

Abstract
Vaccine‐induced CD8+ T‐cell responses can eradicate developing tumors in vivo in mouse models. Translating these successes into approved treatments for cancer patients has been challenging, since many of these models lack expression of clinically proven/relevant tumor antigens. We have shown that mesothelin is a clinically relevant CD8+ T‐cell target in human pancreas cancer, which is also highly conserved among species. Here, we utilize the murine mesothelin‐expressing pancreatic tumor model (Panc02) to identify the immune‐relevant mesothelin‐derived peptides and study interventions that enhance the antitumor response. We first screened overlapping peptides of the entire murine mesothelin protein to identify two new CD8+ mesothelin‐restricted epitopes. These peptides were then evaluated for recognition by vaccine‐induced T cells from mice treated with vaccine in sequence with low‐dose cyclophosphamide (CY) and an anti‐CD25 IL‐2Rα monoclonal antibody (PC61). These treatments are both known to deplete subpopulations of T regulatory cells (Tregs). Our findings demonstrate that combined Treg‐depleting therapies synergize to enhance vaccine efficacy. Furthermore, our data supports mesothelin as a relevant antigen in murine and clinical models and the use of Panc02 as a clinically relevant murine model of pancreatic cancer for evaluating antigen‐targeted immunotherapies in immune‐tolerant hosts.
Keywords: mesothelin, T regulatory cells, depletion, peptide identification, vaccination, tumor therapy
Introduction
The goal of cancer vaccine development is to identify approaches that can recruit and activate CD8+ T cells. Numerous preclinical and clinical studies have demonstrated the recognition of cancer‐associated antigens that are the targets of vaccine‐induced CD8+ T‐cell responses. Transfer of antigen‐specific CD8+ T cells into tumor‐bearing mice and some cancer patients has demonstrated the clinical utility of inducing immune‐relevant CD8+ T cells that can traffic to and lyse tumors expressing the target antigen. However, therapeutic cancer vaccines have been limited by the existence of multiple mechanisms of immune tolerance that prevent the induction and maintenance of activated CD8+ T cells in tumor‐bearing hosts. Therefore, preclinical tumor models with corresponding tumors that express clinically relevant antigens have become an important tool for developing and optimizing vaccine approaches that can overcome multiple mechanisms of immune tolerance. Although current preclinical models have already been employed to evaluate a number of tolerance mechanisms, there is still a deficit of models that utilize antigens known to be immune‐relevant targets expressed by human tumors. Additional models that represent multiple categories of immune‐relevant tumor antigens, as well as multiple tumor types that likely have different tumor microenvironments and mechanisms of T‐cell downregulation, are needed to facilitate progress in this field.
Mesothelin is a recently identified CD8+ T‐cell target that is expressed by the majority of pancreatic and ovarian cancers. Mesothelin is a member of the megakaryocyte‐potentiating factor (MPF) gene family that shares 58% amino acid homology between mice and humans (NP_061345 and NP_005814). The gene encodes a 69‐kDa protein that is processed to a 40‐kDa cell surface glycoprotein (mesothelin), a differentiation antigen expressed on mesothelial cells lining the pleura, the pericardium, and the peritoneum, and a 31‐kDa shed fragment known as the MPF. 1 ” 3 In addition, mesothelin may play a role in the process of pancreatic tumor development, adhesion, and dissemination. 4 The limited expression of mesothelin in normal tissues and its overexpression in many tumor types, especially pancreatic cancers, make it an excellent candidate target for testing in antigen‐specific vaccine strategies. Its utility as a candidate tumor antigen is further emphasized by recent findings that mesothelin is overexpressed by about 20–30% of all tumor types tested. Furthermore, a phase I study of an allogeneic granulocyte‐macrophage colony‐stimulating factor (GM‐CSF)‐transduced pancreatic tumor vaccine demonstrated the postvaccination induction of CD8+ T cells specific for mesothelin that correlated with prolonged disease‐free survival. 5 Although limited in expression on normal cells, it is likely that mesothelin represents a typical tumor‐associated antigen that elicits multiple mechanisms of immune tolerance when targeted by immunization approaches in tumor‐bearing hosts.
Immune tolerance and the failure to generate immunity to tumor‐associated antigens have been attributed to the inhibitory and suppressive function of regulatory T cells (Tregs) in mouse models and patients. 6 , 7 , 8 , 9 The detrimental effects of Tregs on tumor immunity are supported by evidence of increased numbers of CD4+CD25+ Tregs in the blood of patients with ovarian, lung, breast, and pancreatic cancers. In addition, a poor prognosis has been correlated with increased numbers of tumor‐infiltrating Tregs in patients with these cancers. 10 , 11 , 12 , 13 To this end, the inhibition of Treg‐mediated suppression has been evaluated as a mechanism for modulating the host environment to improve vaccine efficacy. Previous studies have demonstrated that combining vaccine treatment with immune‐modulating doses of chemotherapy to diminish tolerance caused by the inhibitory effects of regulatory CD4+CD25+ Tregs can facilitate the induction of antigen‐specific immune responses. 14 , 15 , 16 Specifically, treatment of tumor‐bearing mice with cyclophosphamide (CY), anti‐CD25 IL‐2R (PC61), or anti‐GITR monoclonal antibodies (mAb) has been shown to induce tumor‐specific T cells and enhance the rejection of advanced tumors in subsets of mice. 17 , 18 , 19 , 20 , 21 However, none of these Treg‐depleting agents when given as single agents in sequence with immunotherapeutic strategies have cured the majority of treated mice, nor have cured mice of larger tumor burdens. Thus, tumor models with defined immune‐relevant cancer‐associated antigens are needed to better elucidate the mechanisms of immune tolerance, including tolerance caused by Tregs.
Panc02 is a transplantable tumor line that has similarities to human pancreatic adenocarcinomas. We have found that this tumor line also overexpresses mesothelin. 4 , 22 Here, we report the use of this tumor model to better understand the role of Tregs in inhibiting immunity to mesothelin. Specifically, we developed a screening approach using an overlapping peptide library to identify a panel of H‐2Kb‐ and Db‐restricted mesothelin epitopes that cover the entire protein. We then studied the immune responses induced by combinatorial Treg‐depleting and vaccine approaches that target these epitopes. Our studies demonstrate that a combination of Treg depletion and whole‐cell GM‐CSF vaccine is successful in generating immunity to established Panc02 tumors and can induce a mesothelin‐specific CD8+ T‐cell repertoire that recognizes naturally processed mesothelin epitopes. Identification and characterization of these epitopes in the context of a relevant tumor antigen model are critical steps toward developing a specific mesothelin‐based antigen/peptide vaccine approach to test in patients.
Materials
Cell lines and medium
Panc02 is a highly tumorigenic murine pancreatic tumor cell line with ductal morphology derived from a methylcholanthrene‐treated C57B1/6 mouse. 22 , 23 Panc02 cells were maintained in DMEM (Life Technologies, Rockville, MD, USA) supplemented with 10% Fetalclone II (Hyclone, Logan, UT, USA), 1% L‐glutamine (Life Technologies), and 0.5% Penicillin/Streptomycin (Life Technologies) in a humidified atmosphere at 37°C under 5% C02. The minimum tumorigenic dose (100% tumor outgrowth) for Panc02 was established to be 2.5 × 10 5 cells (data not shown). B78H1‐GM cells are an MHC class I‐negative variant of the B16 melanoma tumor, engineered to secrete GM‐CSF. 24 B78H1‐GM cells were maintained in RPMI (Life Technologies) supplemented with 10% Fetalclone II, 0.5% L‐glutamine, and 1% Pencillin/ Streptomycin. The T‐2Db‐ and T‐2Kb‐expressing cell lines were a gift from Peter Cresswell, and were maintained in IMDM (Life Technologies) supplemented with 10% Fetalclone II, 1% L‐glutamine, 1% sodium pyruvate (Sigma‐Aldrich, St. Louis, MO, USA), 1% nonessential amino acids (Sigma‐Aldrich), 0.5% Penicillin/Steptomycin, and 0.5 mg/mL G418 (LifeTechnologies). MHC class I expression on these cells was confirmed routinely by flow cytometry ( Figure 1A ).
Figure 1.
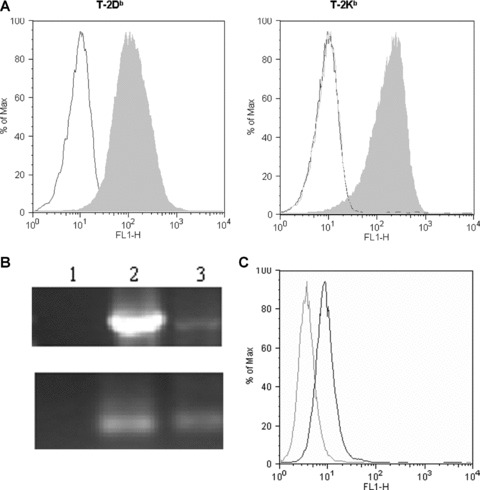
Characterization of cell lines confirms H‐2Kb and H‐2Db expression on T‐2 cells by flow cytometry and mesothelin expression on Panc02 cells by RT‐PCR. (A) T‐2Kb and T‐2Db cells were incubated with mAb to H‐2Kb/H‐2Db followed by FITC‐conjugated secondary Ab to mouse IgG to confirm surface expression of the MHC molecules (solid gray). An H‐2Db‐specific antibody was used to detect H‐2Db on cells. The same antibody was used to demonstrate the lack of H‐2Db on the H‐2Kb‐expressing cells (gray line). Expression and flow cytometric analysis was controlled by the simultaneous staining of these T‐2 cells with a mouse lgG2aK isotype control (black line). (B) RT‐PCR for mesothelin mRNA of the Panc02 murine pancreatic cancer cell line demonstrates strong expression as a 2,000 bp band, compared to the B78H1‐GM cell line (top gel), and no expression on chloramphenicolacetyltransferase‐cDNA, which served as a kit control. GAPDH (bottom gel) served as an RNA control, and PCR products were confirmed by visualization of a band at 100 bp (1 = Control RNA, 2 = Panc02, 3 = B78H1‐GM) (C) CAK‐1 staining (black line) of Panc02 cells after cell permeabilization. Isotype control staining is shown in the gray line.
Primers and RT‐PCR
Panc02 mesothelin expression was confirmed by RT‐PCR [ Figure IB ). Total RNA was isolated using the RNeasy Isolation Kit (Qiagen, Valencia, CA, USA). First‐strand synthesis of cDNA was carried out using the Superscript II kit (Invitrogen, Carlsbad, CA, USA), according to the manufacturers protocols. PCR primers were designed to amplify a 2‐kb cDNA mesothelin fragment as follows: Meso2s:5′‐CGGAATTCTCTACACAGACCATGGCCTTG‐3′ and Meso4a: 5′‐GCCCCAGTAGAGCTGGGACC‐3′. Glyceral‐dehyde‐3‐phosphate dehydrogenase (GAPDH) primers were used as a control. PCR conditions consisted of an initial denaturation (95°C, 2 minutes), followed by 34 cycles of amplification (95°C, 1 minutes; 62°C, 30 seconds; 72°C, 2 minutes and 30 seconds), and a final extension step (72°C, 15 minutes). PCR products were resolved by electrophoresis in a 1% agarose gel and stained with ethidium bromide. Control PCR reactions consisted of cDNA synthesized from B78H1‐GM cells and chloramphenicolacetyltransferase‐cDNA provided in the Superscript II kit { Figure IB ).
Peptides
A panel of 123 peptides was synthesized as a means to identify Kb‐ and Db‐restricted murine mesothelin epitopes. The Peptide Generator Database (PeptGen) (http://hiv‐web.lanl.gov/content/ hiv‐db/PEPTGEN/PeptGenSubmitForm.html) was used to create sets of overlapping peptides of the mouse mesothelin protein sequence obtained from the NCBI Protein database. Consecutive peptides were constructed as 15‐mers that overlapped by 10 (>95% purity) at the Johns Hopkins Oncology Peptide Synthesis C ore Facility. Furthermore, 8‐ and 9‐mers were generated from the 15‐mers that elicited a positive immune response.
PC61 production
Hybridoma clone PC61.5.3 (anti‐CD25 IL‐2R) was purchased from ATCC (catalog number TIB‐222 Manassas, VA, USA). Cells were maintained in DMEM supplemented with 2% L‐glutamine, 1% nonessential amino acids, 0.05 mM 2‐mercaptoethanol, 10% FBS, 0.5% Penicillin/Streptomycin, and 0.02% Gentamycin in a humidified atmosphere at 37°C under 10% C02. For the production of the antibody (Ab), the cells were cultured in protein‐free hybridoma medium (PFHMII; Life Technologies) for approximately 10 days, and the viability was analyzed by trypan blue staining until a majority of cells were dead. Supernatants were collected and large cellular debris was removed by centrifugation for 25 minutes (2,500 rpm). PC61 was concentrated using Centricon‐30 tubes (Millipore, Billerica, MA, USA), according to the manufacturer's instructions, and then dialyzed by three overnight exchanges in PBS at 4°C. SDS‐PAGE (Biorad, Hercules, CA, USA) and Coomassie blue staining confirmed PC61 purity.
Flow cytometry
The cells were stained in FACS buffer (2% FBS in PBS) with the primary Ab for 20 minutes at 4°C. Antibodies to CD4 (Allophycocyanin‐conjugated, APC) and CD25 (Biotin‐conjugated, pure) were purchased from BD Pharmingen (La Jolla, CA, USA). PE‐labeled Ab to Foxp3 was purchased from eBiosciences (San Diego, CA, USA), and intracellular staining was carried out according to the manufacturer's instructions. The secondary Ab for CD25 was Streptavidin‐Alexa Fluor® 488 (Invitrogen, Carlsbad, CA, USA). The cells were stained and analyzed by FACScan (Becton Dickinson, Mountain View, CA, USA) using the CellQuest software. The acquired data were reanalyzed using the Flowjo software (Treestar, Inc., Ashland, OR, USA). For the mesothelin staining, CAK‐1 Ab was used after cell permeabilization ( Figure 1C ).
T‐cell assays
In vitro T‐cell stimulations were performed to increase the precursor frequencies of mesothelin‐specific CD8+ T cells from vaccinated mice prior to epitope‐screening studies. Immunized mice were euthanized by C02 inhalation 7–10 days after vaccination. Splenocytes and axillary lymph nodes (LNs) were isolated, made into single‐cell suspensions using a 100‐μm filter mesh, and cultured in CTL medium (RPMI supplemented with 10% FBS, 0.5% L‐glutamine, 1% Penicillin/Streptomycin, and 0.05 mM 2‐mercaptoethanol). Erythrocytes were removed by ACK lysis (Biosource, Rockville, MD, USA), and lymphocytes were washed, counted, and resuspended at a concentration of 1 × 10 6 cells/mL. Lymphocytes (2 × 106) were stimulated for 7 days with 2.5 μg/mL of specific peptide in a 24‐well plate (2 mL/ well), with medium replacement on day 3. Stimulated CD8+ T cells were harvested and assayed for specific activity on day 7 using GM‐CSF secretion as a surrogate marker of T‐cell effector function. Cultured CD8+ T cells were isolated using the Dynal CD8‐negative isolation kit (Dynal, Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. Isolated CD8+ T cells were counted and resuspended in CTL medium at a concentration of 3 × 106 cells/mL. T‐2Kb and T‐2Db target cells (6 × 105 cells/mL) were pulsed with relevant peptide at 2.5 μg/ mL in CTL medium for 2–5 hours at room temperature, with occasional gentle shaking to prevent cells from settling. The target cells were irradiated with 5,000 rads. The targets and CD8+ T cells were cultured for 24 hours in flat‐bottomed 96‐well plates at a ratio of 3 × 104 T‐2 cells to 3 × 105 CD8+ T cells (0.15 mL final volume). Supernatants were harvested and analyzed using GM‐CSF ELISA kits (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's instructions.
Intracellular cytokine staining (ICS)
ICS was performed as directed using a BD Pharmingen kit for detection of murine interferon‐gamma (IFN‐γ). Briefly, 1 × 106 splenocytes (nylon wool purified or collected from the 1‐week stimulation cultures) were incubated for 6 hours with an equal ratio of indicated targets in the presence of Golgi Stop (1.3 μl;/mL). Cells were washed in FACS buffer and stained with α‐CD8Cy (BD‐Pharmingen) and fixed, permeabilized, and stained with α‐IFN‐γ‐PE (BD‐Pharmingen). For the peptide titration assays, splenocytes were stimulated for 7 days with 2 μM of the specific 8‐mers, collected, and used for the ICS. T‐2Kb pulsed with a range of peptide concentration from 2 μM to 200 pM were used as targets. Control target cells were pulsed with ovum‐derived SINFEKKL peptide (Kb‐restricted) 25 or LCMV GP133_41 (T‐2Db). 26 ’ 27 The percentage of antigen‐specific cells was calculated by subtracting the percentage of IFN‐γ+ T cells in the irrelevant antigen sample from the percentage of IFN‐γ+ T cells in the relevant antigen sample.
Mice and in vivo experiments
C57BL/6 mice (6–8 weeks) were purchased from Taconic Farms (Hudson, NY, USA) for all experiments. For tumor studies, mice received a subcutaneous injection of 2.5 × 10750.1 mL Panc02 cells on the inner flank of the right hind limb on day 0. On day 2, CY (100 mg/kg) and PC61 (50 μg) diluted in PBS were injected intraperitoneally (i.p.) (0.5 mL volume). On day 3, whole‐cell vaccines or mock vaccines were administered. Specific vaccine was composed of a mixture of irradiated (5,000 rads) Panc02 and B78H1‐GM cells at a concentration of 2 × 107/mL of each cell type. Mock vaccine consisted of irradiated B78H1‐GM cells only at a concentration of 1 × 107/mL. Cultured vaccine or mock vaccine were harvested, washed in PBS, irradiated, and administered subcutaneously (s.c.) in equal aliquots in the remaining three limbs (0.1 mL volume). Mice were monitored twice a week for the formation of palpable tumors, defined by a diameter that exceeded 3 mm. Animals were sacrificed by C02 inhalation when tumor diameters exceeded 20 mm or sooner if ulceration of the tumor was evident.
In tumor protection studies, bone marrow dendritic cells (DCs) were generated from C57BL/6 mice, as previously described. 28 On the day of immunization, DCs were collected, resuspended at 4 × 106/mL in AIM‐V medium (Life Technologies), and pulsed with 300 μg/mL peptide for 3 hours. DCs were washed three times in PBS (pH 7.4), resuspended at 2 × 106 cells/mL, and injected s.c. in each hind limb on days 0 and 7 (0.1 mL volume). Mice were subsequently challenged with Panc02 tumor cells in the right hind limb on day 14 and checked twice a week for the development of tumors.
In vivo CTL
Splenocytes from naive C57B1/6 mice were pulsed with 2.5 μg/mL of irrelevant (SIINFEKL) or specific mesothelin peptide for 2 hours at room temperature. Pulsed splenocytes were washed twice with PBS and then labeled with 2.5 or 0.25 μM CFSE (Molecular Probes, Eugene, OR, USA). A total of 5 × 106 CFSE‐labeled mesothelin and irrelevant peptide‐pulsed cells were transferred intravenously (i.v.) into the indicated groups of mice. Eighteen hours later, the splenocytes were isolated as previously described. Target cells were distinguished from recipient cells based on the CFSE staining. The percent killing of the CFSE‐labeled peptide‐pulsed targets was calculated as follows: 100‐[(percentage of relevant peptide pulsed in immunized/percentage of irrelevant peptide pulsed in immunized)/(percentage of relevant peptide pulsed in naive/ percentage of irrelevant peptide pulsed in naive)] x 100. 29
Statistical analysis of Kaplan‐Meier survival plots
Kaplan‐Meier survival plots were generated using time to tumor formation as the outcome with the GraphPad Prism software (GraphPad Software, San Diego, CA, USA). Statistical differences in survival between groups were assessed using the log‐rank test. Comparison data sets were considered statistically significant when p values were <0.05.
Results
Effective treatment of preestablished pancreatic tumors is achieved using a combinatorial approach that sequences T reg cell depletion with a GM‐CSF‐secreting whole tumor cell vaccine
Immune tolerance to preexisting cancer is a state commonly found in cancer patients. Multiple mechanisms of immune tolerance have been characterized and shown to limit the potential effectiveness of cancer vaccine approaches aimed at inducing antigen‐specific T‐cell‐mediated responses. 30 , 31 Previously published studies from our group and others have demonstrated that combining vaccines with immune‐modulating doses of chemotherapeutic agents can enhance the efficacy of vaccine‐induced immunity against progressing tumors. 14 , 16 CY, in particular, has been shown to inhibit Tregs, a T‐cell population that has been reported to inhibit higher avidity antigen‐specific T‐cell responses against some tumors. 15 We have been studying the mechanisms of immune tolerance in the Panc02 model of pancreatic adenocarcinoma. 22 ’ 23 To overcome tolerance in this model, we first evaluated the role of Tregs in inhibiting the induction of immune responses by our GM‐CSF‐secreting whole tumor cell approach. This vaccine approach has been shown to induce effective antigen‐specific T‐cell responses that are associated with clinical responses in multiple mouse models and some patients with cancer. 32
CY and PC61 (an anti‐CD25 IL‐2 Rα mAb that selectively targets Tregs because of their high expression of CD25) were evaluated as single Treg‐depleting agents and in combination to enhance the potency of the whole‐cell Panc02 cells (antigen source) mixed with the bystander GM‐CSF‐secreting B78H1 cell line (GM‐CSF source) as the vaccine. Tumors were established s.c. (right flank) in 10 mice per group that were given a tumorigenic dose (2.5 × 105 cells) of Panc02 cells on day 0. Mice were subsequently treated with either immune‐modulating doses of CY (100 mg/kg), the mAb, PC61 (50 μg), or the combination, i.p. on day 2, followed by three simultaneous subcutaneous injections (left flank and right and left upper limbs) of the GM‐CSF vaccine (1 × 106 B7H1 GM‐CSF cells mixed with 1 × 106 Panc02 cells) or mock vaccine (1 × 106 B7H1 GM‐CSF cells) on day 3. Mice were monitored twice a week for the progression of tumors. As shown in Figure 2, both CY and PC61 administered alone with either the tumor vaccine or the mock vaccine fail to significantly enhance the overall antitumor response. However, 60% of mice had tumor clearance when both CY and PC61 were given in combination with the tumor vaccine, although these data did not reach statistical significance when compared with the tumor clearance observed in mice that were given the combination of CY and PC61 along with the mock vaccine (30% of mice cleared tumor) ( Figure 2 ). The tumor‐free incidence in mice that received CY and PC61 together with the vaccine was, however, significant when compared with mice that were given tumor alone (p < 0.0001), PC61 plus vaccine (p < 0.0001), or CY plus vaccine (p = 0.0006).
Figure 2.
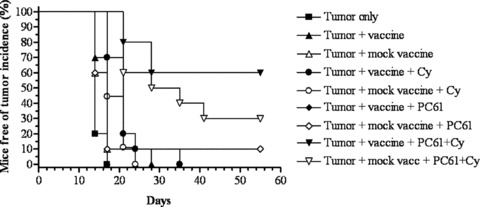
The combination of CY and PC61 given concurrently with the GM‐CSF whole‐cell vaccine is more effective in augmenting antitumor activity when compared with CY or PC61 administered as single agents with the vaccine. C57BL/6 mice were challenged on day 0 with 2.5 × 105 Panc02 tumor cells, as described, followed by administration of 100 mg/kg of CY or 50 μg of PC61 on day 2 and irradiated whole‐cell vaccine on day 3. Mice were treated with vaccine alone (A), vaccine plus CY (·), vaccine plus PC61 (♦), or vaccine plus CY and PC61 (▾). Identical groups were set up with mock vaccine administered as a control (open symbols). Mice left untreated following inoculation of tumor (▪) served as a positive control. The data are presented as Kaplan‐Meier survival curves that indicate the percentage of tumor‐free animals as a function of time after tumor inoculation. Data are represented as results obtained from experiments with at least 10 mice per group. Tumor incidence in mice that received CY and PC61 in addition to vaccine was significantly different by the log‐rank test, when compared with animals that received tumor alone (p < 0.0001), PC61 plus vaccine (p < 0.0001), or CY plus vaccine (p = 0.0006).
It is interesting to note that antitumor activity was also observed in the mice that received the mock vaccine plus CY and PC61, and that the difference in tumor rejection between this group and the mice receiving the mesothelin‐specific vaccine plus CY and PC61 did not reach significance. The antitumor activity associated with the mock vaccine can be explained in several ways. First, both vaccines were given to mice that had preestablished and progressing mesothelin‐expressing tumors. Therefore, it is likely that the administration of the mock vaccine after Treg depletion was able to unleash preexisting tumor‐specific CD8+ T‐cell populations in these cancer‐bearing animals, thus explaining the tumor protection observed. We have previously reported the presence of baseline mesothelin‐specific CD8+ T‐cell responses in pancreatic cancer patients before a similar human vaccine. The baseline T‐cell responses to pancreatic cancer antigens such as mesothelin are likely secondary to the large antigen load from the pretreatment tumor burden. Furthermore, this baseline response to mesothelin was enhanced to a greater degree in patients given CY in combination with vaccine versus vaccine alone. The enhanced mesothelin response in patients treated with CY and vaccine was also associated with longer survival and may suggest that these cells were the ones that were more effectively activated. 33 In addition, in the analysis of the data presented in Figure 2, it is important to note that the cells used as mock vaccine (B78H1‐GM cells) appear to have low levels of mesothelin expression by PCR ( Figure IB ), and we cannot exclude that this low level of mesothelin in the mock vaccine also had contributed to the results observed. In summary, the data presented in Figure 2 are consistent with the idea that inclusion of immune‐modulating doses of Treg‐depleting agents enhances the potency of our GM‐CSF‐secreting tumor vaccine in this pancreatic cancer model of immune tolerance. In addition, these data provide evidence that inhibition of Tregs may require a multitherapeutic approach to achieve a successful outcome.
Panc02‐bearing mice treated with PC61 alone or in combination with Cy demonstrate more durable Treg cell depletion kinetics when compared with mice treated with Cy alone
To evaluate the in vivo effects of the chemotherapeutic agent CY and the mAb PC61 in our mouse Panc02 model system, we investigated the profile of CD4+CD25+Foxp3+ Treg depletion at specific time points following administration of these agents. First, the total number of CD4+CD25+ T cells isolated from the axillary LNs and spleens of treated mice were compared to the total number of CD4+ T cells by flow cytometry, and the percentage of CD4+CD25+ T cells was determined. A significant 3‐fold decrease (p < 0.0001) in the percentage of CD4+CD25+ T cells was observed in both spleen and LN preparations 4 days after treatment of mice with either CY, or PC61, or a combination of both agents ( Figure 3A ). The percentage of CD4+CD25+ T cells was subsequently monitored for 28 days, at which time CD4+CD25+ T cells recovered to levels present on day 0. In contrast, the rate of recovery of CD4+CD25+ T cells in mice treated with CY alone was faster in comparison with mice treated with only PC61 or a combination of both agents. Specifically, mice that were treated with either PC61 or PC61 in combination with CY maintained a reduced percentage of CD4+CD25+ T cells on day 28 ( Figure 3A ). This difference in the proportion of CD4+CD25+ T cells in the LNs following CY treatment was significant when compared with the percentage of CD4+CD25+ T cells present in mice treated with PC61 alone (p= 0.004) or mice treated with both agents (p= 0.0007). Although this difference in the kinetics of CD4+CD25+ T‐cell recovery was more evident in the LNs than in the spleen, combined with the tumor‐free survival data, it is nevertheless an indication that the combination of PC61 and CY is more effective in eliminating Treg cells than either agent given alone.
Figure 3.
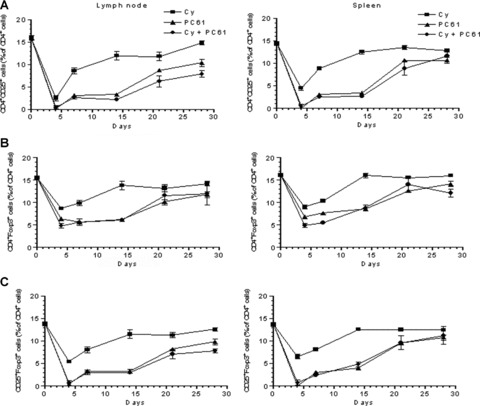
The combination of cyclophosphamide and PC61 is more effective in depleting T regulatory cells than cyclophosphamide alone. Characterization of Treg depletion following vaccination of mice with CY, PC61, or both was carried out to obtain a kinetic profile of CD4+CD25+ or CD4+Foxp3+ cells in mice injected with 100 mg/kg of CY, 50 μg of PC61, or both Treg‐depleting agents. Spleens and axillary lymph nodes were isolated from immunized mice, cells were stained as described, and flow cytometry was performed on days 4, 7, 14, 21, and 28 following immunization. Gates were set on lymphocytes by forward and side scatter, and an analysis was carried out by gating on the CD4+ population to determine the percentages of CD4+ cells that were simultaneously positive for CD25 or Foxp3. Each experimental group consisted of three mice for each time point analyzed. Data points shown are the mean ± SEM from one representative experiment that was repeated at least twice.
In a second set of experiments, CD4+CD25+ T cells expressing the Treg transcription factor, Foxp3, were evaluated in the spleen and LNs of mice treated with Cy, PC61, or the combination. As expected, there was a similar 3‐fold reduction in the percentage of CD4+CD25+Foxp3+ T cells by day 4 in mice treated with CY plus PC61 (p < 0.0001) or PC61 alone (p < 0.0001) compared with mice treated with CY only ( Figure 3B ). In addition, between days 7 and 14, there was a steady increase in the proportion of CD4+CD25+Foxp3+ T cells in both the LNs and the spleen of mice treated with CY alone. In contrast, the proportion of CD4+CD25+Foxp3+ cells observed in mice treated with either PC61 or the combination had a slower recovery, similar to the recovery of the total CD4+CD25+ T‐cell population treated with PC61 or the combination, as shown in Figure 3 A . To correlate the proportion of CD4+CD25+ T cells with the percentage of CD4+CD25+Foxp3+ T cells isolated from the LNs and spleens of treated mice, the number of CD4+CD25+Foxp3+ T cells was also analyzed on the indicated days ( Figure 3C ). Interestingly, the kinetic profile of CD4+CD25+Foxp3+ T cells was equivalent to that obtained for CD4+CD25+ cells ( Figure 3A ), thereby confirming that the CD25+ targeted by these treatment regimes are also Foxp3+. While these treatments did demonstrate an effective reduction in the percentage of CD4+CD25+Foxp3+ cells, the depletion of CD4+ T‐cell subsets with this phenotype was not as marked compared with the decrease observed in the overall CD4+CD25+ subset ( Figure 3B ). This relatively moderate effect on CD4+CD25+Foxp3+ T‐cell numbers could indicate that these treatment regimens may be targeting a specific subset of CD4+CD25+Foxp3+ T cells rather than the total population of CD4+ Foxp3+ T cells. In addition, it is possible that PC61 may affect Treg activity as well as deplete it, or that the differences in Treg depletion between these treatments maybe more evident at the tumor site rather than in the peripheral blood and LNs. Taken together, these data provide evidence to support the conclusion that CY, administered in combination with the anti‐CD25 IL‐2 Rα mAb, PC61, is more effective in initiating and maintaining Treg depletion than CY administered alone.
Treg depletion prior to vaccination increases tumor‐specific CD8+ T‐cell expansion
We previously reported that Treg cell depletion using low‐dose CY is able to uncover HER‐2/neu‐expressing T‐cell responses that are associated with the rejection of Her‐2/neu‐expressing mammary tumors in 20–30% of HER‐2/neu transgenic mice. 15 In this study, we demonstrate that treatment combining CY and PC61, prior to the vaccine, can significantly increase the tumor‐free survival rate in up to 60% of mice, compared with mice that are given either treatment alone. To assess whether this difference in clinical response correlates with cancer‐specific T‐cell responses, mice treated with PC61 plus CY prior to vaccination were compared with mice treated with either Treg‐depleting agent alone and evaluated for the induction of Panc02‐specific CD8+ T cells. Specifically, mice received either CY, or PC61, or the combination on day 0, and vaccine on day 1, as described in Figure 2 . Ten days post vaccination, the animals were sacrificed, and the frequencies of CD8+ cells secreting IFN‐γ in response to the Panc02 tumor cells were quantified by ICS. In accordance with our survival studies, IFN‐γ‐secreting T cells were detected in all animals that had Treg cells depleted prior to vaccination. Furthermore, there was at least a 4‐fold increase in the number of Panc02‐specific T cells in mice that received CY plus PC61 prior to the vaccine when compared with those treated with the vaccine alone (p < 0.001) and all other groups tested ( Figure 4 ). These data demonstrate that Treg cell depletion prior to vaccination significantly increases the number of vaccine‐induced, activated cancer‐specific CD8+ T cells, and that the presence of these T cells correlate with in vivo antitumor responses.
Figure 4.
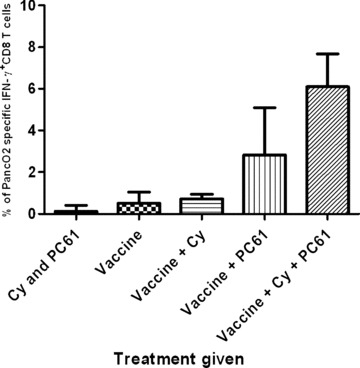
T regulatory cell depletion with cyclophosphamide, PC61, or the combination prior to vaccination results in Panc02‐specific CD8+ T‐cell activation. Five mice per group received adjuvant treatment on day 0 (CY, PC61, or both) and were immunized on day 1 with the GM‐CSF‐secreting whole‐cell vaccine. Ten days later, lymphocytes from the spleens and draining lymph nodes were enriched using nylon wool columns. The frequency of Panc02‐specific CD8+ cells was quantified using IFN‐yintracellular staining and flow cytometry analysis. The frequency of tumor‐specific IFN‐y‐positive CD8+ T cells was significantly higher in the group that received vaccine, cyclophosphamide, and PC61 than in the animals that received other treatments (CY and PC61: p < 0.0001, vaccine alone: p < 0.0001, vaccine + CY: p < 0.0001, vaccine + PC61: p = 0.0297). Data points shown are the mean ± SD from a representative experiment that was repeated three times with similar results.
Identification of epitope‐rich regions of the mesothelin precursor protein that are recognized by CD8+ T cells isolated from mice treated with the Treg‐depleting agents CY and PC61 given in sequence with vaccination
We have previously reported the detection of CD8+ T‐cell responses specific for a new candidate pancreatic tumor antigen, mesothelin, in some patients treated with an allogeneic GM‐CSF‐secreting whole pancreatic tumor cell vaccine. 5 Mesothelin has also been shown to be an adhesion molecule in pancreatic tumors that likely confers metastatic potential. 4 As described above, our in vivo studies demonstrate that combining our murine whole‐cell vaccine with Treg‐depleting agents is more effective in inducing an effective antitumor immune response than vaccine alone. In addition, we have found that Panc02 cells express mesothelin in a similar pattern as that observed with human pancreatic tumors (data not shown). Thus, the identification of mesothelin epitopes would facilitate the elucidation of the role Tregs play in regulating antigen‐specific CD8+ T‐cell responses specific for Panc02.
Previous studies have shown that T‐cell epitopes in a given antigen can be found by scanning the amino acid sequence for peptides that fit the motif of the restricting MHC molecule using several public domain databases. Although these databases could predict several epitopes within mesothelin that bind the H‐2Kb and H‐2Db molecules, significant levels of mesothelin‐specific CD8+ responses specific for these epitopes could not be detected in the animals vaccinated using our combinatorial approach. Therefore, to identify the immunodominant epitopes recognized by the lymphocytes from mice treated with Treg depletion and vaccine, we screened pools of 10 overlapping 15 amino acid (15‐mer) peptides (each overlapping by 10 amino acids) that spanned the entire mesothelin precursor protein. First, 123 overlapping 15‐mer peptides were synthesized to span the entire sequence of the mouse mesothelin precursor protein. Lymphocytes isolated from the spleen and LNs of mice that received CY, PC61, and vaccine were enriched for CD8+ T cells and stimulated in vitro in the presence of the Panc02 cells. The CD8+ T cells from 1‐week cultures were subsequently evaluated for recognition of T‐2 cells expressing H‐2Kb or H‐2Db pulsed with the individual 15‐mers that spanned the whole mesothelin sequence following an overnight incubation. Supernatants from overnight cultures were harvested and evaluated for GM‐CSF levels as a measure of specific CD8+ T‐cell responses to the mesothelin peptide pools. Using this approach, 15‐mer peptides, containing CD8+‐restricted epitopes, that spanned both the MPF and the mesothelin portions of the precursor protein were identified ( Figure 5 A for H‐2Kb and Figure 5B for H‐2Db). Finally, eight 9‐amino acid peptides overlapping by all but one amino acid were synthesized that covered the individual 15‐mer epitopes found to be positive in at least two consecutive ELISA assays. These smaller peptides were again screened using CD8+ T cells isolated from CY‐, PC61‐, and vaccine‐treated mice to identify the specific mesothelin epitopes presented to the T cells by the H‐2Kb and H‐2Db molecules. It has previously been determined that H‐2Kb‐binding peptides are 8‐mers, whereas H‐2Db‐restricted peptides are 9‐mers. 34 Using this strategy, three H‐2Db‐restricted 9‐mers and three H‐2Kb‐restricted 8‐mers were identified for further evaluation. Figure 5B and 5C shows the result of the screening of two H‐2Kb epitopes identified.
Figure 5.
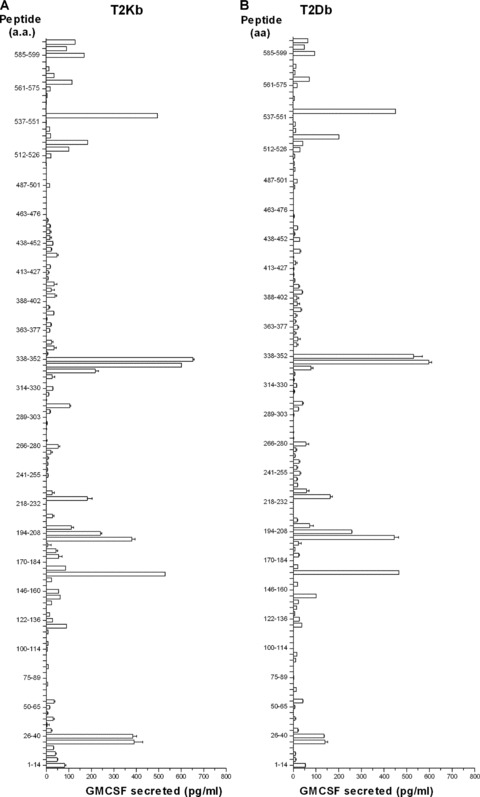
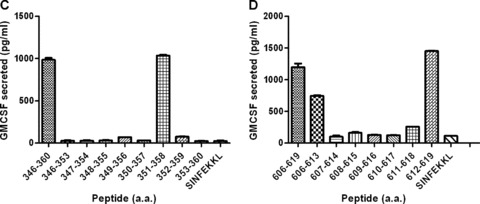
Peptide library screening of the entire mesothelin protein using immunized lymphocytes from mice receiving combinatorial T regulatory cell depletion and the vaccine identifies new mesothelin CD8+ T‐cell epitopes. (A and B) Mice received cyclophosphamide and PC61 on day 0, followed by vaccine on day 1. Spleens and lymph nodes were isolated between days 7 and 10 post vaccine, and single‐cell suspensions were stimulated in vitro in the presence of individual peptides to be tested, as described. The CD8+ T cells from these cultures were isolated in 7 days and co‐incubated overnight in the presence of peptide‐pulsed T‐2 cells expressing H‐2Kb (A) or H‐2Db (B). Culture supernatants were harvested and GM‐CSF ELISAs were performed as a means of identifying the effector CD8+ T‐cell response to the mesothelin peptides. Peptides tested were 15‐mers spanning the entire Megakaryocyte Potentiating Factor/Mesothelin precursor protein. GM‐CSF release was calculated as GM‐CSF release in the presence of T2 targets pulsed with specific peptide less GM‐CSF release in the presence of an irrelevant peptide. Splenocytes from mice immunized with the combinatorial approach were isolated 1 week after immunization and stimulated for a week with the identified 15‐mer peptides. The cells were then harvested and co‐incubated overnight with T‐2Kb pulsed with the respective 8‐mers or irrelevant controls (SINFEKKL). 8‐mers were considered for further testing when the GM‐CSF levels were higher than the ones from the 15‐mer cultures. Panel C shows the result for Meso346_360 8‐mer screening and 5D for Meso606_619 8‐mer screening. Data points shown are the mean ± SD from a representative experiment that was repeated twice with similar results.
Validation of intracellular domain epitopes Meso351–619 and612–619 as the target of CD8+ cells derived from mice that received vaccine in combination with CY and PC61
To characterize the frequency and function of the T cells specific to the mesothelin epitopes identified, we used intracellular IFN‐γas an initial measure of activated antigen‐specific CD8+ T cells. Mice were Treg‐depleted using CY plus PC61,1 day prior to vaccination with the whole tumor GM‐CSF‐secreting vaccine, as described in Figure 2 . Seven days later, spleens and LNs were collected, and isolated lymphocytes were cultured for 1 week in the presence of 2 μM of the respective mesothelin 8‐mer or 9‐mer peptide. T cells expressing IFN‐γ were quantified following an overnight stimulation with MHC‐restricted T‐2 cells pulsed in vitro with titrated concentrations of the mesothelin epitopes or irrelevant control peptides (ranging from 2 μM to 200 pM). From the peptides identified in the initial screening, only two T‐2Kb epitopes could be validated using the frequency of IFN‐γ CD8+ cells as a measurement of activated T‐cell function. IFN‐γ‐secreting CD8+ T cells generated after vaccination could recognize these epitopes even when picomolar concentrations of the 8‐mers were pulsed onto antigen‐presenting cells, demonstrating the sensitivity of these responses ( Figure 6A and 6B ). Furthermore, IFN‐γ‐producing Meso351–358– and Meso612–619‐specific T cells were detected in the LNs and spleen when isolated directly from mice vaccinated with the combinatorial approach, without requiring in vitro stimulation ( Figure 6C ). In addition, CD8+ T cells stimulated for a week with Meso351‐358 were able to recognize the native epitopes presented by the tumor cells, demonstrating that this epitope is naturally processed by the Panc02 cells (data not shown).
Figure 6.
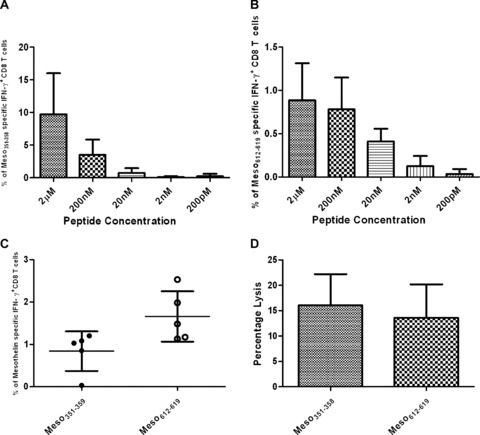
Functional mesothelin‐specific CD8+ T cells are detected in mice following Treg depletion and vaccination. (A and B) Splenocytes from mice vaccinated following Treg depletion (CY and PC61) were stimulated in vitro for a week with the identified 8‐mers or 9‐mers. Lymphocytes were then collected and incubated for 6 hours with either T‐2Kb‐ or T‐2Db‐specific antigen‐presenting cells pulsed with increasing concentrations of peptides Meso351_358 (A) and Meso612–619 (B) from picomolar to micromolar range. Data points shown are the mean ± SD of a representative experiment that was repeated at least twice with similar data. (C) Mice were sacrificed 10 days post vaccination and lymphocytes were enriched by nylon wool purification. The frequency of mesothelin‐specific T cells straight ex vivo was accessed through intracellular IFN‐γ staining. Data points represent the percentage of Meso351_358‐and Meso612–619‐specific IN F‐y‐positive CD8+ T cells. (D) Mice received intravenous peptide‐pulsed, CFSE‐labeled splenocytes on day 21 after Treg depletion and vaccine. After 18 hours, mice were sacrificed and splenocytes harvested. The percentage of mesothelin‐specific lysis (Meso351_358 and Meso612_619) was determined by flow cytometric analysis, as described in Materials and Methods. Data presented are the mean percentage (°/o Meso‐specific lysis ± SD) for measurements in 10 mice per group of a representative experiment.
To confirm that these epitopes are recognized by CD8+ T cells in vivo, mesothelin epitope‐specific CD8+ T cells were measured following the combinatorial vaccine treatment in an in vivo cytolytic assay. Mice were treated with CY plus PC61 on day 0, and the GM‐CSF‐secreting Panc02 vaccine on day 1. Two weeks later, fluorescent‐labeled splenocytes pulsed with the test mesothelin epitopes or irrelevant control epitopes were adoptively transferred into treated mice. An analysis by flow cytometry revealed that vaccination of mice following Treg depletion induced both Meso351‐358– and Meso612‐619 ‐specific CD8+ cytolytic T cells. Specifically, a 15–20% lysis of Meso612‐619‐loaded target cells and a 10–15% lysis of theMeso ‐loaded targets were measured following treatment with Treg depletion in sequence with vaccination ( Figure 6Ό ). Taken together, these data suggest that our immunotherapeutic approach can overcome self‐tolerance and is effective at eliciting functional mesothelin‐specific T cells following vaccine treatment if Tregs are inhibited. In addition, our screening of a mesothelin epitope library was successful in finding two new murine mesothelin‐derived epitopes with specific immunization potential.
DC vaccination using Meso351–358– and Meso612–619‐pulsed cells protects mice in vivo against tumor development
To determine if the identified mesothelin‐derived epitopes are immunogenic candidate epitopes for vaccine development, these epitopes were pulsed onto in vz’iro‐generated DCs and tested for their in vivo immunization potential. Mice received subcutaneous injections of DCs pulsed with each peptide on day 0 in the left flank, followed by a subcutaneous Panc02 tumor challenge 2 weeks later in the right flank. DCs derived from C57BL/6 mice were pulsed in vitro with either the H‐Kb‐restricted Meso612–619 or Meso612–619„„or irrelevant SIINFEKL peptide prior to vaccination. Interestingly, 80% of the mice immunized with DCs pulsed with Meso were protected against tumor challenge, whereas only 20% of mice were protected in the control group (irrelevant peptide) (p=0.0145) ( Figure 7 ). The animals immunized with Meso351–358 also had a promising trend toward protection, although not statistically significant (p = 0.0539). These data confirm immunogenicity of the discovered epitopes and that mesothelin is a potential target for the development of antigen‐specific vaccine approaches for the treatment of pancreatic cancer.
Figure 7.
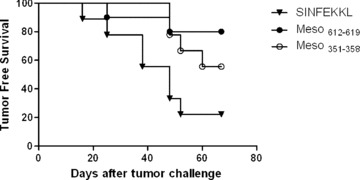
DC vaccination with identified mesothelin epitopes protects animals against the development of pancreatic cancer. Bone marrow‐derived dendritic cells generated from C57BL/6 mice were pulsed with 300 μg/mL of either Meso351358, or Meso612619, or the SIINFEKL peptide. Subcutaneous injections (0.1 ml_) of 2 × 105 cells were given in each hind limb on days 0 and 7. Animals were challenged s.c. with 2.5 × 105 Panc02 cells on day 14. Mice were followed twice per week for the development of tumor. The data are presented as Kaplan‐Meier survival curves that indicate the percentage of tumor‐free animals as a function of time after tumor inoculation. Data are represented as results obtained from experiments with at least 10 mice per group (p= 0.0145 for mice treated with Meso612619 vs. control peptide, and p= 0.0539 for mice treated with Meso351_358 vs. control peptide).
Discussion
We previously reported that immune‐modulating doses of CY given 1 day prior to vaccination with a GM‐CSF‐secreting tumor vaccine depletes Tregs and allows for the induction of tumor‐specific T cells capable of curing up to 20% of treated mice. 15 Building on our previous findings, we now demonstrate a synergistic benefit in combining different Treg cell‐depleting therapies with our whole‐cell GM‐CSF‐secreting vaccine in order to activate a more robust antigen‐specific CD8+ T‐cell response capable of curing Panc02 cancers in the majority of treated mice.
In addition, we demonstrate the utility of mesothelin as a target antigen expressed by Panc02 carcinomas. Importantly, we have employed a functional protein‐scanning approach to identify and validate dominant and immune‐relevant mesothelin epitopes expressed by Panc02 cells that were not predicted by querying multiple peptide‐binding databases. Thus, these findings further demonstrate that mesothelin is an immune‐relevant antigen that can serve as a target of T cells and support the use of the Panc02 model for evaluating antigen‐targeted immunotherapies for the treatment of cancers in immune‐tolerant hosts.
To our knowledge, this is the first report demonstrating the synergistic effect of combining two Treg‐depleting agents to enhance vaccine‐induced antigen‐specific T‐cell responses. A number of studies have compared CY and PC61 as single agents 35 , 37 and found varied levels of Treg depletion for both, depending on the tumor model used. In one study, CY was even shown to deplete effector T cells in addition to Tregs, thereby minimizing the benefit of Treg depletion. 36 In contrast, we previously reported that CY given to HER‐2/neu transgenic mice with HER‐2/neu‐expressing mammary tumors selectively depletes Tregs that are progressing through the cell cycle. In untreated tumor‐bearing mice, Tregs were shown to be the predominant cycling T‐cell population. 15 However, this cycling population represented less than half of the Foxp3+CD4+CD25+ Treg cells in these mice. It is therefore not surprising that Treg depletion with CY prior to vaccination only cured about 20% of tumor‐bearing mice. Interestingly, Sherman and colleagues reported that effector cells that are in the process of being tolerized or deleted are also cycling and proliferating, indicating that depletion of cycling cells with CY may be beneficial in clearing out tolerizing cells. 38 , 40 Since CD25 is upregulated in Tregs, the CD25‐specific Ab, PC61, has been used as an alternative method for depleting Tregs in multiple tumor models. Since CD25 is also upregulated in activated effector T cells, this agent, like CY, is best given to tumor‐bearing hosts, prior to vaccine administration. Viehl and colleagues previously reported that depletion of CD4+CD25+ Tregs using PC61 improved vaccine‐induced T‐cell responses that were capable of preventing Panc02 tumors when the treatment was given prior to tumor challenge. 37 The successful use of PC61 mAb to deplete Tregs is potentially compromised by the possibility that the Ab could also deplete effector T cells. Although the mechanism by which PC61 inhibits Treg function is not yet known, studies by Yewdell and colleagues have demonstrated that the mechanism by which PC61 ‐type antibodies work may not be by T‐cell deletion alone. In fact, they suggest that PC61 selectively inhibits Tregs that suppress T cells specific for immunodominant epitopes, thereby creating a hierarchy of suppression within the T‐cell repertoire targeted at a specific antigen. 41
CD25/IL‐2ROC, which is upregulated in Tregs, is part of the high‐affinity receptor complex for IL‐2. Thus, another possible explanation for Treg depletion of effector T cells is the competition for limited sources of IL‐2 within the tumor's microenvironment. Antony et al. tested this hypothesis using IL‐2Rα knockout mice and found that IL‐2Rα−/− Th cells were fully capable of helping CD8+ T cells, suggesting that competition is part of a more complex interaction between Tregs and the immune system. Others have shown that depriving Tregs of IL‐2 leads to a downregulation of the IL‐2 receptor, leading to possible suppression of Tregs. 42 , 44 We now show that the combination of CY with PC61 allows for a significantly improved T‐cell‐mediated immune response over either agent alone. Since neither agent fully depletes the Foxp3‐expressing CD4+CD25+ population of Tregs, it is likely that each agent targets a different subpopulation of Tregs. Alternatively, it may be that these agents are at suboptimal levels for depleting Tregs, but with increasing doses of either one, there is an increased deletion of the effector T cells that are recruited to the immune response. Thus, this study, together with previously reported data, suggests that CY depletes activated cycling Tregs, while addition of PC61 selectively blocks resting Tregs from being activated. Additional studies are needed to fully define the role of each agent in depleting this population of T cells that have emerged as an important mechanism of T‐cell regulation in cancer patients.
A major advantage of immune‐based therapies is their ability to target specific antigens, thereby increasing specificity of the treatment while maintaining low toxicity. Recent technologic developments have allowed for the rapid identification of candidate tumor antigens, which in turn, require clinically relevant mouse models for elucidating the mechanisms by which the immune system responds to these antigens. We previously identified mesothelin as a potential target antigen in human pancreatic cancer due to its limited tissue expression and relative overexpression in cancerous versus normal tissue. Mesothelin is also overexpressed by Panc02 tumors, the best characterized pancreatic cancer mouse tumor. Screening several public domain databases failed to reveal T‐cell‐recognized mesothelin epitopes presented by the two MHC class I molecules (H‐2Db and H‐2Kb) expressed by Panc02 cells. Consequently, we developed a functional protein‐scanning approach using an overlapping 15‐mer library of the entire mesothelin protein, which ultimately identified two H‐2Kb epitopes, both of which successfully immunize against the Panc02 tumor. The screening of 15‐mer peptide libraries to identify new antigenic epitopes has successfully been used to identify viral epitopes. 45 , 46 Through the use of a number of in vitro and in vivo T‐cell assays, we have validated two new mesothelin epitopes that are recognized by CD8+ T cells following vaccination when Tregs are inhibited. Thus, it is possible and feasible to identify immune‐relevant tumor‐associated T‐cell epitopes using a pan‐protein screening process and immunized lymphocytes from mice treated with Treg depletion and vaccination. Validation of these epitopes included demonstrating the binding of picomolar quantities of peptide to their MHC class I molecule, the recognition by immunized T cells, and the ability to immunize against the Panc02 tumor with DCs loaded with the peptide. Identified peptides that did not meet these criteria were not chosen for further evaluation. In addition, antigen‐specific T cells generated against these epitopes were shown to lyse Panc02 cells, confirming that these epitopes are naturally processed and presented by Panc02 cells. It is important to point out that DCs loaded with these epitopes could only protect against the development of tumors, but did not generate T cells potent enough to cure preexisting tumors. This is not surprising since the DC vaccine approach is likely not inducing tumor‐specific CD4+ T cells, a critical T‐cell population that is required to induce the most effective antitumor immune responses in most mouse models of cancer. In addition, the mechanisms of immune tolerance are expected to be more profound in tumor‐bearing hosts.
In summary, immune‐based therapies are currently under development for the treatment of many cancers. Current immunotherapies are limited by the multiple mechanisms of immune tolerance exhibited by developing cancers, which in turn, actively inhibit the effectiveness of antigen‐specific T‐cell responses. We report here a stringent method for identifying multiple T‐cell epitopes recognized by an immunized T‐cell population. The identification of immune‐relevant tumor antigens and their specific T‐cell epitopes will facilitate the elucidation of the multiple mechanisms of immune cell suppression and allow the future development of interventions that bypass these mechanisms.
Conflict of Interest
Under a licensing agreement between Cell Genesys and the Johns Hopkins University, the University is entitled to milestone payments and royalties on sales of the vaccine product described in this article. Under a licensing agreement between Anza Corporation and the Johns Hopkins University, Dr. Elizabeth M. Jaffee is entitled to a share of royalties received by the University on sales of future products in humans. The terms of these arrangements are being managed by the Johns Hopkins University in accordance with its conflict of interest policies.
Acknowledgment
We are thankful for the support given by the Sol Gold Pancreatic Cancer Center, the Dana and Albert Broccoli Foundation, the Broad Foundation Grant, the GI Spore (P50CA62924), and the NCDDG grants (U19CA113341).
References
- 1. Chang K, Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Nat! Acad Sci USA. 1996; 93: 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Robinson BW; Creaney J, Lake R, Nowak A, Musk AW, De Klerk N, Winzell P, Hellstrom KE, Hellstrom I. Mesothelin‐family proteins and diagnosis of mesothelioma. Lancet. 2003; 362: 1612–1616. [DOI] [PubMed] [Google Scholar]
- 3. Hassan R, Bera T, Pastan I. Mesothelin: a new target for immunotherapy. Gin Cancer Res. 2004; 10: 3937–3942. [DOI] [PubMed] [Google Scholar]
- 4. Li M, Bharadwaj U, Zhang R, et al Mesothelin is a malignant factor and therapeutic vaccine target for pancreatic cancer. Mol Cancer Ther. 2008; 7: 286–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thomas AM, Santarsiero LM, Lutz ER, et al Mesothe I in‐specific CD8(+) T cell responses provide evidence of in vivo cross‐priming by antigen‐presenting cells in vaccinated pancreatic cancer patients. J Exp Med. 2004; 200: 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang HY, Wang RF. Antigen‐specific CD4+ regulatory T cells in cancer: implications for immunotherapy. Microbes Infect. 2005; 7: 1056–1062. [DOI] [PubMed] [Google Scholar]
- 7. Chattopadhyay S, Chakraborty NG, Mukherji B. Regulatory T cells and tumor immunity. Cancer Immunol Immunother. 2005; 54: 1153–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fehervari Z, Sakaguchi S. CD4+ Tregs and immune control. J Gin Invest. 2004; 114: 1209–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shevach EM. Regulatory T cells: introduction. Semin Immunol. 2004; 16: 69–71. [DOI] [PubMed] [Google Scholar]
- 10. Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck‐Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res. 2003; 9: 606–612. [PubMed] [Google Scholar]
- 11. Liyanage UK, Moore TT, Joo HG, et al Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002; 169: 2756–2761. [DOI] [PubMed] [Google Scholar]
- 12. Curiel TJ, Coukos G, Zou L, et al Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004; 10: 942–949. [DOI] [PubMed] [Google Scholar]
- 13. Piccirillo CA, Shevach EM. Naturally‐occurring CD4+CD25+ immunoregulatory T cells: central players in the arena of peripheral tolerance. Semin Immunol. 2004; 16: 81–88. [DOI] [PubMed] [Google Scholar]
- 14. Berd D, Maguire HC Jr, Mastrangelo MJ. Induction of cell‐mediated immunity to autologous melanoma cells and regression of metastases after treatment with a melanoma cell vaccine preceded by cyclophosphamide. Cancer Res. 1986; 46: 2572–2577 [PubMed] [Google Scholar]
- 15. Ercolini AM, Ladle BH, Manning EA, Pfannenstiel LW, Armstrong TD, Machiels JPH, Bieler JG, Emens LA, Reilly RT, Jaffee EM. Recruitment of latent pools of high‐avidity CD8(+) T cells to the antitumor immune response. J Exp Med. 2005; 201: 1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Machiels JP, Reilly RT, Emens LA, et al Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage‐colony stimulating factor‐secreting whole‐cell vaccines in HER‐2/neu tolerized mice. Cancer Res. 2001; 61: 3689–3697 [PubMed] [Google Scholar]
- 17. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti‐CD25 (interleukin‐2 receptor alpha) monoclonal antibody. Cancer Res. 1999; 59: 3128–3133. [PubMed] [Google Scholar]
- 18. Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999; 163: 5211–5218. [PubMed] [Google Scholar]
- 19. Tanaka H, Tanaka J, Kjaergaard J, Shu S. Depletion of CD4+ CD25+ regulatory cells augments the generation of specific immune T cells in tumor‐draining lymph nodes. J Immunother. 2002; 25: 207–217 [DOI] [PubMed] [Google Scholar]
- 20. Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, Yamaguchi T, Shimizu J, Nomura T, Chiba T, Sakaguchi S. Treatment of advanced tumors with agonistic anti‐GITR mAb and its effects on tumor‐infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med. 2005; 202: 885–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004; 34: 336–344. [DOI] [PubMed] [Google Scholar]
- 22. Corbett TH, Roberts BJ, Leopold WR, et al Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BIV6 mice. Cancer Res. 1984; 44: 717–726. [PubMed] [Google Scholar]
- 23. Schmidt T, Ziske C, Marten A, Endres S, Tiemann K, Schmitz V, Gorschluter M, Schneider Q Sauerbruch T, Schmidt‐Wolf IGH. Intratumoral immunization with tumor RNA‐pulsed dendritic cells confers antitumor immunity in a C57BL/6 pancreatic murine tumor model. Cancer Res. 2003; 63: 8962–8967. [PubMed] [Google Scholar]
- 24. Levitsky HI, Lazenby A, Hayashi RJ, Pardoll DM. In vivo priming of two distinct antitumor effector populations: the role of MHC class I expression. J Exp Med. 1994; 179: 1215–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rotzschke O, Falk K, Stevanovic S, Jung G, Walden P, Rammensee HG. Exact prediction of a natural T cell epitope. Eur J Immunol. 1991; 21: 2891–2894. [DOI] [PubMed] [Google Scholar]
- 26. Pircher H, Moskophidis D, Rohrer U, Burki K, Hengartner H, Zinkernagel RM. Viral escape by selection of cytotoxic T cell‐resistant virus variants in vivo. Nature 1990; 346: 629–633. [DOI] [PubMed] [Google Scholar]
- 27. Hudrisier D, Mazarguil H, Laval F, Oldstone MB, Gairin JE. Binding of viral antigens to major histocompatibility complex class I H‐2Db molecules is controlled by dominant negative elements at peptide non‐anchor residues. Implications forpeptide selection and presentation. J Biol Chem. 1996; 271: 17829–17836. [DOI] [PubMed] [Google Scholar]
- 28. Inaba K, Swiggard WJ, Steinman RM, Romani N, Sculer G. Current Protocols in Immunology. Mew York : Wiley; 1998. [Google Scholar]
- 29. Kumaraguru U, Suvas S, Biswas PS, Azkur AK, Rouse BT. Concomitant helper response rescues otherwise low avidity CD8+ memory CTLs to become efficient effectors in vivo. J Immunol. 2004; 172(6): 3719–3724. [DOI] [PubMed] [Google Scholar]
- 30. Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol. 2003; 21: 807–839. [DOI] [PubMed] [Google Scholar]
- 31. Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006; 90: 51–81. [DOI] [PubMed] [Google Scholar]
- 32. Laheru D, Jaffee EM. Immunotherapy for pancreatic cancer—science driving clinical progress. Nat Rev Cancer. 2005; 5: 459–467 [DOI] [PubMed] [Google Scholar]
- 33. Laheru D, Lutz E, Burke J, Biedrzycki B, Solt S, Onners B, Tartakovsky I, Nemunaitis J, Le D, Sugar E, Hege K, Jaffee E. Allogeneic granulocyte macrophage colony‐stimulating factor‐secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: a pilot study of safety, feasibility, and immune activation. Clin Cancer Res. 2008; 14(5): 1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee HG. Allele‐specific motifs revealed by sequencing of self‐peptides eluted from MHC molecules. Nature. 1991; 351: 290–296. [DOI] [PubMed] [Google Scholar]
- 35. Nagayama Y, Hase W, Motoyoshi Y, Saitoh O, Sogawa R, Nakao K. Distinct responses of two hepatocellular carcinoma cell lines of a similar origin to immunotherapies targeting regulatory or effectorT cells. Oncol Rep. 2007; 17: 1269–1273. [PubMed] [Google Scholar]
- 36. Matsushita N, Pilon‐Thomas SA, Martin LM, Riker Al. Comparative methodologies of regulatory T cell depletion in a murine melanoma model. J Immunol Methods. 2008; 333: 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Viehl CT, Moore TT, Liyanage UK, Frey DM, Ehlers JP, Eberlein TJ, Goedegebuure PS, Linehan DC. Depletion of CD4+CD25+ regulatory T cells promotes a tumor‐specific immune response in pancreas cancer‐bearing mice. Ann Surg Oncol. 2006; 13: 1252–1258. [DOI] [PubMed] [Google Scholar]
- 38. Redmond WL, Wei CH, Kreuwel HT, Sherman LA. The apoptotic pathway contributing to the deletion of naive CD8 T cells during the induction of peripheral tolerance to a cross‐presented self‐antigen. J Immunol. 2008; 180: 5275–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hernandez J, Aung S, Marquardt K, Sherman LA. Uncoupling of proliferative potential and gain of effector function by CD8(+) T cells responding to self‐antigens. J Exp Med. 2002; 196: 323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kreuwel HT, Aung S, Silao C, Sherman LA. Memory CD8(+) T cells undergo peripheral tolerance. Immunity. 2002; 17: 73–81. [DOI] [PubMed] [Google Scholar]
- 41. Haeryfar SM, DiPaolo RJ, Tscharke DC, Bennink JR, Yewdell JW. Regulatory T cells suppress CD8+ T cell responses induced by direct priming and cross‐priming and moderate immunodominance disparities. J Immunol. 2005; 174: 3344–3351. [DOI] [PubMed] [Google Scholar]
- 42. Antony PA, Restifo NP. CD4+CD25+ T regulatory cells, immunotherapy of cancer, and interleukin‐2. J Immunother. 2005; 28: 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. De La Rosa M, Rutz S, Dorninger H, Scheffold A. Interleukin‐2 is essential for CD4+CD25+ regulatory T cell function. EurJ Immunol. 2004; 34: 2480–2488. [DOI] [PubMed] [Google Scholar]
- 44. Depper JM, Leonard WJ, Drogula C, Kronke M, Waldmann TA, Greene WC. Interleukin 2 (IL‐2) augments transcription of the IL‐2 receptor gene. Proc NatI Acad Sci USA. 1985; 82: 4230–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maecker HT, Dunn HS, Suni MA, Khatamzas E, Pitcher Li, Bunde T, Persaud N, Trigona W, Fu TM, Sinclair E, Bredt BM, McCune JM, Maino VC, Kern F, Picker LI. Use of overlapping peptide mixtures as antigens for cytokine flow cytometry. J Immunol Methods. 2001; 255: 27–40. [DOI] [PubMed] [Google Scholar]
- 46. Kern F, Faulhaber N, Frommel C, Khatamzas E, Prosch S, Schonemann C, Kretzschmar I, Volkmer‐Engert R, Volk HD, Reinke P. Analysis of CD8 T cell reactivity to cytomegalovirus using protein‐spanning pools of overlapping pentadecapeptides. Eur J Immunol. 2000; 30: 1676–1682. [DOI] [PubMed] [Google Scholar]