

Abstract
S-Adenosylmethionine decarboxylase (AdoMetDC) is a critical enzyme in the polyamine biosynthetic pathway and a subject of many structural and biochemical investigations for anti-cancer and anti-parasitic therapy. The enzyme undergoes an internal serinolysis reaction as a post-translational modification to generate the active site pyruvoyl group for the decarboxylation process. The crystal structures of AdoMetDC from Homo sapiens, Solanum tuberosum, Thermotoga maritima, and Aquifex aeolicus have been determined. Numerous crystal structures of human AdoMetDC and mutants have provided insights into the mechanism of autoprocessing, putrescine activation, substrate specificity, and inhibitor design to the enzyme. The comparison of the human and potato enzyme with the T. maritima and A. aeolicus enzymes supports the hypothesis that the eukaryotic enzymes evolved by gene duplication and fusion. The residues implicated in processing and activity are structurally conserved in all forms of the enzyme, suggesting a divergent evolution of AdoMetDC.
Keywords: polyamines, protein evolution, putrescine activation, cation-pi interactions, pyruvoyl cofactor
Introduction
Decarboxylation reactions in enzymes frequently depend on pyridoxal-5′-phosphate (PLP) or thiamin pyrophosphate as a cofactor for catalysis. S-Adenosylmethionine decarboxylase (AdoMetDC) belongs to a small class of enzymes that depend on a pyruvoyl cofactor for the decarboxylation process (Hackert and Pegg 1997; van Poelje and Snell 1990; Tabor and Tabor 1984a; Pegg et al. 1998). Other examples of pyruvoyl group dependant decarboxylases are aspartate decarboxylase, histidine decarboxylase, and arginine decarboxylase, respectively (Gallagher et al. 1993; Gallagher et al. 1989; Albert et al. 1998; Schmitzberger et al. 2003; Tolbert et al. 2003a; Soriano et al. 2008). AdoMetDC is at a critical inititation point in the polyamine biosynthetic pathway (Tabor and Tabor 1984b; Wallace et al. 2003; Gerner and Meyskens 2004; Gerner et al. 2007). The polyamines spermidine and spermine are aliphatic cations critical for cell growth, differentiation and proliferation. AdoMetDC catalyzes the conversion of S-adenosylmethionine (AdoMet) to S-adenosyl-5′-(3-methylthiopropylamine) (dcAdoMet) (Figure 1), which is completely committed to polyamine biosynthesis; hence the expression and activity of AdoMetDC are closely related to the cellular concentration of the polyamines. In addition, the activity of AdoMetDC depletes the cellular pool of AdoMet for other functions such as methylation of DNA and other essential reactions. The polyamine biosynthetic machinery is upregulated in various types of cancer and parasitic diseases (Gerner and Meyskens 2004; Pegg 1988). The inhibitors of the pathway have shown promise in several clinical trials as chemotherapeutic agents (Casero and Marton 2007; Basuroy and Gerner 2006; Fabian et al. 2002; Meyskens and Gerner 1999; Williams-Ashman and Schenone 1972; Millward et al. 2005). The most successful inhibitor of polyamine biosynthesis is α-difluoromethylornithine (DFMO), an inhibitor of ornithine decarboxylase (ODC), which is used for the treatment of African sleeping sickness (Bacchi et al. 1994).
Figure 1.
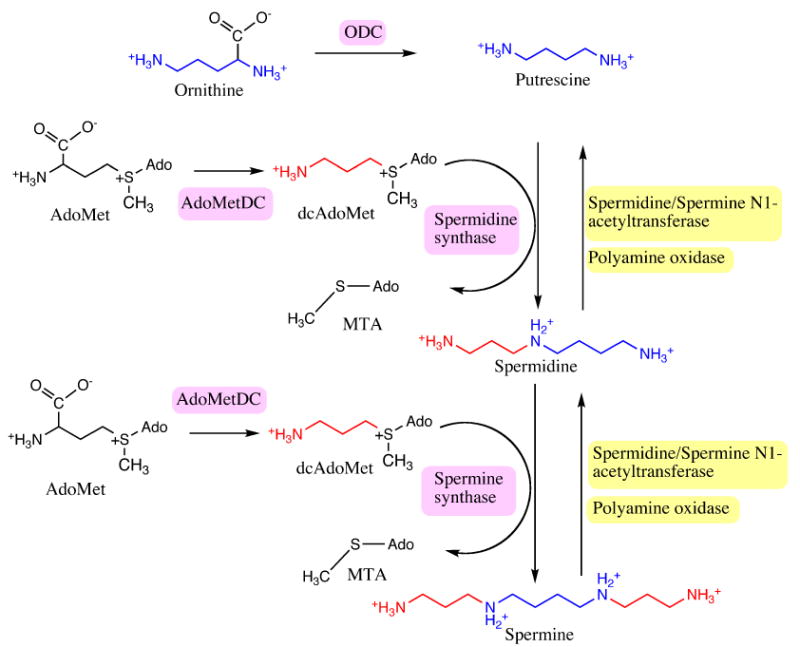
Overview of the polyamine biosynthetic pathway. The biosynthetic enzymes are highlighted in pink and the degradative enzymes are highlighted in yellow. Putrescine is colored blue. The aminopropyl group transferring from dcAdoMet to polyamines is colored red.
ODC and AdoMetDC catalyze early steps in the polyamine biosynthetic pathway, are primary targets for inhibitor design and are subjects of numerous biochemical and structural investigations. AdoMetDC is expressed as an inactive proenzyme that undergoes autoprocessing to the active enzyme. The autoprocessing involves an internal serinolysis reaction leading to the cleavage of the backbone to α and β subunits with the latter being the smaller subunit and to generation of a pyruvoyl group at the N-terminus of the α chain (Xiong and Pegg 1999; Tolbert et al. 2003b; Ekstrom et al. 2001). In humans, the autoprocessing reaction happens spontaneously, while other factors are required for processing in some species. The mechanism of autoprocessing in human AdoMetDC (hAdoMetDC) is shown in Figure 2 and is expected to be similar in other species. In humans the cleavage occurs between residues Glu67 – Ser 68. The backbone of the serine residue attacks the adjacent carbonyl carbon of Glu67 to generate a five-membered ring, the oxyoxazolidine intermediate. The intermediate rearranges to form an ester intermediate. The basic residue His243 abstracts a proton from the Cα carbon of Ser68 resulting in the cleavage of the ester intermediate to two chains. The subunit containing the N-terminal part of the uncleaved chain is called the β-chain and the subunit containing the C-terminal part is called the α-chain. The N-terminus of the α-chain contains a newly formed dehydroalanine residue that tautomerizes to form an imine, which further hydrolyzes to form the pyruvoyl group.
Figure 2.
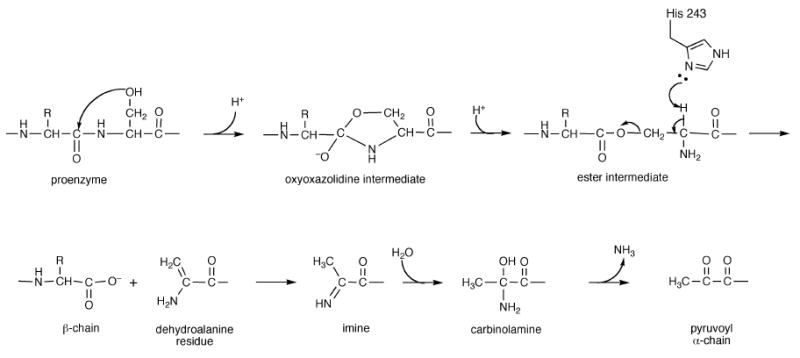
Mechanism of autoprocessing in human AdoMetDC.
The formation of the pyruvoyl group renders AdoMetDC active for the decarboxylation reaction. The substrate AdoMet binds to the enzyme through a Schiff base to the active site puruvoyl group. The decarboxylation reaction proceeds with the pair of electrons from the leaving group (CO2) delocalized into the pyruvoyl group. An acidic residue, likely Cys82, protonates the Cα carbon of the substrate to generate the imine intermediate. The imine is further hydrolyzed to release the product dcAdoMet and regenerate the pyruvoyl group for additional rounds of catalysis. Incorrect protonation of the intermediate would result in transamination and an irreversibly inactive enzyme. Activity assays of human AdoMetDC have indicated that on an average, the enzyme performs ∼15,000 turnover reactions in vitro before being transaminated. The turnover to inactivation number of the C82A mutant is ∼143 suggesting that Cys82 is critical for the activity of AdoMetDC (Xiong et al. 1999).
The diamine putrescine is known to regulate the activity of AdoMetDC by affecting the rates of autoprocessing and decarboxylation; however, the effects of putrescine on AdoMetDC are species specific. Putrescine activates the autoprocessing reactions in humans as well as the decarboxylation reaction in humans, Trypanosoma brucei, Trypanosoma cruzi, and Caenorhabditis elegans (Beswick et al. 2006; Clyne et al. 2002; Ndjonka et al. 2003). In Neurospora crassa, putrescine is essential for the decarboxylation reaction but there is no effect on the processing reaction (Hoyt et al. 2000).
AdoMetDC proteins can be classified into two groups (Table 1). The bacterial and archeal enzymes fall in group 1 and the eukaryotic enzymes fall in group 2. In each group there are two subclasses. Group 1a comprises proteins from gram negative bacteria that require a metal ion for activity (Lu and Markham 2007). A prototype of this group is AdoMetDC from Eschericia coli which requires a metal (presumably Mg2+) ion for activity. Group 1b comprises enzymes from gram positive bacteria and archaea that do not require a metal ion for activity (Sekowska et al. 2000; Kim et al. 2000). The group 1b AdoMetDCs are generally smaller than the group 2 enzymes. Group 2a comprises monomeric enzymes from eukaryotes that are not activated by putrescine (as represented by plant AdoMetDC) (Bennett et al. 2002). The buried charge site in group 2a enzymes contains two arginine residues that mimic the role of putrescine for constitutive processing and decarboxylation rates as in other eukaryotic enzymes. Group 2b comprises dimeric eukaryotic AdoMetDCs, which include the enzymes from humans and parasites (Willert et al. 2007; Beswick et al. 2006). The human enzyme represents the group of AdoMetDCs (group 2b-I) that are activated by putrescine and distinguished by a conserved Lys80 that connects the putrescine binding site and the active site through a hydrogen bonding network. The parasitic enzymes represent a novel class of AdoMetDCs (Group 2b-II) that are fully active in the form of a heterodimer formed between the active protomer and a structurally homologous inactive regulatory subunit called the prozyme. The parasitic enzymes have a substitution of isoleucine at the position of Lys80 and activation of putrescine is predicted to be due to allosteric effects (Beswick et al. 2006).
Table 1.
Classification of AdoMetDCs
| Type | Representative species | Oligomeric state | Activation factor | |
|---|---|---|---|---|
| 1a | Gram negative bacteria, archaea | E. coli. | tetramer | Metal ion |
| 1b | Gram positive bacteria, archaebacteria | Bacillus subtilis, T. maritima, A. aeolicus | dimer | Not known |
| 2a | Eukaryotes (plants) | Potato | monomer | None |
| 2b-I | Eukaryotes | Human | dimer | Putrescine |
| 2b-II | Eukaryotes (parasites) | T. brucei, T. cruzi | heterodimer | Prozyme |
In this review we summarize findings from more than a decade of structural studies on AdoMetDC. These include structures of the human, plant and microbial AdoMetDCs. We also describe structural studies that unraveled the importance of the sulfonium center in determining substrate specificity, the structural basis for putrescine activation and the structure-based design of AdoMetDC inhibitors. We describe the discovery of AdoMetDC prozyme in certain parasites, which has important implications for structure-based inhibitor design. Finally we present new studies characterizing the active site of microbial AdoMetDC and we present an analysis of the structural evidence for the evolution of AdoMetDCs.
Structure of human AdoMetDC
The crystal structures of AdoMetDC from Homo sapiens, Solanum tuberosum, Thermotoga maritima and Aquifex aeolicus (PDB code 2III) have been determined (Ekstrom et al. 1999; Toms et al. 2004; Bennett et al. 2002). The use of the human enzyme as an attractive anti-cancer target has been the subject of numerous structural investigations. Human AdoMetDC crystallizes as a dimer with the two protomers related by a two-fold rotation. The overall structure of the protomer is a four layer αββα sandwich. The central β sheet comprises eight antiparallel β strands each flanked by α and 310 helices on either side (Figure 3). The pyruvoyl group is formed at the N-terminus of the α-chain and ispositioned between the β sheets. The crystal structure also reveals a molecule of putrescine bound between the β sheets 16 – 20 Å away from the active site.
Figure 3.
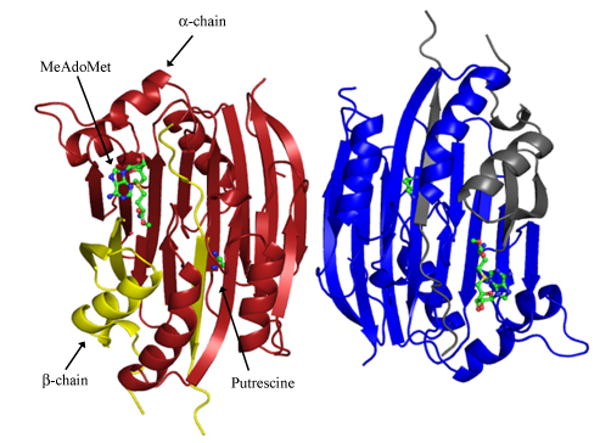
Dimeric structure of processed human AdoMetDC (PDB code 1I7B) viewed down the two-fold axis. The α-chain is colored dark red and β-chain is colored yellow in one of the protomers. Putrescine and MeAdoMet are shown in ball and stick.
The crystal structures of the non-processing S68A mutant (PDB code 1MSV) and H243A mutant (PDB code 1JL0) were determined to better understand the mechanism of autoprocessing and residues involved in the processing (Ekstrom et al. 2001; Tolbert et al. 2003b). The S68A mutant lacks the hydroxyl group necessary for processing and traps the enzyme in a form similar to the proenzyme. The crystal structure of the S68A mutant reveals that residues Leu65-Ser68 form a type II β turn that connects two adjacent antiparallel β strands. Residues His243, Ser229, and Cys82 are located in close proximity to the β turn. Modeling studies of the proenzyme suggest that these residues play a role in stabilizing the oxyoxazolidine intermediate in the autoprocessing reaction. The crystal structure of the H243A mutant trapped the enzyme in the ester form. The trapping of the ester intermediate provides evidence that residue His243 acts as a base in the autoprocessing reaction and abstracts a proton from the Cα atom of the ester intermediate. A superposition of the processed enzyme, proenzyme, and the ester intermediate along with the residues implicated in the processing of hAdoMetDC is shown in Figure 4.
Figure 4.
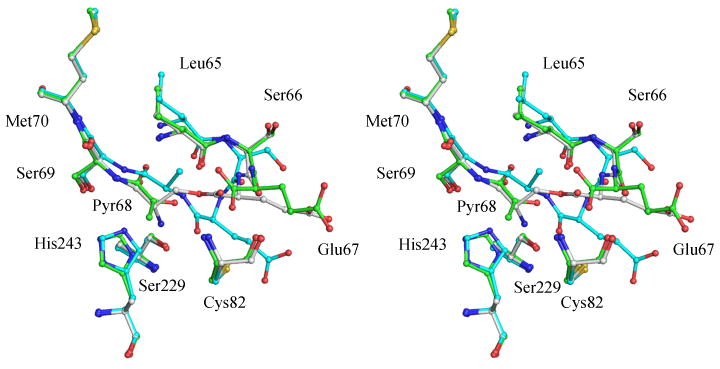
Stereoview of the superposition of the cleavage site in processed enzyme, proenzyme, and ester intermediate of hAdoMetDC (PDB codes 1JEN, 1MSV, and 1JL0). The carbon atoms of the processed enzyme are colored green, those of the proenzyme are colored cyan, and those of the ester intermediate are colored gray. Resides His243, Cys82 and Ser229 that are implicated in the processing mechanism are also shown.
Active site of hAdoMetDC
The active site of hAdoMetDC is characterized by the pyruvoyl group and consists of residues from both β sheets. The structure of hAdoMetDC with the substrate analog S-adenosylmethionine methyl ester (MeAdoMet: PDB code 1I7B) provided the relevant details of substrate binding and active site of the enzyme (Tolbert et al. 2001). MeAdoMet binds covalently to hAdoMetDC through Schiff base formation at the active site pyruvoyl group. The ribose of MeAdoMet makes two hydrogen bonds to Glu247 and the adenine base stacks between Phe223 and Phe7 in an unusual syn conformation (Figure 5). Catalytic residues Cys82, His243, and Ser229 are located near the 5′-linker of the ligand. The carboxymethyl group of MeAdoMet is buried in a cavity formed by Leu65, Cys82, Cys49, Thr81, Thr85, Glu67, and the adenine base. The binding mode of the substrate to AdoMetDC has been the subject of detailed investigations. NMR studies in solution suggest that AdoMet adopts primarily an anti conformation rather than the higher energy syn conformation (Markham et al. 2002). Numerous structural and modeling studies suggest that hAdoMetDC binds ligands in the higher energy conformation with the following stabilizing interactions: (a) π-π stacking between the adenine ring and Phe223 and Phe7; (b). Hydrogen bonds between the adenine base and Glu67; and, (c) Electrostatic interaction between the sulfonium ion and the N3 atom of the adenine ring. The crystal structures with substrate analog inhibitors 5′-deoxy-5′-[N-methyl-N-(3-hydrazino-propyl)amino]adenosine (MHZPA) and 5′-deoxy-5′-[N-methyl-N-[(2-aminooxy)-ethyl]amino]adenosine (MAOEA) reveal that the inhibitors bind to the enzyme by making a covalent bond to the pyruvoyl group and with the adenine base in the syn conformation (Tolbert et al. 2001). The crystal structure of the F223A mutant of hAdoMetDC with MeAdoMet revealed that the ligand still bound in the syn conformation, despite lacking the Phe223 stacking interaction with the adenine base (McCloskey et al. 2009).
Figure 5.
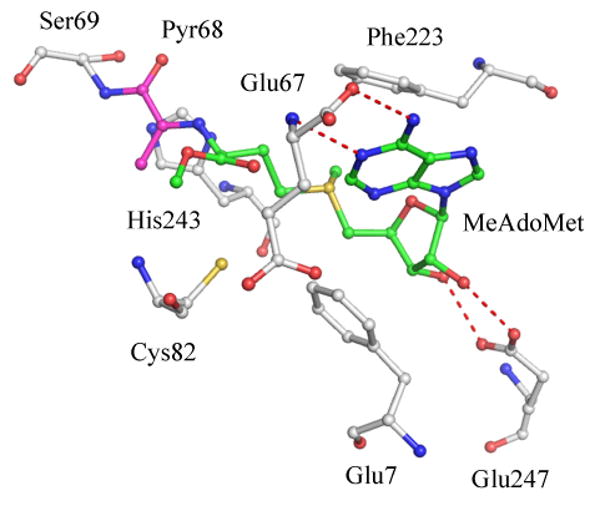
Active site of human AdoMetDC with MeAdoMet bound (PDB code 1I7B). The carbon atoms of MeAdoMet are colored green and those of the pyruvoyl group are colored cyan. Hydrogen bonds are shown as red dashed lines. MeAdoMet makes a Schiff base linkage with the pyruvoyl group and the adenine ring stacks against Phe223 and Phe7 in the syn conformation.
Role of the sulfonium center
Human AdoMetDC provides an interesting case of substrate specificity, ligand binding mode and inhibitor design. Activity assays done in vitro suggest that a positive charge at the position of the sulfonium ion is essential for ligands to bind to AdoMetDC (Pankaskie and Abdel-Monem 1980). Substrate analogs lacking the positive charge did not bind to the enzyme (Pegg and Jacobs 1983; Bale et al. 2009). Alternatively, substrate analogs containing a nitrogen atom at the position of the sulfonium ion maintain the positive charge at physiological pH and show inhibitory activity by binding to the enzyme (McCloskey et al. 2009).
The substrate and inhibitor specificity of AdoMetDC was a puzzling issue since there were no negative charges in the active site to interact with the positive charge on the substrate. Recent crystal structures with inhibitors 5′-deoxy-5′-(dimethylsulfonio)adenosine (MMTA) and 5′-deoxy-5′-(N-dimethyl)amino-8-methyl adenosine (DMAMA) reveal that the specificity arises due to a favorable cation - π interaction between the sulfonium ion and the adjacent carbon atoms with the aromatic rings Phe223 and Phe7. Quantum mechanical calculations reveal that the cation - π interactions in hAdoMetDC provide a stabilization of ∼ 4.5 kcal/mol. In addition, stopped flow kinetic experiments demonstrate that aromatic residues Phe223 and Phe7 in addition to Glu247 are critical for substrate binding (Bale et al. 2009). The importance of these residues is further supported by the complexes with inhibitors methylglyoxal bis(guanylhydrazone) (MGBG) and 4-amidinoindan-1-one-2′-amidinohydrazone (CGP48664A). These inhibitors of AdoMetDC bind to the enzyme by stacking between the aromatic phenyl groups and hydrogen bonding to Glu247, Ser229, Leu65 and the pyruvoyl group (Tolbert et al. 2001).
Putrescine activation of human AdoMetDC
The crystal structure of hAdoMetDC reveals the active site and the binding pocket of putrescine in the enzyme (Ekstrom et al. 2001). Putrescine binds 16 – 20 Å from the active site (Figure 6) and makes extensive interactions with the enzyme. The terminal nitrogen of putrescine farther from the active site is hydrogen bonded to Asp174, Thr 176 and Glu15. The other terminal nitrogen is hydrogen bonded to Glu178, Glu256, Glu15, and Ser113 through water molecules. The aliphatic carbon chain of putrescine stacks against the aromatic rings of Phe111 and Phe285. Human AdoMetDC exists as a dimer and biochemical studies reveal positive cooperativity in putrescine and substrate binding to the dimeric form of the enzyme.
Figure 6.
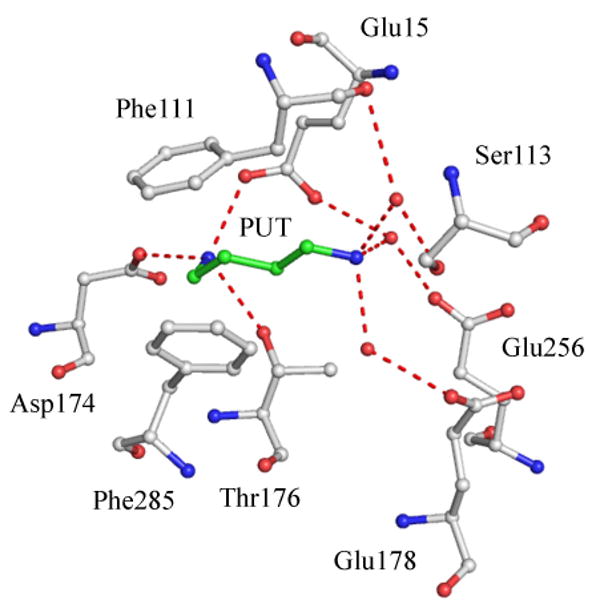
Putrescine binding site in hAdoMetDC (PDB code 1I7B). Putrescine and interacting residues are shown in ball and stick. Water molecules are shown as red spheres and hydrogen bonds are shown as red dashed lines.
The crystal structures of the putrescine free wild type enzyme and the acid to amide mutants of Asp174, Glu178, and Glu256 have been determined (Bale et al. 2008). Putrescine was added to the E178Q and E256Q mutants and the structures obtained. The acid to amide mutations modulate the effects of putrescine on the enzyme. The crystal structures of all the variants mentioned above bound to S-Adenosylmethionine methyl ester (MeAdoMet) were also obtained. The structures of putrescine free AdoMetDC showed key conformational changes when compared to AdoMetDC with putrescine. The binding of putrescine closes the loop Ser312-Phe320 that shields putrescine from external solvent. The closing of the loop is associated with the concerted rearrangement of aromatic residues Phe285, Tyr318, Phe315, and Phe320 (Figure 7a). The crystal structure of the E256Q mutant reveals an alternate conformation of active site residues His243, Glu11, and Ser229 (Figure 7b). The pKa and orientation of active site residues are regulated by a hydrogen bonding network connecting the putrescine binding site and active site (Figure 7c). The hydrogen-bonding network is mediated by Lys80, which is disordered in most of the structures, suggesting that the side chain is shuttling between both the sites. In summary, the crystal structures of putrescine free AdoMetDCs suggest that the mechanism of activation of putrescine is due to following reasons: (a) Conformational changes in loop 312-320 shield putrescine from external solvent; (b) The hydrogen bonding network between putrescine binding site and active site is mediated by Lys80; and, (C) Key catalytic residues are oriented for activity.
Figure 7.
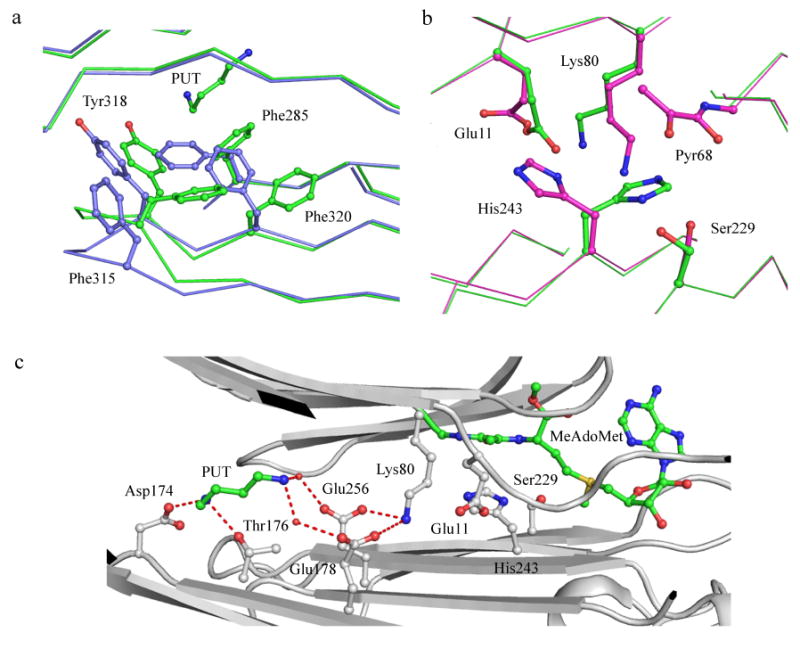
(a) Conformational changes upon putrescine binding in hAdoMetDC (PDB codes 1I7B and 3EP9). Protein with putrescine is colored green and without putrescine is colored light blue. Oxygen atoms are colored red and nitrogen atoms are colored dark blue). Putrescine and aromatic residues undergoing conformational and positional change are shown in ball and stick. (b) Alternate conformation of catalytic residues in the E256Q mutant (PDB codes 3EP4 and 3EPB). The carbon atoms of residues in active conformation are colored green and those of the inactive conformation are colored cyan. (c) Hydrogen bonding network between the putrescine binding site and the active site. All interacting residues are shown in ball and stick. Putrescine and MeAdoMet carbon atoms are colored green. Water molecules are shown as spheres and hydrogen bonds are shown as dashed lines.
The dimer interface plays a key role in the positive cooperativity of putrescine and substrate binding to the dimer. The dimer interface of hAdoMetDC is primarily hydrophilic and held together by hydrogen bonding between Ser312-Ser312, Phe315-Arg307, and Gln311-Gln311. Loop 312-320 that undergoes conformational change upon putrescine binding is located at the dimer interface. The dimer interface provides a possible mechanism by which the binding of putrescine in one protomer enhances the affinity in the other. Other unknown factors are also involved that result in cooperativity in binding.
Inhibitor design for AdoMetDC
The crystal structures revealed molecular details of multiple targets for inhibitor design in hAdoMetDC viz. the active site, proenzyme, and the putrescine binding site. In addition, the crystal structures with inhibitors MGBG, CGP48664A, MHZPA, MAOEA, MMTA, and DMAMA revealed the interactions of the inhibitors with the active site.
The preference of binding the syn conformation has been exploited to make more potent inhibitors to hAdoMetDC. Ligands with a methyl substitution at the C8 position on the adenine ring favor a syn conformation and are 5-18 fold more potent inhibitors than the unsubstituted ligands. In addition, the ligands synthesized with C8 substitutions have a variety of terminal groups to investigate the ability to form a covalent bond with the active site pyruvoyl group (McCloskey et al. 2009). Compounds MMTA and DMAMA are competitive inhibitors of AdoMetDC since they lacked terminal groups for Schiff base linkage. They inhibit hAdoMetDC with an IC50 in the low micromolar range and are ideal candidates for further development.
The crystal structure of hAdoMetDC was also used to virtually screen the active site for novel inhibitors (Brooks et al. 2007). The docking studies were done using the National Cancer Institute's diversity set of compounds and one of the compounds (NSC 354961) showed inhibitory activity with IC50 in low micromolar range.
Structure of plant AdoMetDC
The crystal structure of AdoMetDC from Solanum tuberosum (pAdoMetDC) has been obtained to 2.3 Å (PDB code 1MHM). The enzyme crystallizes in the physiologically relevant monomeric form in the fully processed state. The overall structure of pAdoMetDC is very similar to the hAdoMetDC protomer. The key difference between the enzymes is in the buried charge site. The plant enzyme contains most of the residues similar to the putrescine binding site in humans, but there are key mutations. Residues Asp174, Phe285, Phe111, and Leu13 in the human enzyme are mutated to Val181, His294, Arg114, and Arg18, respectively, in the potato enzyme. Residue Asp174 is critical to binding putrescine and as expected, pAdoMetDC does not bind putrescine. The positively charged arginine residues Arg114 and Arg18 are positioned similarly to the terminal nitrogen atoms of putrescine. There is a hydrogen bonding network between the buried charge site and the active site mediated by Lys85. The arginine residues in the active site mimic putrescine for constitutive rates of processing and activity for plant AdoMetDCs. The active site of pAdoMetDC is very similar to hAdoMetDC with the exception of Phe12 being disordered (equivalent of Phe7 in humans).
Structure of T. maritima AdoMetDC
The structure of AdoMetDC from the T. maritima (TmAdoMetDC) proenzyme provided the first structural insights into group 1b AdoMetDCs (PDB code 1TLU). The protomer of TmAdometDC is a two layer αβ sandwich. The central antiparallel β strand is flanked by two α helices and a short 310 helix. TmAdoMetDC crystallizes as dimer in the asymmetric unit. The protomers are related by a two-fold noncrystallographic rotation and interact face-to-face across the β sheets. The dimer interface comprises charged residues His68, Glu11, Arg112, Asp79, His64, His7, and His110. The charged residues are part of a hydrogen bonded network connecting the active sites located on the opposite edges of the β sheets. The active site comprises residues from both the protomers. indicating that dimerization is necessary for the activity of class 1b enzymes. Residues His68′, Cys83, and Ser55′ are responsible for processing in TmAdoMetDC and are located near the β turn where cleavage occurs (′ denotes residues from the other protomer). The crystal structure of processed TmAdoMetDC and complexes with MeAdoMet and MMTA have been recently obtained and provide more details into substrate binding in group 1b AdoMetDCs (unpublished data).
Discovery of the AdoMet prozyme
The parasitic AdoMetDCs are group 2b enzymes with a recently discovered allosteric mechanism of activation. The activity of the parasitic AdoMetDC from Trypanosoma brucei (TbAdoMetDC) is stimulated ∼1200 fold upon the formation of a heterodimer between the active enzyme and a catalytically dead homolog called the prozyme (Willert et al. 2007). The heterodimers are not further activated by putrescine. The gene encoding for the prozyme is also present in other trypansomatid parasites. The prozymes lack the critical “ES” motif as well as the acidic cysteine residue for processing and activity. In addition to the key mutations that render the prozyme inactive, the sequence of the prozyme has diverged significantly from the enzyme (∼ 30% sequence identity between the prozyme and the enzyme. The parasites of the trypanosomatid family evolved the prozyme through a gene duplication event of the enzyme followed by a significant mutational drift. It was recently shown that the T. cruzi AdoMetDC is activated 110 fold by the prozyme and is further activated by putrescine. In addition, AdoMetDCs are also activated by the formation of cross species heterodimers and are further activated by the addition of putrescine (Willert and Phillips 2009).
The allosteric activation of TbAdoMetDC by prozyme shows similarity to the cooperativity seen in hAdoMetDC. As discussed earlier, the dimer interface holds the key to the cooperativity in substrate binding to hAdoMetDC. The activation by cooperativity is minimal in hAdoMetDC, but suggests that the parasitic AdoMetDC might be allosterically activated by similar mechanism through the dimer interface. In addition, the prozyme might be structurally homologous to the enzyme to bring about similar changes for activation. The cross species activation also suggests that the prozymes from different parasitic species have a similar structure or bring about similar changes to the enzyme. Further activation of the cross species heterodimers by putrescine suggests that other factors are involved for full activity.
Evolution of AdoMetDC
The crystal structures of various AdoMetDCs available present a strong case of protein evolution between group 1 and group 2 enzymes. The prokaryotic AdoMetDC protomer is structurally identical to the C-terminal and N-terminal halves of hAdoMetDC despite the low sequence identity. In addition, the topology of the two β sheets in hAdoMetDC is similar, suggesting that the eukaryotic enzyme has evolved by gene duplication and fusion from the prokaryotic enzyme. A superposition between the human, T. maritima, and A. aeolicus enzymes is shown in Figure 8. The superposition reveals that the human enzyme has two extra β strands in the N-terminal (β7, β8) and C-terminal chains (β15, β16). Strands β7 and β15 are at the dimer interface of hAdoMetDC and interact with identical strands from the other protomer. The monomeric enzyme from potato has equivalent strands β7 and β15 but the dimerization is hindered by key amino acid mutations at the dimer interface. In addition, an insertion of an amino acid in the β strand leads to a bulge in the strand and steric clashes at the proposed dimer interface (Bennett et al. 2002).
Figure 8.
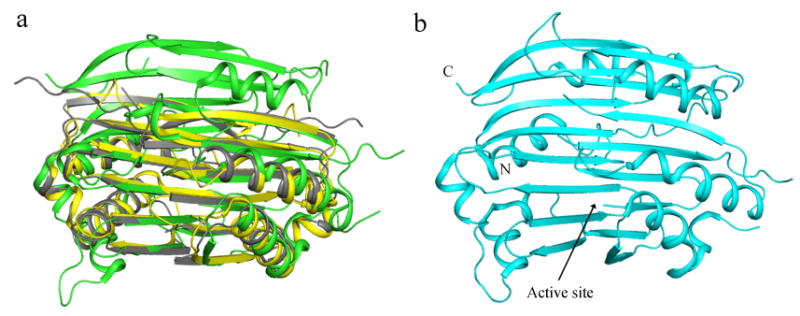
(a) Superposition of human (green), dimeric T. maritima (yellow), and dimeric A. aeolicus (gray) AdoMetDC. (b) Ribbon diagram of monomeric potato AdoMetDC.
The group 1 and 2 AdoMetDCs have diverse sequences but the residues involved in processing and substrate binding are structurally conserved (see Table 2 for details). The conservation suggests a strong case of divergent evolution of eukaryotic AdoMetDC preserving the function. The processing in the second “active site” of group 2 AdoMetDCs is lost due to mutations at the processing site and additional residues in the β turn where processing occurs. In addition, residues responsible for substrate binding and decarboxylation are also missing in the other half of group 2 enzymes.
Table 2.
Comparison of structurally conserved residues in AdoMetDCs
| Role | Human | Potato | T. maritima | A. aeolicus |
|---|---|---|---|---|
| Processing | His243, Cys82, Ser229 | His249, Cys87, Ser236 | His68, Cys83, Ser55 | His69, Cys84, Thr56 |
| Substrate binding | Glu247, Phe223, Phe6, Glu67 | Glu253, Phe230, Phe12, Glu72 | Glu72, Phe49, Trp70, Glu62 | Glu73, Phe50, Trp71, Glu63 |
| Buried charge site | Putrescine, Glu178, Glu256, Glu15, Thr176, Lys80, | Arg18, Arg114, Glu185, Glu262, Glu20, Thr183, Lys85 | - | - |
Conclusions and future directions
Human and parasitic AdoMetDCs are promising targets for the design of anti-cancer and anti-parasitic drugs, respectively. The crystal structures of various AdoMetDCs have helped understand in detail various aspects of the enzyme viz. processing, activation, decarboxylation, substrate specificity, and binding mode. The structural information from complexes of hAdoMetDC with various ligands was vital in the design of more potent inhibitors for the active site of the enzyme. In addition, the structures of AdoMetDCs from various species reveal the evolutionary link between the prokaryotic and eukaryotic enzymes. No structures are available for the heterodimeric parasitic AdoMetDCs, the determination of which would provide insights into the mechanism of activation of parasitic enzymes and aid in the design of inhibitors for anti-parasitic activity.
Acknowledgments
We thank Ms. Leslie Kinsland for assistance in the preparation of this manuscript. This work was supported by National Institutes of Health grant CA-94000.
Abbreviations
- AdoMetDC
S-adenosylmethionine decarboxylase
- dcAdoMet
S-adenosyl-5′-(3-methylthiopropylamine)
- AdoMet
S-adenosylmethionine
- MeAdoMet
S-adenosylmethionine methyl ester
- hAdoMetDC
human AdoMetDC
- pAdoMetDC
potato AdoMetDC
- TmAdoMetDC
Thermotoga maritima AdoMetDC
- ODC
ornithine decarboxylase
- MGBG
methylglyoxal bis(guanylhydrazone)
- CGP48664A
4-amidinoindan-1-one-2′-amidinohydrazone
- MMTA
5′-deoxy-5′-(dimethylsulfonio)adenosine
- DMAMA
5′-deoxy-5′-(N-dimethyl)amino-8-methyl adenosine
- MHZPA
5′-deoxy-5′-[N-methyl-N-(3-hydrazinopropyl)amino]adenosine
- MAOEA
5′-deoxy-5′-[N-methyl-N-[(2-aminooxy)ethyl]amino]adenosine
- TbAdoMetDC
Trypanosoma brucei AdoMetDC
References
- Albert A, Dhanaraj V, Genschel U, Khan G, Ramjee MK, Pulido R, Sibanda BL, von Delft F, Witty M, Blundell TL, Smith AG, Abell C. Crystal structure of aspartate decarboxylase at 2.2 Å resolution provides evidence for an ester in protein self-processing. Nat Struct Biol. 1998;5:289–293. doi: 10.1038/nsb0498-289. [DOI] [PubMed] [Google Scholar]
- Bacchi CJ, Nathan HC, Yarlet N, Goldberg B, McCann PP, Sjoerdsma A, Saric M, Clarkson AB. Combination chemotherapy of drug-resistant Trypanosoma brucei rhodesiense infections in mice using DL-a-difluoromethylornithine and standard trypanocides. Antimicrob Agents Chemother. 1994;38:563–569. doi: 10.1128/aac.38.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale S, Brooks W, Hanes JW, Mahesan AM, Guida WC, Ealick SE. Role of the Sulfonium Center in Determining the Ligand Specificity of Human S-Adenosylmethionine Decarboxylase. Biochemistry. 2009;48:6423–6430. doi: 10.1021/bi900590m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale S, Lopez MM, Makhatadze GI, Fang Q, Pegg AE, Ealick SE. Structural basis for putrescine activation of human S-adenosylmethionine decarboxylase. Biochemistry. 2008;47:13404–13417. doi: 10.1021/bi801732m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basuroy UK, Gerner EW. Emerging concepts in targeting the polyamine metabolic pathway in epithelial cancer chemoprevention and chemotherapy. J Biochem. 2006;139:27–33. doi: 10.1093/jb/mvj022. [DOI] [PubMed] [Google Scholar]
- Bennett EM, Ekstrom JE, Pegg AE, Ealick SE. Monomeric S-Adenosylmethionine Decarboxylase from Plants Provides an Alternative to Putrescine Stimulation. Biochemistry. 2002;41:14509–14517. doi: 10.1021/bi026710u. [DOI] [PubMed] [Google Scholar]
- Beswick TC, Willert EK, Phillips MA. Mechanisms of allosteric regulation of Trypanosoma cruzi S-adenosylmethionine decarboxylase. Biochemistry. 2006;45:7797–7807. doi: 10.1021/bi0603975. [DOI] [PubMed] [Google Scholar]
- Brooks WH, McCloskey DE, Daniel KG, Ealick SE, Secrist JA, 3rd, Waud WR, Pegg AE, Guida WC. In silico chemical library screening and experimental validation of a novel 9-aminoacridine based lead-inhibitor of human S-adenosylmethionine decarboxylase. J Chem Inf Model. 2007;47:1897–1905. doi: 10.1021/ci700005t. [DOI] [PubMed] [Google Scholar]
- Casero RA, Jr, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov. 2007;6:373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- Clyne T, Kinch LN, Phillips MA. Putrescine activation of Trypanosoma cruzi S-adenosylmethionine decarboxylase. Biochemistry. 2002;41:13207–13216. doi: 10.1021/bi026541d. [DOI] [PubMed] [Google Scholar]
- Ekstrom JE, Matthews II, Stanley BA, Pegg AE, Ealick SE. The crystal structure of human S-adenosylmethionine decarboxylase at 2.25 Å resolution reveals a novel fold. Structure. 1999;7:583–595. doi: 10.1016/s0969-2126(99)80074-4. [DOI] [PubMed] [Google Scholar]
- Ekstrom JL, Tolbert WD, Xiong H, Pegg AE, Ealick SE. Structure of a human S-adenosylmethionine decarboxylase self-processing ester intermediate and mechanism of putrescine stimulation of processing as revealed by the H243A mutant. Biochemistry. 2001;40:9495–9504. doi: 10.1021/bi010736o. [DOI] [PubMed] [Google Scholar]
- Fabian CJ, Kimler BF, Brady DA, Mayo MS, Chang CH, Ferraro JA, Zalles CM, Stanton AL, Masood S, Grizzle WE, Boyd NF, Arneson DW, Johnson KA. A phase II breast cancer chemoprevention trial of oral alpha-difluoromethylornithine: breast tissue, imaging, and serum and urine biomarkers. Clin Cancer Res. 2002;8:3105–3117. [PubMed] [Google Scholar]
- Gallagher T, Rozwarski DA, Ernst SR, Hackert ML. Refined structure of the pyruvoyl-dependent histidine decarboxylase from Lactobacillus 30a. J Mol Biol. 1993;230:516–528. doi: 10.1006/jmbi.1993.1168. [DOI] [PubMed] [Google Scholar]
- Gallagher T, Snell EE, Hackert ML. Pyruvoyl-dependent histidine decarboxylase. Active site structure and mechanistic analysis. J Biol Chem. 1989;264:12737–12743. [PubMed] [Google Scholar]
- Gerner EW, Meyskens FL., Jr Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer. 2004;4:781–792. doi: 10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- Gerner EW, Meyskens FL, Jr, Goldschmid S, Lance P, Pelot D. Rationale for, and design of, a clinical trial targeting polyamine metabolism for colon cancer chemoprevention. Amino Acids. 2007;33:189–195. doi: 10.1007/s00726-007-0515-2. [DOI] [PubMed] [Google Scholar]
- Hackert ML, Pegg AE. In: Pyruvoyl-dependent enzymes, Comprehensive Biological Catalysis. Sinnott ML, editor. London: Academic Press; 1997. pp. 201–216. [Google Scholar]
- Hoyt MA, Williams-Abbott LJ, Pitkin JW, Davis RH. Cloning and expression of the S-adenosylmethionine decarboxylase gene of Neurospora crassa and processing of its product. Mol Gen Genet. 2000;263:664–673. doi: 10.1007/s004380051215. [DOI] [PubMed] [Google Scholar]
- Kim AD, Graham DE, Seeholzer SH, Markham GD. S-Adenosylmethionine decarboxylase from archaeon Methanococcus jannaschii: Identification of a novel family of pyruvol enzymes. J Bacteriol. 2000;182:6667–6672. doi: 10.1128/jb.182.23.6667-6672.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZJ, Markham GD. Metal ion activation of S-adenosylmethionine decarboxylase reflects cation charge density. Biochemistry. 2007;46:8172–8180. doi: 10.1021/bi6025962. [DOI] [PubMed] [Google Scholar]
- Markham GD, Norrby PO, Bock CW. S-adenosylmethionine conformations in solution and in protein complexes: conformational influences of the sulfonium group. Biochemistry. 2002;41:7636–7646. doi: 10.1021/bi025682c. [DOI] [PubMed] [Google Scholar]
- McCloskey DE, Bale S, Secrist JA, Tiwari A, Moss TH, Valiyaveettil J, Brooks WH, Guida WC, Pegg AE, Ealick SE. New Insights into the Design of Inhibitors of Human S-Adenosylmethionine Decarboxylase: Studies of Adenine C(8) Substitution in Structural Analogues of S-Adenosylmethionine. J Med Chem. 2009;52:1388–1407. doi: 10.1021/jm801126a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyskens FL, Jr, Gerner EW. Development of difluoromethylornithine (DFMO) as a chemoprevention agent. Clin Cancer Res. 1999;5:945–951. [PubMed] [Google Scholar]
- Millward MJ, Joshua A, Kefford R, Aamdal S, Thomson D, Hersey P, Toner G, Lynch K. Multi-centre Phase II trial of the polyamine synthesis inhibitor SAM486A ( CGP48664) in patients with metastatic melanoma. Invest New Drugs. 2005;23:253–256. doi: 10.1007/s10637-005-6734-z. [DOI] [PubMed] [Google Scholar]
- Ndjonka D, Da'dara A, Walter RD, Luersen K. Caenorhabditis elegans S-adenosylmethionine decarboxylase is highly stimulated by putrescine but exhibits a low specificity for activator binding. Biol Chem. 2003;384:83–91. doi: 10.1515/BC.2003.009. [DOI] [PubMed] [Google Scholar]
- Pankaskie M, Abdel-Monem MM. Inhibitors of polyamine biosynthesis 8: Irreversible inhibition of mammalian S-adenosyl-L-methionine decarboxylase by substrate analogs. J Med Chem. 1980;23:121–127. doi: 10.1021/jm00176a004. [DOI] [PubMed] [Google Scholar]
- Pegg AE. Polyamine metabolism and its importance in neoplastic growth and as a target for chemotherapy. Cancer Res. 1988;48:759–774. [PubMed] [Google Scholar]
- Pegg AE, Jacobs G. Comparison of inhibitors of S-adenosylmethionine decarboxylase from different species. Biochem J. 1983;213:495–502. doi: 10.1042/bj2130495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegg AE, Xiong H, Feith D, Shantz LM. S-adenosylmethionine decarboxylase: structure, function and regulation by polyamines. Biochem Soc Trans. 1998;26:580–586. 526. doi: 10.1042/bst0260580. [DOI] [PubMed] [Google Scholar]
- Schmitzberger F, Kilkenny ML, Lobley CM, Webb ME, Vinkovic M, Matak-Vinkovic D, Witty M, Chirgadze DY, Smith AG, Abell C, Blundell TL. Structural constraints on protein self-processing in L-aspartate-a-decarboxylase. Embo J. 2003;22:6193–6204. doi: 10.1093/emboj/cdg575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekowska A, Coppée JY, Le Caer JP, Martin-Verstraete I, Danchin A. S-adenosylmethionine decarboxylase of Bacillus subtilis is closely related to archaebacterial counterparts. Mol Microbiol. 2000;36:1135–1147. doi: 10.1046/j.1365-2958.2000.01930.x. [DOI] [PubMed] [Google Scholar]
- Soriano EV, McCloskey DE, Kinsland C, Pegg AE, Ealick SE. Structures of the N47A and E109Q mutant proteins of pyruvoyl-dependent arginine decarboxylase from Methanococcus jannaschii. Acta Crystallogr D Biol Crystallogr. 2008;64:377–382. doi: 10.1107/S0907444908000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor CW, Tabor H. Methionine adenosyltransferase (S-adenosylmethionine synthetase) and S-adenosylmethionine decarboxylase. Advan Enzymol Related Areas Mol Biol. 1984a;56:251–282. doi: 10.1002/9780470123027.ch4. [DOI] [PubMed] [Google Scholar]
- Tabor CW, Tabor H. Polyamines. Annu Rev Biochem. 1984b;53:749–790. doi: 10.1146/annurev.bi.53.070184.003533. [DOI] [PubMed] [Google Scholar]
- Tolbert DW, Ekstrom JL, Mathews II, Secrist JAI, Kapoor P, Pegg AE, Ealick SE. The structural basis for substrate specificity and inhibition of human S-adenosylmethionine decarboxylase. Biochemistry. 2001;40:9484–9494. doi: 10.1021/bi010735w. [DOI] [PubMed] [Google Scholar]
- Tolbert WD, Graham DE, White RH, Ealick SE. Pyruvoyl-dependent arginine decarboxylase from Methanococcus jannaschii: crystal structures of the self-cleaved and S53A proenzyme forms. Structure. 2003a;11:285–294. doi: 10.1016/s0969-2126(03)00026-1. [DOI] [PubMed] [Google Scholar]
- Tolbert WD, Zhang Y, Cottet SE, Bennett EM, Ekstrom JL, Pegg AE, Ealick SE. Mechanism of human S-adenosylmethionine decarboxylase proenzyme processing as revealed by the structure of the S68A mutant. Biochemistry. 2003b;42:2386–2395. doi: 10.1021/bi0268854. [DOI] [PubMed] [Google Scholar]
- Toms AV, Kinsland C, McCloskey DE, Pegg AE, Ealick SE. Evolutionary Links as Revealed by the Structure of Thermotoga maritima S-Adenosylmethionine Decarboxylase. J Biol Chem. 2004;279:33837–33846. doi: 10.1074/jbc.M403369200. [DOI] [PubMed] [Google Scholar]
- van Poelje PD, Snell EE. Pyruvol-dependent enzymes. Ann Rev Biochem. 1990;59:29–59. doi: 10.1146/annurev.bi.59.070190.000333. [DOI] [PubMed] [Google Scholar]
- Wallace HM, Fraser AV, Hughes A. A perspective of polyamine metabolism. Biochem J. 2003;376:1–14. doi: 10.1042/BJ20031327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert EK, Fitzpatrick R, Phillips MA. Allosteric regulation of an essential trypanosome polyamine biosynthetic enzyme by a catalytically dead homolog. Proc Natl Acad Sci U S A. 2007;104:8275–8280. doi: 10.1073/pnas.0701111104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert EK, Phillips MA. Cross-species activation of trypanosome S-adenosylmethionine decarboxylase by the regulatory subunit prozyme. Mol Biochem Parasitol. 2009 doi: 10.1016/j.molbiopara.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Ashman HG, Schenone A. Methylglyoxal bis(guanylhydrazone) as a potent inhibitor of mammalian and yeast S-adenosylmethionine decarboxylases. Biochem Biophys Res Commun. 1972;46:288–295. doi: 10.1016/0006-291x(72)90661-4. [DOI] [PubMed] [Google Scholar]
- Xiong H, Pegg AE. Mechanistic studies of the processing of human S-adenosylmethionine decarboxylase proenzyme. Isolation of an ester intermediate. J Biol Chem. 1999;274:35059–35066. doi: 10.1074/jbc.274.49.35059. [DOI] [PubMed] [Google Scholar]
- Xiong H, Stanley BA, Pegg AE. Role of cysteine-82 in the catalytic mechanism of human S-adenosylmethionine decarboxylase. Biochemistry. 1999;38:2462–2470. doi: 10.1021/bi9825201. [DOI] [PubMed] [Google Scholar]