, ATP was observed to occupy the adenine pocket of the NAD substrate (1Z0Z), and it was proposed that free ATP displaces the adenosine portion of NAD prior to phosphoryl transfer (Liu et al., 2005). However, such a mechanism appears unlikely based on the identification of the ATP pocket within domain 1 of DgkB. In DgkB, the adenine ring engages a platform created by the β2-α2 loop (Figure 1A, 1C), and NAD kinases display sequence variation in this region that may relate to whether or not adenine binding is required. Archael NAD kinases that use polyphosphate would not be expected to have the DgkB-like adenine-binding pocket that is predicted to occur in the ATP-dependent eukaryotic NAD kinases. In fact, sequence comparison of ATP- versus polyphosphate-dependent NAD kinases reveals a sequence insertion in the former that is suitably positioned to form an adenine binding pocket similar to that of DgkB.
The DgkB Dimerization Interface
Purified DgkB has an apparent subunit molecular weight of 42 kDa as determined by SDS gel electrophoresis (Figure 3A, inset), which is slightly higher than the predicted size of 37,333 Da based on the DNA sequence plus the His-tag. DkgB is a well-behaved soluble protein that exhibited a Stoke's radius of 41 Å by gel filtration chromatography (Figure 3A). This radius translates into a molecular weight of 112 kDa using a set of standard globular proteins as calibrators (Figure 3A, inset), and indicates that DgkB exists either as a compact trimer or extended dimer in solution. To resolve this uncertainty, sedimentation velocity experiments were employed to unequivocally determine the oligomerization state and shape of DgkB (Figure 3B). These data show that DkgB is an extended cigar-shaped 73 kDa dimer in solution with a frictional coefficient (f/f0) of 1.53.
Figure 3.
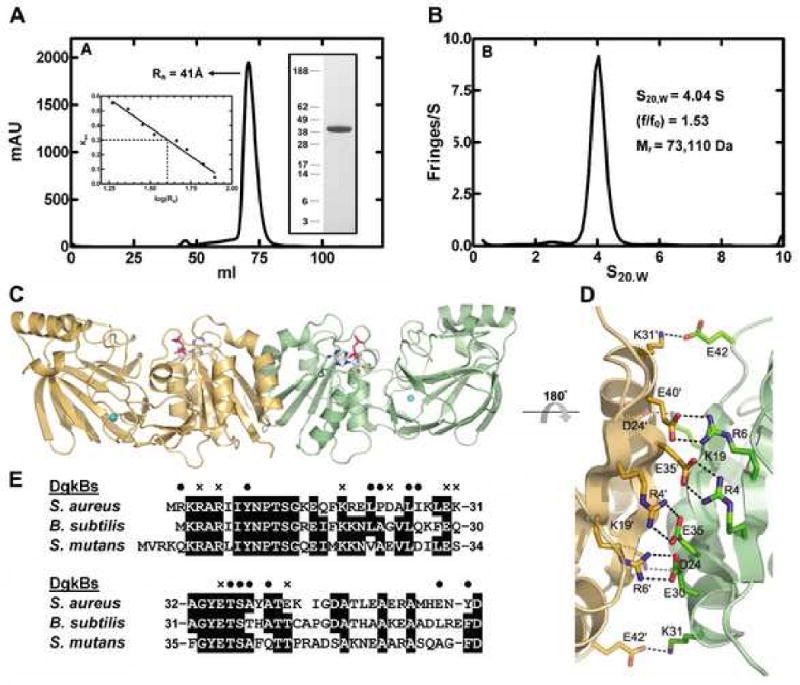
Properties of the DgkB dimer. (A) Gel filtration chromatography of DgkB using a Sephadex-200 10/300 GL column. DgkB eluted with a Stokes radius (Rs) of 41 Å based on calibration of the column with 8 protein standards (left inset). SDS gel electrophoresis (right inset) showed the presence of a single protein with an apparent subunit molecular weight of 42 kDa. The molecular weight calculated from the DNA sequence was 37,333. (B) Sedimentation velocity analysis of DgkB. The protein characteristics determined from the velocity sedimentation experiment are shown as an inset in the figure. (C) The structure of the DgkB dimer. The green monomer is the observed molecule in the asymmetric unit shown in Fig. 1A, tilted backwards 45°. The sand colored monomer is generated by 2-fold symmetry. ADP carbons and Mg1 (sphere) are colored cyan. (D) The conserved DgkB dimerization interface. Only the residues involved in salt bridges are shown as sticks. Hydrogen bonds are indicated with broken lines. (E) A representative sequence alignment of the amino-terminal residues in known bacterial DgkBs involved in dimer formation. Residues responsible for salt bridges and van der Waals interactions are indicated with an “x” and black spheres (•), respectively.
A cigar-shaped dimer, consistent with the hydrodynamic data, is generated by a crystallographic 2-fold symmetry axis in our structure (Figure 3C). The active site locale is distant from the dimer interface, and each monomer can presumably support independent substrate binding and catalysis. The dimer interface is formed by the first 60 residues in the sequence and by secondary structure elements β1, α1, β2 and α2. The buried surface area is 1463 Å2. The interaction surface is mediated by a combination of van der Waals packing between helices α1 and α1′ and a distinctive constellation of salt bridges. A magnified view of the dimer interface is illustrated in Figure 3D. Eight salt bridges are formed at the interface that represents 2 × 4 reciprocal interactions around the two-fold crystal axis (Arg4-Glu35′, Arg6-Glu30′, Lys19-Asp24′, Lys31-Glu42′). A sequence alignment of the N-terminal dimerization region in 3 bacterial members of Pfam00781 that are known to be DgkBs is shown in Figure 3E, and this reveals that the core Arg6-Glu30′ and Arg4-Glu35′ interactions are conserved. The 3KRAR6 and 30ExxGYETS37 motifs are two regions of DgkB proteins that are absent from other bacterial family members that are not DgkBs, notably YegS. Thus, the presence or absence of these two dimerization signature motifs may predict which bacterial family members are DgkBs and which are not. Consistent with this analysis, the E. coli YegS has been reported to be a monomer (Bakali et al., 2007), but the S. typhimurium YegS is stated to be a dimer (Nichols et al., 2007) and may employ an alternative dimerization strategy.
The Structural Divalent Cation Binding Site (Mg1)
A distinctive feature of the DgkB structure is a divalent cation binding site occupied by a Mg2+ ion (Mg1) within the β10-β11 loop in domain 2 (Figure 4A). The hexacoordinate Mg1 complex is formed by the side chain of conserved Asp216, the backbone carbonyl oxygens of Lys213 and Tyr218, and three water molecules oriented by their interactions with the protein. Mg1 is ∼24 Å from the β-phosphate of ADP, and is not the magnesium ion required to coordinate the ATP γ-phosphate within the P-loop. However, a notable feature of the Mg1 binding site evident from Figure 4A is that it connects to the active site via a network of conserved Asp residues (D124 and D271) and well-ordered water molecules. Asp124 has a key structural role and tethers the β10-β11 loop, the domain interface, and the active site residues via four hydrogen bonding interactions (Figure 4A). These interactions involve a Mg1-bound water, the backbone amides of Gly217 and Arg287, and a network water molecule linked to the backbone carbonyl of Ile134. The carboxylate of Asp271 forms hydrogen bonds with the amide nitrogens of conserved domain interface residues Ile134 and Asn135, fixing the orientation of Glu273 on a loop adjacent to the active site. Asn135 lies directly across from Asp68, the active site residue predicted to chelate Mg2 in the P-loop.
Figure 4.
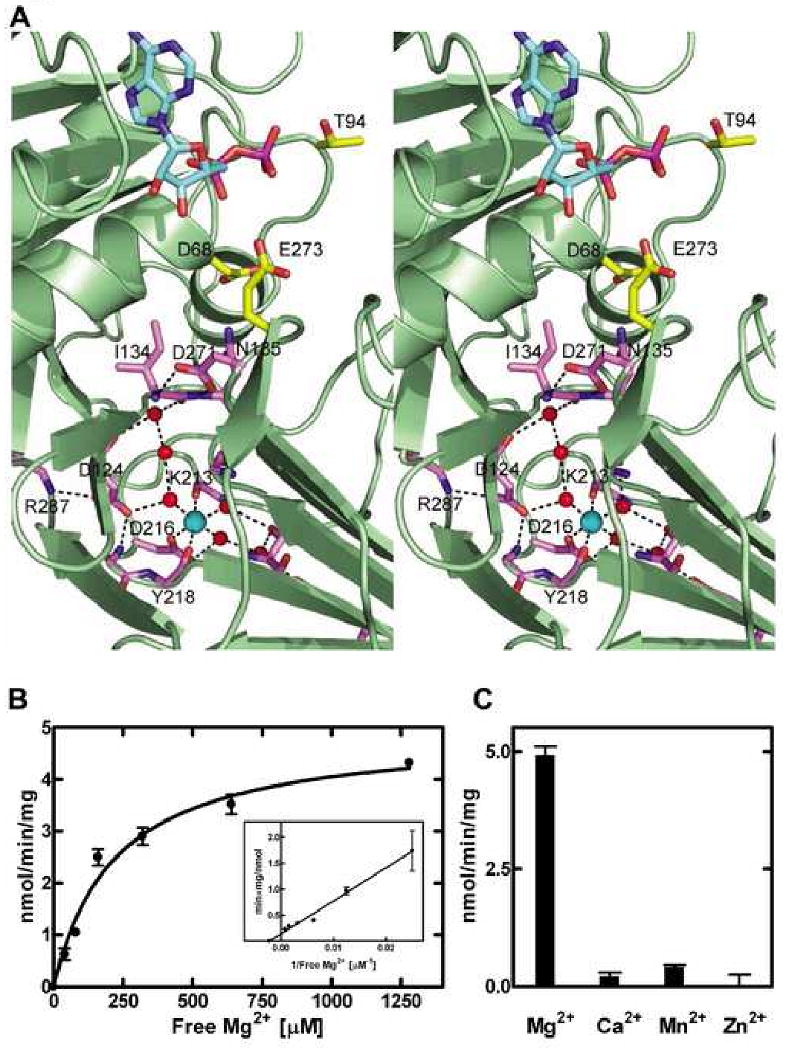
The conserved Asp•water•Mg1 network. (A) A stereo view of the conserved Asp•water•Mg1 network is depicted. The Mg1 and water molecules are shown as cyan and red spheres, respectively. ADP carbons are colored cyan, key active site residues are yellow, and residues connecting the Mg1 to the active site are purple. Hydrogen bonds are indicated by broken lines. (B) The structural Mg1 is required for catalytic activity. Assays were performed at a fixed 5 mM ATP•Mg2+ concentration and the free Mg2+ varied as indicated using the approach outlined by Nakayama et al. (Nakayama et al., 2002). DgkB activity was strictly dependent on a Mg2+ ion independent of the Mg2+ bound to ATP with an apparent Kd of 219 ± 34 μM. The extrapolated X-intercept is marked by the symbol ●. (C) Comparison of divalent cations as DgkB activators.
Mutagenesis to Probe Mechanism
Mutagenesis studies of DgkB based on the crystal structure were performed to further examine three aspects of the catalytic mechanism: 1) to confirm the importance of residues involved in ATP binding; 2) to identify the acidic residue that acts as a general base that abstracts a proton from DAG to facilitate catalysis; and 3) to investigate whether the structural Mg1 site is important for catalysis. DAG is not water soluble, and the assay uses micelles to deliver the substrate to the enzyme as originally described (Walsh and Bell, 1986). The results are summarized in Table 2.
Table 2. Catalytic activities of DgkB missense mutants.
| DgkBa | Activity |
|---|---|
| WT | 4.93 ± 0.06 |
| D68A | <0.4 |
| P91A | <0.4 |
| T94A | <0.4 |
| N96A | <0.4 |
| D97A | <0.4 |
| D124A | <0.4 |
| E168A | 4.99 ± 0.08 |
| D216A | 0.96 ± 0.02 |
| D271A | <0.4 |
| E273A | <0.4 |
DgkB mutants were purified using the same method described for the wild-type protein.
The most important residue in the ATP binding signature motif is Asp68. DgkB(D68A) had no detectable activity consistent with the proposed role of Asp68 in mediating the interaction of Mg2 with the γ-phosphate of ATP (see above). The ADP•Mg2+ product complex structure of E. coli PFK (1PFK) reveals how Mg2+ engages this motif via Asp103, and superposition of our P-loop structure with PFK places Asp68 in a position to coordinate the Mg2+ ion bound to the ATP γ-phosphate. However, in our structure, the side chain of Asp68 makes hydrogen bonds to the backbone amides of Asp97 and Phe98 suggesting that an approximate 120° rotation of the side chain is necessary for Asp68 to function in Mg2+ chelation.
The inability to detect activity in the DgkB(E273A) makes Glu273 a prime candidate for the catalytic base. Most kinases utilize an acidic amino acid side-chain extending into the active site to facilitate the reaction by abstracting a proton from the substrate hydroxyl and lining up the substrate for in-line attack on the ATP γ-phosphate (Mildvan, 1997). Glu273 is firmly fixed at the active site and is ideally located to fulfill this role in the DgkB structure (Figure 4A). Its importance is emphasized by the conservation of this residue in almost every member of the Pfam00781 family including all of the human DGKs (Figure 2). Glu168 was another potential candidate, but mutation of this residue did not affect activity and it is not conserved in the superfamily (Figure 2).
The structural Mg1 site, located approximately 24 Å from the active site, and the associated Asp•water•Mg2+ network are critical for catalysis. The dependence of DgkB activity on the free Mg2+ concentration (Figure 4B) clearly illustrates that two ions are required for full activity. The apparent Kd for free Mg2+ was 219 ± 34 μM (Figure 4B, inset). Neither Ca2+ Mn2+ nor Zn2+ was able to effectively replace Mg2+, although Ca2+ and Mn2+ did support a few percent activity of DgkB (Figure 4C). However, we cannot discriminate whether the loss of catalytic activity observed is due to an inability of these metals to substitute for Mg1 or Mg2. Mutagenesis of the conserved Asp residues involved in the Mg1-water network also compromised activity, most notably DgkB(D124A) and DgkB(D271A). Asp216 is within the coordination shell of Mg1 and furthest from the active site, and DgkB(D216A) shows a modest decrease in activity. Taken together, these data suggest that the Mg1-water network extending toward the active site side is important for optimum catalysis.
Discussion
Our structural studies indicate that DgkB partially extracts the water insoluble DAG from the bilayer and presents the 3-hydroxyl of the glycerol backbone to ATP bound to domain 1 for catalysis at the domain interface. In the dimer structure shown in Figure 3C, the paired active site entrances are both located on the ‘upper’ surface and appropriately positioned to mediate simultaneous interfacial catalysis on membrane-associated DAG substrate. In addition, this orientation would appropriately position the putative β8-α6 substrate recognition loop to identify DAG. This loop distinguishes DgkB from the structurally related YegS and NAD kinases, consistent with its proposed role in substrate recognition. This orientation of the DgkB dimer also places three conserved positively-charged surface residues, Lys15, Arg20 and Lys165 in the correct position to interact with an anionic membrane. Although a standard mixed micelle assay is used in our studies to measure DAG activity, our preliminary results do show that DgkB efficiently catalyzes the phosphorylation of DAG presented in the context of lipid bilayers containing anionic phopholipids (unpublished data).
The Mg1•DgkB•ADP product complex structure represents a catalytically incompetent conformation of the enzyme; the ATP- and DAG-binding pockets are incomplete, and the side chain of Asp68 is in an orientation that prevents interaction with Mg2 at the P-loop. This suggests that a conformational change is required to promote formation of a high-affinity ATP pocket. We predict that this involves a hinge-like motion between the two DgkB domains that is typically observed in kinases to eliminate water molecules from the active site (Taylor et al., 1990). Normal Mode Analysis (Suhre and Sanejouand, 2004) of DgkB predicts that the protein can assume “open” and “closed” conformations, with the DgkB crystal structure representing the mid-range of rotation (Figure 5). To support this, the predicted “closed form” brings the catalytic base, Glu273, and the predicted substrate binding loop from domain 2 into close proximity with ADP and Thr94, the amino acid predicted to bind the γ-phosphate of ATP-Mg2+ in the active form of the enzyme.
Figure 5.
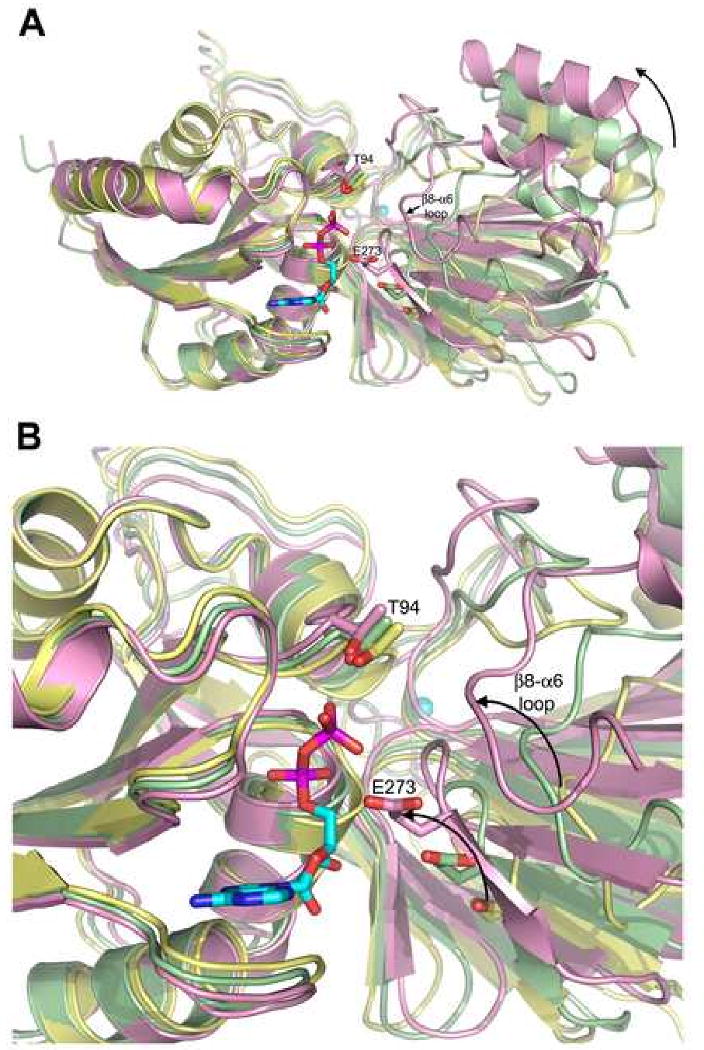
Predicted hinge-like motions in DgkB. (A) The crystal structure of Mg1•DgkB•ADP, shown in palegreen, represents the mid-range of rotation suggested by Normal Mode Analysis (Suhre and Sanejouand, 2004). The two conformational extremes are shown in yellow (“open”) and plum (“closed”). Active site residues Thr94 and Glu273 are shown as sticks for all three models. Protein models were superimposed using domain 1. The curved arrow indicates the direction of rotation of domain 2 relative to Domain 1. (B) A close-up view of panel A highlighting the predicted movement of Glu273 and the putative substrate binding loop upon adopting a “closed” conformation.
Our study clarifies the importance of the structural Mg1 binding pocket within domain 2 in DAG kinase function. The significance of Mg1 is not only in stabilizing the overall fold of the protein, but also in fixing the position of critical active site residues located over 20 Å away. Although Mg1 clearly stabilizes a local region of the fold within an extended loop, a comparison of the DgkB and Mg1•DgkB•ADP structures shows no significant changes in the overall protein conformation indicating that Mg1 binding is not essential to establishing the fold. However, mutagenesis data clearly show that Mg1 binding independent of P-loop ATP-Mg2+ binding is important to support catalysis. There are two conserved aspartic acid residues that participate in forming the connection between the Mg1 and the active site via a structured network of water molecules. Mutation of either Asp124 located in the inter-domain linker or Asp271 at the domain interface yield significantly compromised enzymes. Thus, the Asp•water•Mg1 network is structurally positioned to influence both the orientation of the general base and the residues that bind the ATP γ-phosphate•Mg2+ complex in the active site thereby providing a rationale for the dependence of DgkB activity on the presence of this structural Mg1.
The DgkB structure is directly relevant to the human enzymes that function in cell signaling because the key structural and active site residues are conserved in the sequences of both enzymes (Figure 2). Figures 1A and 2 also reveal that the three major inserts in the human DAG kinases compared to S. aureus DgkB (IN1, IN2 and IN3) all occur in loop regions between the conserved structural elements. The most striking homology occurs in the ATP-binding P-loop, which is the signature motif that identifies the Pfam00781 proteins. Accordingly, mutation of Asp434 in pig DGKα (analogous to Asp68 in DgkB) inactivated the enzyme (Abe et al., 2003). The mammalian DAG kinase catalytic cores also contain the aspartic acid residues that form the Mg1-water network in DgkB that is necessary for optimal catalysis. Mutagenesis of the aspartate residues in pig DGKα that are equivalent to Asp124, Asp216 and Asp271 in DgkB all compromise DGKα catalysis (Abe et al., 2003). In DGKα, Ca2+ is an activating metal due to the presence of an E-F hand domain located outside the catalytic core (Jiang et al., 2000). Although DGKα has not been carefully studied with respect to the cation dependence of the isolated catalytic core, Ca2+ does stimulate the activity of the DGKα catalytic core, and this activation is abolished by mutation of the conserved aspartate residues forming the metal binding site (Abe et al., 2003). Finally, the proposed general base, Glu273, is also conserved in mammalian DAG kinases (Glu699 in pig DGKα). This residue has not been mutated in the mammalian system, but its consistent positioning adjacent to the conserved Asp697 (Asp271 in DgkB) predicts that it plays the same role in both enzymes.
Experimental Procedures
Purification, Properties and Assay of DgkB
The S. aureus SAR1989 gene (DgkB) was amplified by PCR using primer pairs containing an XhoI site upstream of the initiating codon and BamHI site after the stop codon to create an amino terminal His-tagged protein in the pEt-15b expression vector pAJ015 The E. coli BL21 (DE3) strain (Novagen) harboring pAJ015 was grown in LB medium, supplemented with 100 μg/mL carbenicillin and 30 μg/mL chloramphenicol, at 37°C with rotary shaking until the A600 reached 0.8. Then, 400 μM IPTG was added to induce expression for 16 h at 25°C. Lysed cell extract was loaded onto a Ni-NTA affinity column (Qiagen) and the resin was washed with 10 column volumes of 20 mM Tris, pH 7.9, 500 mM NaCl, 1 mM β-mercaptoethanol, 10 mM imidazole and 10% (v/v) glycerol, followed by 10 column volumes of the same buffer containing 50 mM imidazole. DgkB was eluted from the column in the same buffer containing 500 mM imidazole. Fractions containing DgkB were concentrated and applied to a Superdex-200 16/60 GL column. Aliquots (150 μl) were flash-frozen in liquid nitrogen and stored at −80°C. Selenomethionine-labeled DgkB was expressed in an E. coli methionine auxotroph strain B834 grown in M9 minimal medium supplemented with glucose and an amino acid mix containing selenomethionine (Price et al., 2001). Protein was measured by the Bradford method (Bradford, 1976).
Sedimentation velocity experiments were conducted at 50,000 rpm and 4°C using a ProteomeLab XL-I analytical ultracentrifuge, equipped with both absorbance and interference optical detection systems, an eight-hole Beckman An-50 Ti rotor, and cells containing sapphire windows and charcoal-filled Epon double-sector centerpieces. The sedimentation velocity profiles of the protein (50 μg/ml, 400 μl) were collected at 230 nm every 220 sec. The SEDNTERP program (Laue et al., 1992) was used to estimate the molecular weight and the partial specific volume. Data were modeled as a superposition of Lamm equation solutions, c(s), with the software SEDFIT 9.2 (www.analyticalultracentrifugation.com) (Schuck, 2000; Schuck et al., 2002). The sedimentation coefficient distribution c(s) was calculated with maximum entropy regularization, the molecular weight distribution, c(M) was calculated using the value determined from the c(s) distribution, and the f/f0 value was determined from the c(s) analysis (Schuck et al., 2002).
The DgkB micelle assay was modeled after the assay developed by Walsh and Bell (Walsh and Bell, 1986). Assays contained 50 mM MOPS, pH 7.0, 10 mM MgCl2, 1 mM EGTA, 150 mM LiCl, 50 μM [14C]diacylglycerol (specific activity = 55 mCi/mmol), 5 mM ATP, and 100 mM octyl-β-d-glucopyranoside in a final volume of 50 μl. The reactions were initiated by the addition of protein and incubated at 25°C for 5 minutes. Reactions were terminated by spotting 30 μl aliquots onto a Whatmann DE81 filter disc that was immediately placed in hexane:ethyl ether (1/1,v/v), the discs were washed 3 × 30 min with 10 ml/disc and the bound [14C]PtdOH was quantitated by liquid scintillation counting in triplicates.
Crystallography and Structure Determination
Selenomethionine-labeled DgkB was crystallized at 4°C by the vapor diffusion method. Upon setup, the drop contained 0.1 M Tris pH 8.5, 9% polyethylene glycol 2000 monomethylether (PEG 2KMME), 1 mM EDTA, 1 mM DTT and 5 mg/ml protein. The well solution contained 0.1 M Tris pH 8.5 and 18% PEG 2KMME. Tetragonal crystals with space group P42212 appeared in about 3 days. Mg1•DgkB•ADP crystals were formed by soaking DgkB crystals with 8 mM ADP and 50 mM MgCl2. Crystals were flash-frozen in mother liquor containing 20% glycerol for cryoprotection. A 2.4 Å DgkB single anomalous dispersion (SAD) dataset at the ‘peak’ wavelength (0.9794 Å) was measured at the SER-CAT 22ID beamline of the Advanced Photon Source (APS). The data were integrated and scaled using HKL-2000 (Otwinowski and Minor, 1997). A sub-structure solution was found using Solve-v2.13 (Terwilliger and Berendzen, 1999). Nine of the 11 possible Se sites were found automatically and input into Resolve (Terwilliger, 2000; Terwilliger, 2003) for density modification, and model building. Roughly 60% of the asymmetric unit was traced automatically. The model was refined using REFMAC (Murshudov et al., 1997). Model building was performed using COOT (Emsley and Cowtan, 2004). A 2.3 Å Mg1•DgkB•ADP dataset was measured at SER-CAT 22ID of the APS, and processed using HKL2000. The final DgkB model, without waters, was used for rigid body refinement in REFMAC. Manual building and refinement were performed using COOT and REFMAC. Data measurement, phasing and refinement statistics are listed in Table 1. Atomic coordinates and structure factors have been deposited with Protein Data Bank accession codes 2QVL (DgkB) and 2QV7 (Mg1•DgkB•ADP). Figure 2 was generated with ESPript (Gouet et al., 1999). All other figures were generated with PyMOL (DeLano, 2002).
Acknowledgments
We thank Chitra Subramanian and Amanda Nourse for technical assistance, and SER-CAT staff scientists Zheng-Qing Fu and Zhongmin Jin. This work was supported by NIH grant GM34496, Cancer Center Support Grant CA 21765, and the American Lebanese Syrian Associated Charities. SER-CAT supporting institutions may be found at www.ser.anl.gov/new/index.html. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe T, Lu X, Jiang Y, Boccone CE, Qian S, Vattem KM, Wek RC, Walsh JP. Site-directed mutagenesis of the active site of diacylglycerol kinase α: calcium and phosphatidylserine stimulate enzyme activity via distinct mechanisms. Biochem J. 2003;375:673–680. doi: 10.1042/BJ20031052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakali HMA, Herman MD, Johnson KA, Kelly AA, Wieslander Å, Hallberg BM, Nordlund P. Crystal structure of YegS, a homologue to the mammalian diacylglycerol kinases, reveals a novel regulatory metal binding site. J Biol Chem. 2007;282:19644–19652. doi: 10.1074/jbc.M604852200. [DOI] [PubMed] [Google Scholar]
- Berger SA, Evans PR. Site-directed mutagenesis identifies catalytic residues in the active site of Escherichia coli phosphofructokinase. Biochemistry. 1992;31:9237–9242. doi: 10.1021/bi00153a017. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Cabacungan E, Pieringer RA. Mode of elongation of the glycerol phosphate polymer of membrane lipoteichoic acid of Streptococcus faecium ATCC 9790. J Bacteriol. 1981;147:75–79. doi: 10.1128/jb.147.1.75-79.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL molecular graphics system. DeLano Scientific; Palo Alto, CA: 2002. [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Garavaglia S, D'Angelo I, Emanuelli M, Carnevali F, Pierella F, Magni G, Rizzi M. Structure of human NMN adenylyltransferase. A key nuclear enzyme for NAD homeostasis. J Biol Chem. 2002;277:8524–8530. doi: 10.1074/jbc.M111589200. [DOI] [PubMed] [Google Scholar]
- Goldberg DE, Rumley MK, Kennedy EP. Biosynthesis of membrane-derived oligosaccharides: a periplasmic phosphoglyceroltransferase. Proc Natl Acad Sci U S A. 1981;78:5513–5517. doi: 10.1073/pnas.78.9.5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- Jerga A, Lu YJ, Schujman GE, de Mendoza D, Rock CO. Indentification of a soluble diacylglycerol kinase required for lipoteichoic acid production in Bacillus subtilis. J Biol Chem. 2007;282:21738–21745. doi: 10.1074/jbc.M703536200. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Qian W, Hawes JW, Walsh JP. A domain with homology to neuronal calcium sensors is required for calcium-dependent activation of diacylglycerol kinase α. J Biol Chem. 2000;275:34092–34099. doi: 10.1074/jbc.M004914200. [DOI] [PubMed] [Google Scholar]
- Kawai S, Mori S, Murata K. Primary structure of inorganic polyphosphate/ATP-NAD kinase from Micrococcus flavus, and occurrence of substrate inorganic polyphosphate for the enzyme. Biosci Biotechnol Biochem. 2003;67:1751–1760. doi: 10.1271/bbb.67.1751. [DOI] [PubMed] [Google Scholar]
- Koch HU, Haas R, Fischer W. The role of lipoteichoic acid biosynthesis in membrane lipid metabolism of growing Staphylococcus aureus. Eur J Biochem. 1984;138:357–363. doi: 10.1111/j.1432-1033.1984.tb07923.x. [DOI] [PubMed] [Google Scholar]
- Koga Y, Nishihara M, Morii H. Products of phosphatidylglycerol turnover in two Bacillus strains with and without lipoteichoic acid in the cells. Biochim Biophys Acta. 1984;793:86–94. [PubMed] [Google Scholar]
- Labesse G, Douguet D, Assairi L, Gilles AM. Diacylglyceride kinases, sphingosine kinases and NAD kinases: distant relatives of 6-phosphofructokinases. Trends Biochem Sci. 2002;27:273–275. doi: 10.1016/s0968-0004(02)02093-5. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, McArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the quality of protein structures. J Appl Crystallogr. 1993;26:282–291. [Google Scholar]
- Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Analytical Centrifugation in Biochemistry and Polymer Science. Cambridge: The Royal Society of Chemistry; 1992. [Google Scholar]
- Liu J, Lou Y, Yokota H, Adams PD, Kim R, Kim SH. Crystal structures of an NAD kinase from Archaeoglobus fulgidus in complex with ATP, NAD, or NADP. J Mol Biol. 2005;354:289–303. doi: 10.1016/j.jmb.2005.09.026. [DOI] [PubMed] [Google Scholar]
- Mildvan AS. Mechanisms of signaling and related enzymes. Proteins: Struct Funct Genet. 1997;29:401–416. [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nakayama S, Nomura H, Smith LM, Clark JF. Simultaneous estimation of intracellular free Mg2+ and pH by use of a new pH-dependent dissociation constant of MgATP. Jpn J Physiol. 2002;52:323–326. doi: 10.2170/jjphysiol.52.323. [DOI] [PubMed] [Google Scholar]
- Neuhaus FC, Baddiley J. A continuum of anionic charge: structures and functions of D-alanyl-teichoic acids in gram-positive bacteria. Microbiol Mol Biol Rev. 2003;67:686–723. doi: 10.1128/MMBR.67.4.686-723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CE, Lamb HK, Lockyer M, Charles IG, Pyne S, Hawkins AR, Stammers DK. Characterization of Salmonella typhimurium YegS, a putative lipid kinase homologous to eukaryotic sphingosine and diacylglycerol kinases. Proteins. 2007;68:13–25. doi: 10.1002/prot.21386. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Oxenoid K, Sonnichsen FD, Sanders CR. Topology and secondary structure of the N-terminal domain of diacylglycerol kinase. Biochemistry. 2002;41:12876–12882. doi: 10.1021/bi020335o. [DOI] [PubMed] [Google Scholar]
- Price AC, Zhang YM, Rock CO, White SW. The structure of β-ketoacyl-[acyl carrier protein] reductase from Escherichia coli: negative cooperativity and its structural basis. Biochemistry. 2001;40:12772–12781. doi: 10.1021/bi010737g. [DOI] [PubMed] [Google Scholar]
- Sakane F, Shin-ichi I, Masahiro K, Satoshi Y, Hideo K. Diacylglycerol kinases: Why so many of them? Biochim Biophys Acta. 2007;1771:793–806. doi: 10.1016/j.bbalip.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P, Perugini MA, Gonzales NR, Howlett GJ, Schubert D. Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys J. 2002;82:1096–1111. doi: 10.1016/S0006-3495(02)75469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman H, Kennedy EP. Relation of turnover of membrane phospholipids to synthesis of membrane-derived oligosaccharides of Escherichia coli. J Biol Chem. 1977;252:4250–4255. [PubMed] [Google Scholar]
- Smith RL, O'Toole JF, Maguire ME, Sanders CR., II Membrane topology of Escherichia coli diacylglycerol kinase. J Bacteriol. 1994;176:5459–5465. doi: 10.1128/jb.176.17.5459-5465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhre K, Sanejouand YH. ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32:W610–W614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taron DJ, Childs WC, III, Neuhaus FC. Biosynthesis of D-alanyl-lipoteichoic acid: role of diglyceride kinase in the synthesis of phosphatidylglycerol for chain elongation. J Bacteriol. 1983;154:1110–1116. doi: 10.1128/jb.154.3.1110-1116.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Buechler JA, Yonemoto W. cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annu Rev Biochem. 1990;59:971–1005. doi: 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- Terwilliger T, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:501–505. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr. 2003;59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Blitterswijk WJ, Houssa B. Properties and functions of diacylglycerol kinases. Cell Signal. 2000;12:595–605. doi: 10.1016/s0898-6568(00)00113-3. [DOI] [PubMed] [Google Scholar]
- Walsh JP, Bell RM. sn-1,2-Diacylglycerol kinase of Escherichia coli. Mixed micellar analysis of the phospholipid cofactor requirement and divalent cation dependence. J Biol Chem. 1986;261:6239–6247. [PubMed] [Google Scholar]