

Abstract
The muted (mu) mouse is a model for Hermansky–Pudlak Syndrome (HPS), an inherited disorder of humans causing hypopigmentation, hemorrhaging and early death due to lung abnormalities. The mu gene regulates the synthesis of specialized mammalian organelles such as melanosomes, platelet dense granules and lysosomes. Further, balance defects indicate that it controls the synthesis of otoliths of the inner ear. The mu gene has been identified by a positional/candidate approach involving large mouse interspecific backcrosses. It encodes a novel ubiquitously expressed transcript, specifying a predicted 185 amino acid protein, whose expression is abrogated in the mu allele which contains an insertion of an early transposon (ETn) retrotransposon. Expression is likewise expected to be lost in the mu J allele which contains a deletion of a single base pair within the coding region. The presence of structurally aberrant melanosomes within the eyes of mutant mice together with localization of the muted protein within vesicles in both the cell body and dendrites of transfected melan-a melanocytes emphasizes the role of the mu gene in vesicle trafficking. The mu gene is present only in mice and humans among analyzed genomes. As is true for several other recently identified mouse HPS genes, the mu gene is absent in lower eukaryotes. Therefore, the mu gene is a member of the novel gene set that has evolved in higher eukaryotes to regulate the synthesis/function of highly specialized subcellular organelles such as melanosomes and platelet dense granules.
INTRODUCTION
The synthesis and/or processing of melanosomes, lysosomes and platelet dense granules is under common genetic control. A principal source of evidence for this is the existence of a series of at least 15 genetically defined mouse hypopigmentation mutants which have associated prolonged bleeding times due to platelet storage granule deficiencies and reduced secretion of lysosomal enzymes from several cell types (1,2). Several of these gene products regulate intracellular vesicle trafficking, encoding subunits of the AP-3 adaptor complex (3–6) (pearl and mocha), the Rab geranylgeranyl transferase enzyme (7) (gunmetal), Rab27a (8) (ashen) or a syntaxin13-interacting protein (9) (pallid). All mutants are models for the genetically heterogeneous inherited disease Hermansky–Pudlak Syndrome (HPS) which presents with hypopigmentation, hemorrhaging and early death due to lung abnormalities (10,11). Three genetically distinct forms of the disease, HPS1, HPS2 and, more recently, HPS3 (12), have been described, and additional human loci are likely. The HPS1, HPS2 and HPS3 genes are orthologs of the mouse pale ear, pearl and cocoa genes, respectively.
The muted mouse was found in 1969 in a stock carrying t-alleles (13); it causes light eyes at birth and fur of a muted brown shade. Subsequent studies established it as an appropriate animal model for human HPS (14). In addition to the common HPS symptoms of hypopigmentation, platelet storage pool deficiency and lysosomal hyposecretion (1), muted mice suffer from a balance and postural defect due to a deficiency of otoliths of the inner ear (13). In this respect, muted mice resemble two other mouse HPS mutations, pallid (15) and mocha (16). Early mapping studies (13) placed the mu gene on the proximal portion of mouse chromosome 13, a position that was refined in subsequent higher resolution mapping in 528 backcross progeny (17).
We now report the identification and expression patterns of the muted gene as an entry into knowledge of the mechanisms of action of this and related genes in the synthesis of specialized mammalian organelles and into future therapies of this debilitating inherited disease.
RESULTS
The mu gene (13) causes obvious hypopigmentation of the coat, producing fur of an obviously light muted brown shade, compared to wild-type agouti littermates (Fig. 1). In young (<5 days of age) mutant mice hypopigmentation of the eyes is likewise apparent (data not shown).
Figure 1.

Hypopigmentation of the coat in adult muted mice. Homozygous mu/mu mice and heterozygous mu/+ littermates on the agouti CHMU/Le strain background are depicted.
Positional/candidate cloning of the muted gene
Previous mapping (17) of the initial 528 progeny of this back-cross had positioned the mu gene within proximal mouse chromosome 13, 0.4 cM distal to microsatellite D13Mit38 and non-recombinant with three microsatellites (D13Mit87, D13Mit88 and D13Mit137). Extension of this interspecific backcross to 1340 progeny produced a high-resolution genetic map (Fig. 2A) of the region of chromosome 13 immediately surrounding the muted gene. Several markers including microsatellites D13Mit38, D13Mit87, D13Mit88, D13Mit137, D13Mit177 and D13Mit296, together with several molecular markers (Fig. 2A), were found recombinant with, though still genetically closely linked to, mu. The mu gene was ultimately localized within a 0.14 cM critical genetic region flanked by the sequence tagged site (STS) marker M-09827 proximally and microsatellite D13Mit87 distally.
Figure 2.

High resolution genetic (A) and physical maps (B) of the region of mouse chromosome 13 containing the mu gene. The centromere (circle) is at left; distances between adjacent markers are given in map units (cM). Numbered markers are D13Mit microsatellites. Other molecular markers include BAC end clones and STSs. STSs M-09827 (also known as 26.MMHAP67FRA11.seq) and M-01611 (also known as MHAa38d12.seq) are described within the contig WC13.11 (http://www-genome.wi.mit.edu/). The mu critical genetic region spanning the 0.14 cM interval between markers M-09827 and D13Mit87 is indicated. A contig of 13 overlapping BACs, represented by horizontal lines (not drawn to scale) and containing the indicated molecular markers is depicted in (B). BACs are located at their approximate chromosomal positions, and their sizes in kb are indicated at the right. Circles indicate T7 BAC ends; squares indicate SP6 BAC ends. BACs rp23-347f7 and rp23-317e13, which span the critical genetic interval and which were sequenced and used for subsequent candidate gene searches, are highlighted. C9, E4, G9 and G6 are BACs from the RPCI-22 library.
A high resolution physical map (Fig. 2B) was constructed from 13 bacterial artificial chromosome (BAC) clones selected initially with the markers M-09827 and D13Mit87 and completed with selected BAC ends as probes for directed walking. Two overlapping BAC clones (rp23-347f7 and rp23-317e13) completely spanned the mu critical genetic region, which was estimated by comparative restriction digests of the two BACs to contain maximally 400 kb. These two BACs were utilized in exon trapping experiments and were completely sequenced (GenBank accession nos AC068907 and AC069562, respectively).
Four known genes (bone morphogenetic protein 6, bone morphogenetic protein 5, elongation factor p18 and a nuclease-sensitive element DNA-binding protein) and 12 novel expressed sequence tags (ESTs) were identified within the sequences of these two BACs by BLAST searching, demonstrating that mu is in a segment of mouse chromosome 13 homologous to human chromosome 6p24–p23.
The muted (mu) mouse was tested for mutations in these genes and ESTs by several techniques including Southern blotting with a battery of restriction enzymes, northern blotting and direct sequencing. EST AA475929 (IMAGE clone 876461), identified both by BAC sequencing and exon trapping of BAC 317e13, became a likely candidate as it displayed quantitative and qualitative differences of RT–PCR kidney products among mu/mu, mu/+ and +/+ mice (Fig. 3). Amplification of transcripts of +/+ and mu/+ produced the expected 401 bp product using primers derived from exon 3 and the 3′-untranslated region (3′-UTR). However, this product was absent in similar amplifications of mu/mu kidney cDNA, which yielded instead a very weak larger band at 584 kb.
Figure 3.
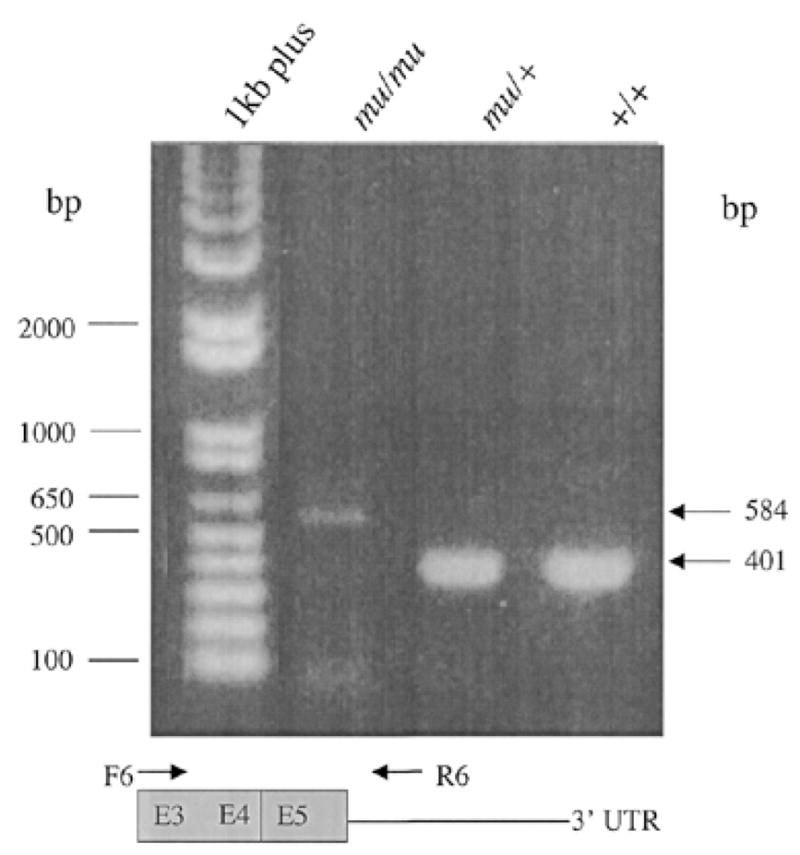
RT–PCR products are abnormal in muted mice. A forward primer (F6) derived from exon 3 of the muted gene and a reverse primer (R6), derived from the 3′-UTR of the muted gene, were used in RT–PCR amplifications of cDNA of kidney of mu/mu, mu/+ and +/+ mice.
Altered expression of this gene in mutant tissues was confirmed by northern blotting (Fig. 4). Several tissues (brain, bone marrow, kidney and liver) of normal C57BL/6J (+/+) mice contained a principal transcript at 1.8 kb which was not visible in total RNA from all tissues of mu/mu mice and was intermediate in quantity in heterozygous mu/+ mice (Fig. 4A). A hybridizing transcript was visible in mu/mu samples when poly(A)+ RNA was blotted (Fig. 4B). However, it presented as a very small quantity of a larger 2.0 kb band. The status of EST AA475929 as a candidate for the mu gene was additionally supported by abnormal Southern blotting patterns (data not shown) and by the observation that a microsatellite polymorphism within this gene completely co-segregated with the mu locus in the 1340 progeny backcross.
Figure 4.

Transcripts are altered in size and expressed at significantly reduced levels in muted mice. (A) Total RNAs of brain, bone marrow, kidney and liver of homozygous muted, heterozygous mu/+ and control C57BL/6J +/+ mice were northern blotted and probed with a 671 bp probe derived from nucleotides 673–1344 of the mouse muted cDNA. (B) Northern blots of poly(A)+ RNA isolated from kidneys of homozygous muted, heterozygotes and control C57BL/6J +/+ mice were similarly probed. The same blots were reprobed with β-actin and glucose 3-phosphate dehydrogenase as loading controls.
Sequencing of the larger 584 bp PCR product (Fig. 3) derived from mu/mu cDNA revealed a 183 bp insertion between exons 3 and 4 at nucleotide +319 (Fig. 5B). This insertion sequence, by BLAST analysis, matched 100% to the mouse early transposon (ETn) (GenBank accession no. Y17107) sequence. The position of the partial ETn sequence between exons 3 and 4 suggested that a partial or complete ETn might be located within intron 3 of the muted gene in mu/mu mice. Indeed an ~9.0 kb product was amplifiable, by long range PCR, from genomic DNA of mu/mu mice, compared to a 1.48 kb product from control C57BL/6J (data not shown). Direct sequencing of muted genomic DNA revealed that the ETn is inserted into intron 3 between 2362A and 2363G (numbering begins at the first base of intron 3) (Fig. 5B). A 6 bp sequence (GGTGCA), which forms nucleotides 2357–2362 in the normal gene, is repeated at the other end of the ETn.
Figure 5.

Summary of the genomic, cDNA and predicted protein structures of the mu gene in (A) normal mice and (B) mutant mu/mu and muJ/muJ mice.
The 183 bp insertion in the mu transcript, corresponding to nucleotides 248–430 in the ETn, is likely generated by activation of cryptic splice sites within the ETn. This leads to an in-frame insertion in the translated protein at position 106, predicting a 246 amino acid protein (Fig. 5B). However, it is doubtful that significant levels of this protein are produced given the very large reductions in mutant transcript levels (Fig. 4).
An obvious pathological mutation likewise was found within mutant mice containing an independent mutation at the muted gene (muJ). Mutants with this allele have a single base pair deletion (del +60G) within the protein coding region in exon 1 (Fig. 5B) of the muted gene. The resulting frameshift predicts a truncated protein of 58 amino acids produced by a premature termination codon at nucleotide +175.
Structure of the muted gene and gene products
The full length muted cDNA sequence of 1791 nt (GenBank accession no. AF426433) was obtained by 5′-rapid amplification of cDNA ends (5′-RACE) extension of clone 876461 using C57BL/6J mouse kidney poly(A)+ RNA. The cDNA contains a small 5′-UTR of 68 nt, a large 3′-UTR of 1142 nt and a poly(A) tail of 23 bp. An open reading frame, predicted to encode a protein of 185 amino acids, is found between nucleotides 1 and 555.
Neither mouse nor human (see below) muted proteins contain an N-terminal signal sequence or transmembrane domains or in fact any other recognizable protein domains. A possible vacuolar targeting motif (TLPK at position 90) is present in the human muted protein, indicating possible involvement in lysosomal or early endosomal traffficking, but this motif is absent in the mouse sequence.
The muted transcript is expressed ubiquitously in the mouse, with relatively lower expression in skeletal muscle (Fig. 6).
Figure 6.
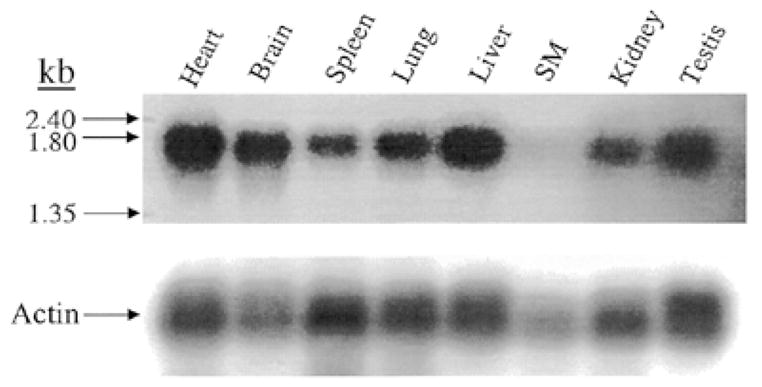
The muted gene is ubiquitously expressed in mouse tissues. A Clontech multiple tissue northern blot was probed with muted cDNA. The blot was reprobed with β-actin as a loading control.
The genomic structure (Fig. 5A) of the mouse muted gene was determined by alignment of the muted cDNA sequence with the sequence of BAC rp23-317e13. The coding sequence is distributed among five exons and four introns, and consensus splicing sequences were found at all exon/intron boundaries. The muted gene contains 52 kb. Primer sequences for amplification of exon/intron boundaries of both mouse and human genes are available upon request.
A sequence corresponding to the mouse muted gene occurs on human chromosome 6p24.3–25.1 in human BACs 303A1 (GenBank accession no. AL096800) and 511E16 (GenBank accession no. AL023694). The human muted cDNA sequence, obtained by RT–PCR and RACE analyses (accession no. AF426434) is 2282 bp and is 79% identical to the mouse sequence. The additional size compared to the mouse transcript derives mainly from an extended 3′-UTR (1680 bp). The genomic structure of the human muted gene, determined by alignment of the cDNA sequence with the sequences of BACs 303A1 and 511E16, is similar to that of the mouse muted gene. The predicted mouse and human muted proteins (Fig. 7) are 76% identical and 86% similar with an additional two amino acids in the human protein due to insertion of the dipeptide GS at amino acid position 19.
Figure 7.

Alignment of mouse mu polypeptide sequence with homologous human sequence. Black-shaded residues are identical; gray-shaded residues are similar.
Analyses of HPS patients with no apparent mutations in known HPS genes have thus far revealed no molecular abnormalities in the muted gene (M. Huizing, W. Gahl and R. Spritz, personal communication).
The muted gene is novel and found only in higher eukaryotes
Searches of the BLASTn, BLASTp and Swiss Pro databases with the mouse muted cDNA and/or predicted protein sequences revealed no significant homologies with the exception of the corresponding human sequence. Only very weak homologies to two other mammalian proteins, α-actinin [30% identity and 46% similarity over a small (72 amino acid) region] and human transcription regulator protein BACH1 (33% identity and 50% similarity over a 69 amino acid region) were apparent.
Melanosomal abnormalities of muted mice
Although the eyes of adult mice appear normal in pigmentation (Fig. 1), closer ultrastructural examination reveals that the eyes of adults have significant quantitative and qualitative abnormalities of melanosomes (Fig. 8). Total numbers of melanosomes are reduced severely within the retinal pigment epithelium (RPE) and significantly reduced within the choroid. The few remaining mutant RPE melanosomes are smaller and often contain unusual inclusions and lamellar bodies. Mutant choroidal melanosomes often are less dense than their normal counterparts and may exhibit irregular outlines.
Figure 8.
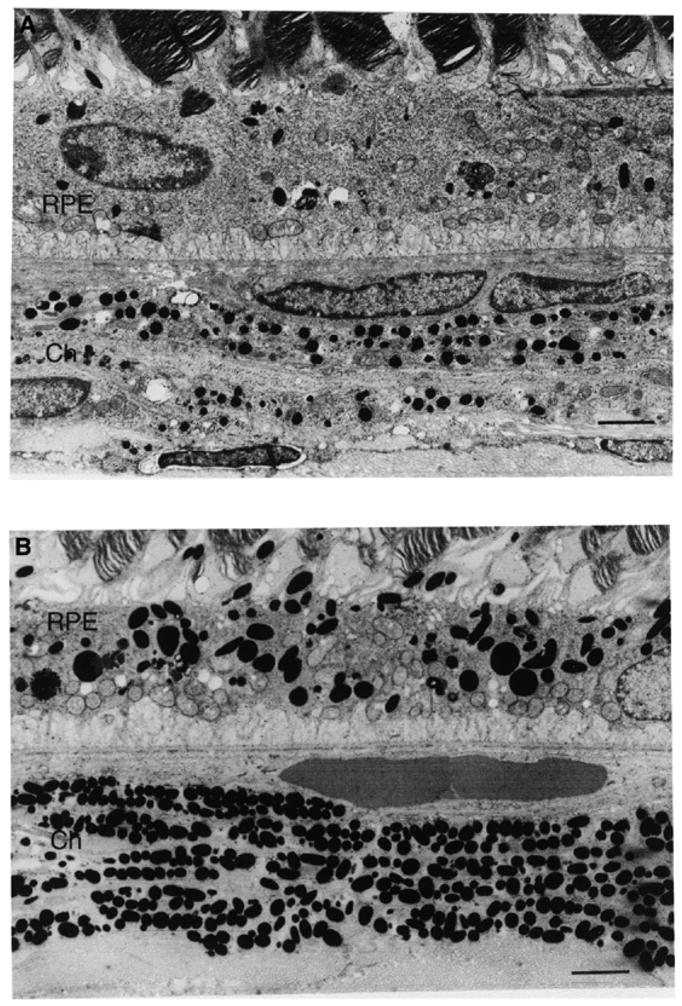
Melanosomes of the retinal pigment epithelium (RPE) and choroid (Ch) of muted mice are qualitatively and quantitatively abnormal. (A) Muted and (B) normal mice. Calibration bars are 2 μm.
Transfection of melan-a melanocytes
Melan-a cells transfected with epitope-tagged muted constructs expressed the muted protein in vesicles distributed throughout both the cell body and dendrites (Fig. 9). Comparison with bright field images established that the vesicles were not coincident with melanosomes. The results were not influenced by the nature of the epitope or the epitope position as similar subcellular distributions were obtained after transfection of other constructs including green fluorescent protein at the C-terminus and the Myc epitope at the N-terminus of the muted protein. Transfected COS-7 cells exhibited a similar vesicular pattern throughout the cell body.
Figure 9.
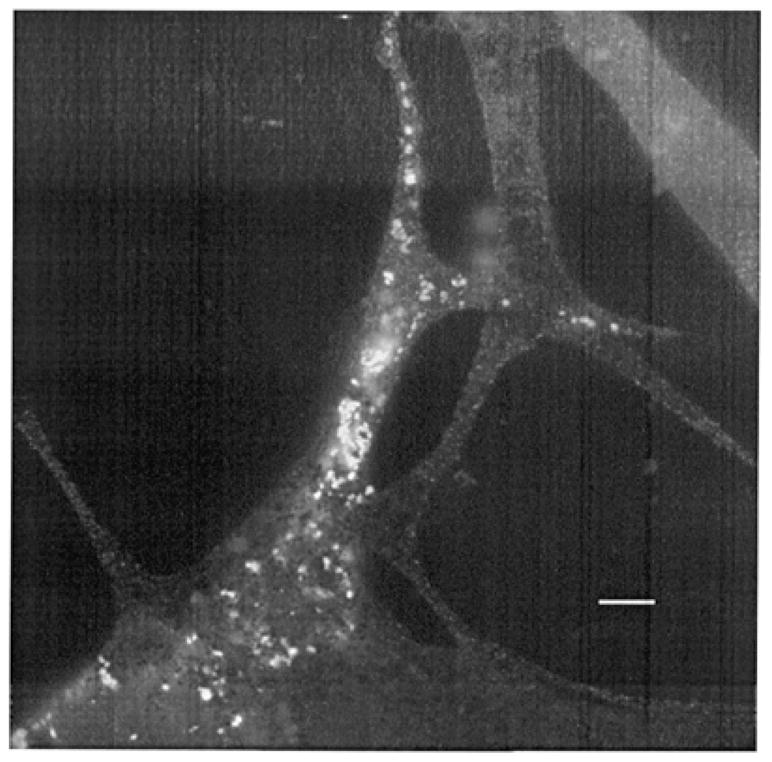
Expression of the mouse muted-FLAG protein in transfected mouse melan-a melanocytes. Note that both the cell body and dendrites of the transfected cells contain organelles which are positive for the muted protein and that these organelles are not visible in non-transfected cells. Bar, 5 μm.
DISCUSSION
The muted gene sequence predicts a novel 185 amino acid protein which has no homology to other mouse models of HPS or with other known gene products involved in vesicle trafficking in any organism. That the bona fide muted gene has been identified is supported by the fact that deleterious mutations were found within it in two independently derived muted alleles. The greatly reduced expression of the corresponding transcript in a wide variety of tissues of mu/mu mice is likewise consistent with this hypothesis.
The muted gene not only is novel, but also is apparently found only in higher eukaryotes. Several lower eukaryotes whose genomes are wholly or largely sequenced, including yeast, Caenorhabditis elegans and Drosophila, contain no similar protein. This result is important since it suggests that novel genes have evolved in higher eukaryotes to affect the synthesis/function of very specialized organelles such as melanosomes and platelet dense granules, which of course are found only in higher eukaryotes. Further, the principal effect of the muted gene and related mouse HPS genes on lysosomes is on secretion (1), a specialized function of this organelle (1,18–20), which is now recognized as important in plasma membrane repair (21,22) in mammalian cells. Supporting the idea that distinct genes have evolved in higher eukaryotes to regulate the function of specialized organelles are the findings that homologs of at least three other HPS genes, pale ear (23,24), pallid (9) and cocoa (25), are not evident in yeast. On the other hand, other identified mouse HPS mutants encode genes, including subunits of the AP-3 adaptor complex (3,5) (pearl and mocha), a subunit of Rab geranylgeranyl transferase (7) (gunmetal) and Rab27a (8) (ashen), which are found in common among all eukaryotes. Clearly, the HPS phenotype can arise from both ‘specialized’ and ‘common’ organellar genes, and the normal production of melanosomes, platelet dense granules and lysosomes requires overlapping contributions from both. Identification of the mechanism(s) of action of the muted gene together with other novel mouse HPS genes, should allow characterization of novel pathways for organelle synthesis in mammals.
Like all HPS genes thus far identified, the muted gene is ubiquitously expressed, and the muted mouse is deficient in expression of the muted transcript in all tissues surveyed. Nevertheless, the phenotypes of the mouse and human HPS mutants are relatively tissue specific, most notably affecting pigmentation and bleeding times. These effects may be related to the fact that melanocytes and platelets are specialized to produce very high quantities of organelles of the lysosome/endocytic pathway as compared with other cells and thus require greater concentrations of HPS proteins. The present experiments document that the muted gene regulates, in both qualitative and quantitative senses, production of melanosomes, not only of the coat, but also those of the RPE and choroid of the eye. It is possible that closer examination will reveal that the muted gene controls organellar production in other tissues. Consistent with the ubiquitous expression of the muted gene, abnormalities of lysosomal secretion have been noted in kidney of muted mutant mice (14). Whether the balance defects caused by absence of inner ear otoliths in muted mice (13) are likewise due to abnormal organellar trafficking is uncertain. However, it is intriguing that calcium-containing granules, observable within vestibular supporting cells, may be important in otolith formation (26). Also, it is noteworthy that balance problems have been described in selected HPS patients (10). Thus far, other in vivo pathologies are not obvious (27) in muted mice.
The muted gene sequence offers no clues to its subcellular targeting as no specific targeting domains, which are maintained in both mouse and human muted proteins, were detected. However, the muted protein is expressed in vesicles, which are not melanosomes, throughout both the cell body and dendrites of transfected melanocytes. The vesicular location is consistent with the observation that several proteins encoded by other HPS genes regulate vesicle trafficking. Clearly, additional studies are required to further define the subcellular site of the muted protein and its interacting protein partners.
Insertions of retroviral elements, such as the ETn found within the mu allele, are associated with other mouse HPS mutations such as those of alleles of the pale ear (23,24) and pearl (4) genes and are a common cause of a large number and variety of mouse mutations (28). In fact, the identical 183 bp partial ETn insertion found in mu transcripts has been detected in transcripts of the Gli3 zinc finger protein-encoding gene in the Pdn mouse (29), an allele of the nude mouse mutant (30) and the Fas gene in the lpr mouse mutant (31). Its insertion into a wide variety of transcripts is likely due to flanking consensus splice signals. The ETn insertion in the mu allele is within an intron at a location that would not necessarily be expected to lead to loss of gene expression. Nevertheless, it is clear from RT–PCR, northern and sequencing analyses that this insertion causes large reductions in gene expression and that mu transcripts contain an additional in-frame183 bp insertion between exons 3 and 4. Reduced expression may result from inefficient splicing of this insertion or from transcription interference by other long range effects of retroviral insertion such as competition for the influence of enhancers or other general effects such as usurping the transcriptional machinery in a locus (28).
Identification and the analyses of the mechanism(s) of action of the muted gene should: (i) increase our understanding of the biogenesis of specialized mammalian organelles such as melanosomes, platelet dense granules and lysosomes and (ii) as an appropriate animal model, eventually allow development of therapies for HPS, which presently has no cure (10,11).
MATERIALS AND METHODS
Mice and backcrosses
Mice carrying the muted (mu) allele on the CHMU/Le ch+/+mu stock background were obtained from the Jackson Laboratory and were subsequently bred at Roswell Park Cancer Institute. Frozen genomic DNA from the independently derived muted-J (muJ) allele was obtained from the Jackson Laboratory. The muJ mutation occurred spontaneously on the T18H stock (the Jackson Laboratory mouse online DNA resource catalog).
Genetic and physical maps
Mapping of the mu gene was conducted simultaneously with that of the pearl (pe) gene (17), utilizing a stock segregating mutant forms of both genes together with the satin (sa) gene. This stock was crossed with the inbred, wild-derived Mus musculus musculus (PWK) inbred strain to obtain a high degree of polymorphism for molecular markers.
High resolution genetic and BAC-based physical maps of the mu critical region were generated as described by Feng et al. (3). An interspecific cross was performed between the mu/mu stock and PWK mice, and 1340 backcross progeny were typed for coat color and molecular markers at 6 weeks using microsatellite markers D13Mit38 and D13Mit138, which flank mu proximally and distally, respectively. Informative mice were typed for all markers within the muted critical region. BACs were selected by screening the RPCI-22 and RPCI-23 BAC libraries (Roswell Park Cancer Institute, Buffalo, NY) with molecular probes and BAC end probes derived from the mu critical genetic region. Positive BACs were subjected to restriction enzyme (NotI) digestion and pulsed-field gel electrophoresis to determine the size of the insert. The degree of BAC overlap was estimated by determination of shared fragments on 0.8% agarose gels following EcoRI digestion. BAC ends were sequenced with SP6 and T7 promoter primers. BAC contigs were constructed by determination of overlap with primer pairs designed at BAC end sequences.
BAC sequencing
BACs rp23-347f7 and rp23-317e13 (Fig. 2) were subjected to complete DNA sequence analysis (32,33) and have GenBank accession nos AC068907 and AC069562, respectively. BAC sequences were annotated by subjecting the working draft sequences to BLAST searches (http://www.ncbi.nih.gov/BLAST) after masking repetitive sequences (ftp.genome.washington.edu/cgi-bin/RepeatMasker).
Other molecular procedures
Total RNA was isolated from mouse tissues with RNAzol B reagent (Tel-Test, Friendswood, TX). Poly(A)+ RNA was obtained with the PolyATract mRNA Isolation System I Kit (Promega). Reverse transcription of RNA was performed with the Superscript Preamplification System for RT–PCR (Gibco BRL). Primers mm-F1 (5′-AACTGGTCGGGATGAGTGG-3′) and mm-R1 (5′-CAGTTAGGTGCTGAAGCACG-3′) amplified the complete open reading frame of the muted cDNA. Primers mm-F6 (5′-CCCAAATGCAGAGAGACCAT-3′) and mm-R6 (5′-GTGCCCCTGAGTGTACTGCT-3′) amplified the region of the muted cDNA containing the ETn insertion. Primers In3F2 (5′-GCTGGAAGGAGGTTGAAAAG-3′) and In3R2 (5′-CCTTTGGCTTTGAATGGGTA-3′) identified the ETn insertion in mu genomic DNA using long-range PCR amplification conditions (Advantage 2 Kit, Clontech). Primers mm-1F (5′-AACACCTCCCATACGCAGAT-3′) and mm-1R (5′-TAGTTCCCATTCTCCGCTTG-3′) identified the mutation within exon 1 of the muted gene in the muJ allele by amplification of exon 1 and its adjacent exon/intron boundaries in genomic DNA. PCR amplification conditions were 94°C for 30 s, 59°C for 30 s, 72°C for 1–2 min, for 35 cycles. PCR products were treated with the Pre-sequencing kit (USB) and then sequenced in an ABI-377 DNA sequencer (Applied Biosystems Inc.) using the BigDye Terminator Cycle Sequencing Ready Reaction Kit.
Full length muted cDNA was obtained by 5′-RACE (SMART kit, Clontech), using primers mm5′RACE1 (5′-CGCAATTCTCGAAGGCCACGTTTT-3′) and mm5′RACE2 (5′-GTCTGTGATCCAGCAGCCTGGAATG-3′). The 3′ end of the muted gene sequence was determined by alignment and sequencing of matched EST IMAGE clones.
Two pairs of primers, hm-5′F (5′-AACTGGTCGGGATGAGTGG-3′)/hm-5′R (5′-GAGTGCCACTGAATATATTGCTAA-3′) and hm-3′F (5′-GCTGAAGTGGATGAAGAGCA-3′)/hm-3′R (5′-TTACACAGGCAGGCATTGTC-3′) were designed from human muted sequences in BACs 303A1 and 511E16 and used in RT–PCR amplifications from human placental poly(A)+ RNA. Full length human muted cDNA was obtained by 5′- and 3′-RACE (SMART kit, Clontech).
For northern blotting, 20 μg total RNA or 2 μg poly(A)+ RNA was electrophoresed on 1.5% agarose gels, transferred to Hybond+ membranes and hybridized with a 400 bp muted cDNA probe generated by RT–PCR amplification of C57BL/6J kidney RNA using the primers mm-F6 and mm-R6.
Electron microscopy
Eye tissues were fixed for 18 h at 4°C in 3% glutaraldehyde and 0.1 M phosphate buffer, pH 7.2. Post-fixation was in 1% osmium tetroxide and 0.1 M phosphate buffer for 1 h. Tissues were dehydrated in serial alcohol and acetone incubations and embedded in Spurr resin. Tissues were sectioned to 80 nm in a Sorvall MT-1B ultramicrotome, and sections were stained with uranyl acetate and lead citrate. Grids were viewed on a Siemans 101 electron microscope at an accelerating voltage of 80 kV.
Bioinformatics
Protein sequences and domain structures were analyzed by the NPS@ web server (Network Protein Sequence Analysis, http://pbil.ibcp.fr/NPSA) and by the UK HGMP Resource Centre (http://www.hgmp.mrfc.ac.uk).
Cell transfection and immunofluorescence
A cDNA of 526 bp, derived by RT–PCR amplification of C57BL/6J kidney RNA and containing the complete coding region, was inserted in-frame into the pCMV-Tag4 expression vector (Stratagene), to produce a FLAG epitope tag at the C-terminus of the muted protein. The fidelity and orientation of the constructed fusion vector was verified by sequencing. Melan-a or COS-7 cells were seeded at 2 × 105 cells/coverslip and transiently transfected with the above muted-Flag fusion plasmid or a pCMV-Tag 4 express (Stratagene) control plasmid (which produces cytoplasmically expressed and FLAG-tagged luciferase) at 2 μg/coverslip using LipofectAMINE (Gibco BRL) in DMEM. After 48 h, cells were fixed with 3.0% formaldehyde in phosphate-buffered saline (PBS) for 20 min and permeabilized in 0.5% Triton X-100 in PBS for 10 min at room temperature. Cells were blocked with 0.5% Triton X-100 and 0.3% normal goat serum in PBS for 30 min, and incubated for 1.5 h with mouse monoclonal anti-FLAG M2 antibody (Stratagene). Coverslips were washed with PBS and incubated for 30 min in blocking solution followed by Alexa488 or 568-conjugated secondary antibody (Molecular Probes) diluted 1:1000 in blocking solution for 1.5 h. Coverslips were washed four times with 0.5% Triton X-100 and 1mg/ml BSA in PBS, rinsed with water and mounted in anti-fade solution. Fluorescent images were acquired and processed using the fluorescent microscope (Nikon, Optiphot) equipped with digital camera (RT color, Diagnostic Instruments Inc.).
Acknowledgments
We thank Y. Jiang, D. Tabaczynski, D. Poslinski and E. Hurley for expert technical assistance. We thank M. Huizing, W. Gahl and R. Spritz for analyses of the muted gene in HPS patient DNAs. This work was supported by grants HL51480, EY12104 and HL31698 (R.T.S.), grant HG02153 (B.A.R.) and the Roswell Park Cancer Institute Cancer Center Support Grant CA 16056.
Footnotes
References
- 1.Swank RT, Novak EK, McGarry MP, Rusiniak ME, Feng L. Mouse models of Hermansky-Pudlak syndrome: a review. Pigment Cell Res. 1998;11:60–80. doi: 10.1111/j.1600-0749.1998.tb00713.x. [DOI] [PubMed] [Google Scholar]
- 2.Swank RT, Novak EK, McGarry MP, Zhang Y, Wei L, Zhang Q, Feng Q. Abnormal vesicular trafficking in mouse models of Hermansky-Pudlak syndrome. Pigment Cell Res. 2000;13:59–67. doi: 10.1034/j.1600-0749.13.s8.12.x. [DOI] [PubMed] [Google Scholar]
- 3.Feng L, Seymour AB, Jiang S, To A, Peden AA, Novak EK, Zhen L, Rusiniak ME, Eicher EM, Robinson MS, et al. The β3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky–Pudlak syndrome and night blindness. Hum Mol Genet. 1999;8:323–330. doi: 10.1093/hmg/8.2.323. [DOI] [PubMed] [Google Scholar]
- 4.Feng L, Rigatti BW, Novak EK, Gorin MB, Swank RT. Genomic structure of the mouse Ap3b1 gene in normal and pearl mice. Genomics. 2000;69:370–379. doi: 10.1006/geno.2000.6350. [DOI] [PubMed] [Google Scholar]
- 5.Kantheti P, Qiao X, Diaz ME, Peden AA, Meyer GE, Carskadon SL, Kapfhamer D, Sufalko D, Robinson MS, Noebels JL, et al. Mutation in AP-3 δ in the mocha mouse links endosomal transport to storage deficiency in platelets, melanosomes and synaptic vesicles. Neuron. 1998;21:111–122. doi: 10.1016/s0896-6273(00)80519-x. [DOI] [PubMed] [Google Scholar]
- 6.Zhen L, Jiang S, Feng L, Bright NA, Peden AA, Seymour AB, Novak EK, Elliott R, Gorin MB, Robinson MS, et al. Abnormal expression and subcellular distribution of subunit proteins of the AP-3 adaptor complex lead to platelet storage pool deficiency in the pearl mouse. Blood. 1999;94:146–155. [PubMed] [Google Scholar]
- 7.Detter JC, Zhang Q, Mules EH, Novak EK, Mishra VS, Li W, Mcmurtrie EB, Tchernev VT, Wallace MR, Seabra MC, et al. Rabgeranylgeranyl transferase α mutation in the gunmetal mouse reduces Rab prenylation and platelet synthesis. Proc Natl Acad Sci USA. 2000;97:4144–4149. doi: 10.1073/pnas.080517697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson SM, Yip R, Swing DA, O Sullivan TN, Zhang Y, Novak EK, Swank RT, Russel LB, Copeland NG, Jenkins NA. A mutation Rab27a causes the vesicle transport defects observed in ashen mice. Proc Natl Acad Sci USA. 2000;97:7933–7938. doi: 10.1073/pnas.140212797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang L, Kuo YM, Gitschier J. The pallid gene encodes a novel, syntaxin 13-interacting protein involved in platelet storage pool deficiency. Nat Genet. 1999;23:329–332. doi: 10.1038/15507. [DOI] [PubMed] [Google Scholar]
- 10.Huizing M, Anikster Y, Gahl WA. Hermansky-Pudlak syndrome and related disorders of organelle formation. Traffic. 2000;1:823–835. doi: 10.1034/j.1600-0854.2000.011103.x. [DOI] [PubMed] [Google Scholar]
- 11.Spritz RA. Multi-organellar disorders of pigmentation: tied up in traffic. Clin Genet. 1999;55:309–317. doi: 10.1034/j.1399-0004.1999.550503.x. [DOI] [PubMed] [Google Scholar]
- 12.Anikster Y, Huizing M, White JG, Shevchenko YO, Fitzpatrick DL, Touchman JW, Compton JG, Bale SJ, Swank RT, Gahl WA, et al. Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat Genet. 2001;28:376–380. doi: 10.1038/ng576. [DOI] [PubMed] [Google Scholar]
- 13.Lyon MF, Meredith R. Muted, a new mutant affecting coat colour and otoliths of the mouse, and its position in linkage group XIV. Genet Res. 1969;14:163–166. doi: 10.1017/s0016672300002007. [DOI] [PubMed] [Google Scholar]
- 14.Swank RT, Reddington M, Howlett O, Novak EK. Platelet storage pool deficiency associated with inherited abnormalities of the inner ear in the mouse pigment mutants muted and mocha. Blood. 1991;78:2036–2044. [PubMed] [Google Scholar]
- 15.Lyon MF. Absence of otoliths in the mouse: an effect of the pallid mutant. J Genet. 1953;51:638–650. [Google Scholar]
- 16.Rolfsen RM, Erway LC. Trace metals and otolith defects in mocha mice. J Hered. 1984;75:158–162. [PubMed] [Google Scholar]
- 17.O’Brien EP, Novak EK, Zhen L, Manly KF, Stephenson D, Swank RT. Molecular markers near two mouse chromosome 13 genes, muted and pearl, which cause platelet storage pool deficiency (SPD) Mamm Genome. 1995;6:19–24. doi: 10.1007/BF00350888. [DOI] [PubMed] [Google Scholar]
- 18.Holtzman E. Lysosomes. Plenum Press; NY: 1989. [Google Scholar]
- 19.Skudlarek MD, Novak EK, Swank RT. Processing of lysosomal enzymes in macrophages and kidney. In: Dingle JT, Dean RT, Sly W, editors. Lysosomes in Biology and Pathology. Vol. 7. Elsevier Science Publishers; NY: 1984. pp. 3–479. [Google Scholar]
- 20.Stinchcombe JC, Griffiths GM. Regulated secretion from hemopoietic cells. J Cell Biol. 1999;147:1–6. doi: 10.1083/jcb.147.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andrews NW. Regulated secretion of conventional lysosomes. Trends Cell Biol. 2000;10:316–321. doi: 10.1016/s0962-8924(00)01794-3. [DOI] [PubMed] [Google Scholar]
- 22.Mcneil PL, Terasaki M. Coping with the inevitable: how cells repair a torn surface membrane. Nat Cell Biol. 2001;3:E124–E129. doi: 10.1038/35074652. [DOI] [PubMed] [Google Scholar]
- 23.Feng GH, Bailin T, Oh J, Spritz RA. Mouse pale ear (ep) is homologous to human Hermansky–Pudlak syndrome and contains a rare ‘AT-AC’ intron. Hum Mol Genet. 1997;6:793–797. doi: 10.1093/hmg/6.5.793. [DOI] [PubMed] [Google Scholar]
- 24.Gardner JM, Wildenberg SC, Keiper NM, Novak EK, Rusiniak ME, Swank RT, Puri N, Finger JN, Hagiwara N, Lehman AL, et al. The mouse pale ear (ep) mutation is the homologue of human Hermansky-Pudlak syndrome (HPS) Proc Natl Acad Sci USA. 1997;94:9238–9243. doi: 10.1073/pnas.94.17.9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki T, Li W, Zhang Q, Novak EK, Sviderskaya EV, Wilson A, Bennett DC, Roe BA, Swank RT, Spritz RA. The gene in cocoa mice, carrying a defect of organelle biogenesis, is a homologue of the human Hermansky–Pudlak syndrome-3 gene. Genomics. 2001;78:30–37. doi: 10.1006/geno.2001.6644. [DOI] [PubMed] [Google Scholar]
- 26.Harada Y, Kasuga S, Mori N. The process of otoconia formation in guinea pig utricular supporting cells. Acta Otolaryngol. 1998;118:74–79. doi: 10.1080/00016489850155161. [DOI] [PubMed] [Google Scholar]
- 27.McGarry MP, Reddington M, Novak EK, Swank RT. Survival and lung pathology of mouse models of Hermansky-Pudlak Syndrome and Chediak-Higashi Syndrome. Proc Soc Exp Biol Med. 1999;220:162–168. doi: 10.1046/j.1525-1373.1999.d01-24.x. [DOI] [PubMed] [Google Scholar]
- 28.Whitelaw E, Martin DI. Retrotransposons as epigenetic mediators of phenotypic variation in mammals. Nat Genet. 2001;27:361–365. doi: 10.1038/86850. [DOI] [PubMed] [Google Scholar]
- 29.Thien H, Ruther U. The mouse mutation Pdn (Polydactyly Nagoya) is caused by the integration of a retrotransposon into the Gli3 gene. Mamm Genome. 1999;10:205–209. doi: 10.1007/s003359900973. [DOI] [PubMed] [Google Scholar]
- 30.Hofmann M, Harris M, Juriloff D, Boehm T. Spontaneous mutations in SELH/Bc mice due to insertions of early transposons: molecular characterization of null alleles at the nude and albino loci. Genomics. 1998;52:107–109. doi: 10.1006/geno.1998.5409. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi S, Hirano T, Kakinuma M, Uede T. Transcriptional repression and differential splicing of FAS mRNA by early transposon (ETn) insertion in autoimmune LPR mice. Biochem Biophys Res Commun. 1993;191:617–624. doi: 10.1006/bbrc.1993.1262. [DOI] [PubMed] [Google Scholar]
- 32.Bodenteich A, Chissoe S, Wang YF, Roe BA. Shotgun cloning as the strategy of choice to generate templates for high throughput dideoxynucleotide sequencing. In: Adams MD, Fields C, Venter C, editors. Automated DNA Sequencing and Analysis Techniques. Academic Press; London, UK: 1994. pp. 42–50. [Google Scholar]
- 33.Chissoe SL, Wang YF, Clifton SW, Ma N, Sun JS, Lobsinger SM, Kenton SM, White JD, Roe BA. Strategies for rapid and accurate DNA sequencing. Methods: A Companion to Methods Enzymol. 1991;3:55–65. [Google Scholar]