

Abstract
Fluorescent-tagging and digital imaging are widely used to determine the subcellular location of proteins. An extensive publicly available collection of images for most proteins expressed in the yeast S. cerevisae has provided both an important source of information on protein location but also a testbed for methods designed to automate the assignment of locations to unknown proteins. The first system for automated classification of subcellular patterns in these yeast images utilized a computationally expensive method for segmentation of images into individual cells and achieved an overall accuracy of 81%. The goal of the present study was to improve on both the computational efficiency and accuracy of this task. Numerical features derived from applying Gabor filters to small image patches were implemented so that patterns could be classified without segmentation into single cells. When tested on 20 classes of images visually classified as showing a single subcellular pattern, an overall accuracy of 87.8% was achieved, with 2330 images out of 2655 images in the UCSF dataset being correctly classified. On the 4 largest classes of these images, 95.3% accuracy was achieved. The improvement over the previous approach is not only in classification accuracy but also in computational efficiency, with the new approach taking about 1 h on a desktop computer to complete all steps required to perform a 6-fold cross validation on all images.
INTRODUCTION
Green Fluorescent Protein (GFP) and its variants are widely used in biological imaging because they can be linked with virtually any protein to visualize location in vivo. GFP-tagging is used both to confirm conjectured localizations and to determine them for previously uncharacterized proteins (although the localization can potentially be altered by the tagging). Traditionally, the assignment of a location is done by visual inspection. However, it is often difficult to insulate against the influence of prior experience or hypotheses on those assignments in order to rely exclusively on the tagged protein images themselves. In addition, visual inspection is not well suited to efficiently handling proteome-scale tasks such as classifying thousands of different GFP-tagged protein images.
Automated classification of subcellular patterns in such images is a viable alternative, and a number of systems for this task have been described (1–3). These typically start by calculating numerical features from the microscope image that are designed to capture essential characteristics of the pattern without being sensitive to the position, orientation and brightness of individual cells. Machine learning algorithms are then trained to predict subcellular location labels from the numerical features. The classification problem generally consists of four steps: 1) image preprocessing, 2) feature extraction, 3) feature selection, and 4) classifier training and evaluation. Among these steps, the first two steps are commonly most important because the steps decide essential qualities of features that influence the entire process. In order to provide information on subcellular location for the many proteins about which little is known, efforts to create proteome-scale image collections have been described (4–7). The most comprehensive coverage to date has been of the yeast proteome, for which Huh et al. (4) collected images of over 4,000 GFP-tagged proteins encoded by cDNAs (a more recent collection for over 1,000 proteins was created by chromosomal tagging with GFP by Hayashi et al. (8)). The Human Protein Atlas (9) has collected images for over 6,000 proteins to date using mono-specific antisera.
The availability of such collections has permitted automated classification systems to be applied on a scale not previously possible. In the first such application, Chen et al. (10) developed an automated system capable of recognizing the patterns in the UCSF yeast image collection. The system used a graphical model method (11) to segment each image into single cell regions (using parallel images of differential image contrast images (DIC) and a DNA-binding probe). For each cell, a feature set containing Zernike moment features, morphological features, wavelet features, DNA overlap features, edge features, and Haralick texture features was calculated. The system showed 81% agreement with visual assignments for proteins having a single location. Results using this approach depend on the accuracy of cell segmentation, and additional methods for segmentation of yeast cells have since been presented (12,13). Systems for performing other kinds of analyses of yeast images have also been described, including systems for analyzing cell morphology (14), counting peroxisomes (15), quantifying protein and RNA expression (16), and carrying out image-based screens (17,18). Some approaches to classifying subcellular patterns do not require segmentation into single cell regions (3,19,20), but while these approaches offer reduced computation time they often sacrifice some accuracy of classification.
In this paper, we present a framework for subcellular pattern classification in yeast that shows both improved accuracy and computational efficiency compared to the previously reported results for this image collection. The approach does not require segmentation of images into single cell regions, eliminating the need for parallel DIC and DNA images. This makes it applicable to datasets for which images of only a single (protein) channel are available.
METHODS
UCSF yeast GFP fusion localization database
The UCSF yeast GFP fusion localization database contains 4156 sets of three 535×512 grayscale images (for DIC, DAPI, and GFP) where yeast cells in each set express different GFP-tagged proteins (4). The DAPI channel reflects the DNA distribution, and the DIC channel shows the boundaries of the cells. The original web site through which the images were made available, http://yeastgfp.ucsf.edu, is not currently online; therefore the images are currently being made available at http://murphylab.web.cmu.edu/data.
One or more labels have been assigned to each GFP image by visual examination (and in some cases using additional information) by two evaluators; a total of 22 labels were used (4). As in the previous work (10), we restricted our automated analysis to 2655 images by selecting images assigned to a single location but eliminating those labeled as “ambiguous” and “composite punctate” which do not correspond to any particular subcellular location. In the resulting set, each image belongs to one of 20 classes such as nucleus, cytoplasm and mitochondrion. The number of images in each class is uneven, ranging from 6 to 823. The names and sizes of the 20 classes are shown in Table 1.
Table 1.
Images with a unique subcellular location in the UCSF dataset
| Class | Number of images | Class | Number of images |
|---|---|---|---|
| Cytoplasm | 823 | Endosome | 34 |
| Nucleus | 496 | Late_Golgi | 33 |
| Mitochondrion | 485 | Actin | 27 |
| ER | 267 | Peroxisome | 21 |
| Vacuole | 121 | Lipid_particle | 19 |
| Nucleolus | 69 | Golgi | 15 |
| Cell_periphery | 57 | Bud_neck | 15 |
| Vacuolar_membrane | 54 | Early_Golgi | 11 |
| Nuclear_periphery | 53 | Microtubule | 10 |
| Spindle_pole | 39 | ER_to_Golgi | 6 |
Intensity adjustment
Due to the different levels of tagged protein expression and varying positions of cells relative to the focal plane, the intensities of yeast cells with the same class label can vary significantly. Thus, we applied intensity adjustment to reduce the intra-class variance. To do this, we first took the 0.05th percentile of intensity distribution as the lowest intensity and the 99.95th percentile as the highest intensity. Then, we linearly adjusted each intensity between 0.05th percentile and 99.95th percentile to the percentile of the entire intensity range. We also mapped every intensity lower than 0.05th percentile to the minimum value and every intensity higher than 99.95th percentile to the maximum value of the entire intensity range. The rationale for using the 0.05th percentile and 99.95th percentiles instead of maximum and minimum was to avoid outlier pixels (e.g., resulting from dust particles or fluorescent debris).
After intensity adjustment, we smoothed each image with a 3×3 rectangular average convolution filter (3×3 matrix with all elements 1/9) without zero padding. We then subtracted the most common pixel value as background and applied a global threshold for each image using the Ridler-Calvard method as described previously (1).
Rotation-invariant feature extraction
Yeast cells float freely and thus it is possible for them to rotate in the media. Different cells in a given field are therefore expected to show different orientations. Features to describe the patterns in such images should therefore be invariant to local rotation. A simple approach to doing this is to calculate texture features in different orientations and average them. This approach was used in the previous automated analysis of the yeast images. Several alternative methods to achieve rotation invariant features have been described.
For example, Varma and Zisserman (21) presented the maximum response (MR) filter sets. The main idea of this filtering method is to apply multiple orientation filters but use only the maximum filter response across all orientations. Though this method is computationally cheap and extracts a small number of features, it achieved high classification accuracy on the Columbia-Utrecht reflectance and texture database (21,22).
Another method to gain rotation-invariant features is to use rotationally invariant filters such as Gaussian filters or Laplacian of Gaussian filters (23). In recent work, Lazebnik et al. (24), also introduced rotationally invariant descriptors, i.e., SPIN and RIFT. Although these descriptors extract rotation-invariant features, they sacrifice the spatial information that may be important to distinguish different classes (25).
Lowe (26) suggested a method to find the dominant gradient orientation of each patch or region. SIFT descriptors with this method showed higher classification accuracies than rotation-invariant descriptors (25); however, the method to find the dominant gradient is computationally expensive (24).
In order to extract local patterns, we used Gabor filters that can catch local frequency and orientation information in the given image. We defined a non-background patch as a 7×7 pixel region that does not include any background pixels and applied Gabor filters with 20 scales and 16 orientations. We obtained rotation invariant features by applying the method discussed in detail in the next paragraph. Then, we calculated the mean and the variance of the energy distribution corresponding to each filter. These means and variances are used to construct 2×20×16=640 Gabor features for each image. The patch size, the number of scales and the number of orientations were determined empirically to obtain the highest cross-validation accuracy, and the other variables of the Gabor filter bank were set to reduce redundant information (27).
We added rotation invariance to the Gabor features as follows. First, we find the first major orientation and the second major orientation corresponding respectively to the maximum value and the second maximum value among all 20×16 filtered values from each patch. After finding these two major orientations, each patch is rotated to have its first major orientation align toward the same direction as all the other patches (Figure 2a). Then, each patch is flipped along the first major axis if needed, to align its second major orientation to form an acute angle on the counterclockwise side from its first major orientation (Figure 2b).
Fig. 2.
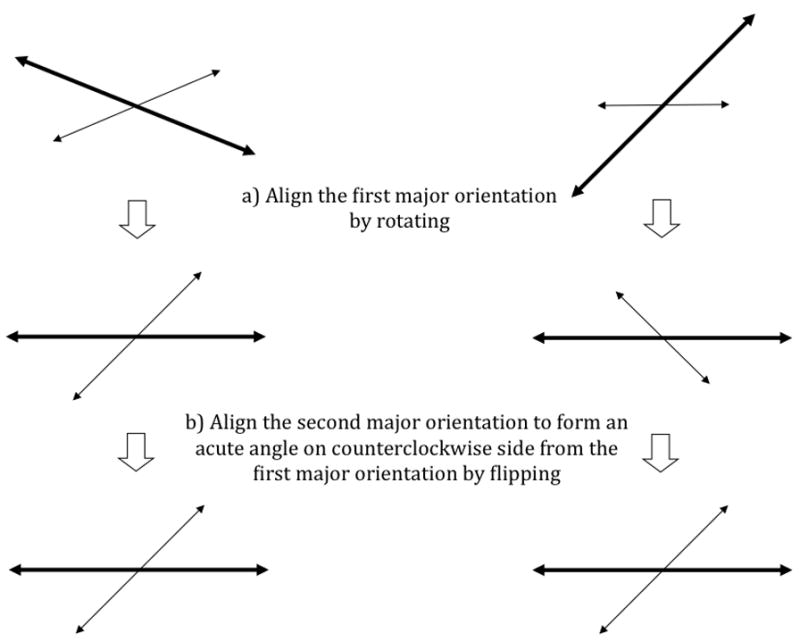
Illustration of the orientation adjustment scheme. The long thick arrows represent the major orientation and the short thin arrows represent the second major orientation.
Directly convolving all possible patches with Gabor filters is often wasteful when a significant number of pixels belong to background. Thus, we first partition the image into rectangular regions, and test whether each region contains any non-background patches. Only when at least one non-background patch is found, the rectangular region is convolved with Gabor filters. We reduce the computation time significantly by using 30×30 rectangular regions.
To evaluate the performance of our rotation invariance method, we implemented a maximum response method and a dominant gradient orientation method as well. The maximum response method adopts the main idea suggested by Varma and Zisserman (21). After convolving all 7×7 patches with 20×16 Gabor filters, we adopt only the maximum response for each scale; then calculate the mean and the variance of the energy distribution of the maximum response corresponding to each scale. As a result, we obtain 40 rotation invariant features for each image.
For the gradient orientation method, we apply the same 20×16 Gabor filters, extract Gabor features, and apply an adaptation of Lowe’s method used for SIFT features (26) to obtain the major orientations with which orientation adjustment is performed. We precompute the gradient magnitude m(x,y) and orientation θ(x,y) for each pixel as
where I(x,y) is the intensity of a pixel. Then for each patch, we form an orientation histogram that has 32 bins. Each sample added to the histogram is weighted by its gradient magnitude, and then a Gaussian circular weight is applied. We find the orientation whose bin has the maximum weight and the second maximum weight, and set them as the major orientation and the second major orientation. Then, these two orientations are applied to adjust the Gabor features’ orientation in the same manner outlined above to obtain rotation invariance.
Feature Selection
To create a compact set of features for use in classification, we used a state-of-the-art extension to Linear Discriminant Analysis, Spectral Regression Discriminant Analysis (SRDA) (28). SRDA is known to save both time and memory compared with other Linear Discriminant Analysis extensions. We used the SRDA source code provided by Cai et al. (28) with regularization and with the default value for the regularization parameter (0.1).
When the number of classes is small compared to the number of images, SRDA tends to overreduce features so that classification does not work well. Thus, when considering only 4 classes, we did not apply feature selection.
Support vector machine classification
We used the LIBSVM implementation of a support vector machine (SVM) classifier (http://www.csie.ntu.edu.tw/~cjlin/libsvm) with a radial basis function (RBF) kernel. Since SVMs are binary classifiers, we adopted the one-against-one approach (29) so that the most frequently predicted class from all possible one-versus-one classifiers is selected to be the predicted class of each image. We evenly split the data into six folds (since the smallest class contains only six samples). Using each fold in turn as a test set, we used four of the remaining folds for training and the last fold as an internal test set for choosing optimal SVM parameters (the slack penalty and the RBF kernel parameter) for the training folds by a grid search. The test accuracy was calculated by aggregating the predictions on all six test sets using the independently chosen parameters.
We measure the confidence of each prediction by calculating the sum of decision values of the prediction. For each prediction, LIBSVM can generate a decision value that is proportional to the distance from decision boundary, and each prediction gets n−1 decision values in an n-class classification. For each image, the n−1 decision values are summed up, representing how confident the prediction is. Varying values of a threshold on this confidence were used to determine the dependence of classification accuracy (precision) on confidence.
Implementation and Availability
All components of our approach were implemented in Matlab except the LIBSVM package which was invoked through a Matlab interface. The source code and data used in this study will be made available upon publication at http://murphylab.web.cmu.edu/software.
RESULTS AND DISCUSSION
Classification Performance
As described in the Methods, our approach consists of image preprocessing (intensity adjustment, background correction), rotation-invariant feature extraction, feature selection, and classification using a support vector machine with a radial basis function kernel. We applied our approach to the 2655 images from the UCSF collection that show proteins assigned to a single location by visual examination. Our system classified 2330 images correctly, an overall accuracy of 87.8%. This is a 6.8% improvement from our previous work that achieved 81.0% accuracy (10). The confusion matrix obtained from 6-fold cross validation is shown in Table 2. Table 3 shows the comparison of the accuracies for each class with the previous work. Significant improvement is observed for some of the classes that were poorly recognized previously, and endosome, late Golgi and actin are now recognized with greater than 50% accuracy. As a result of improvements in the lower frequency classes, the accuracy when weighting by class (rather than by image) is significantly higher than it was previously.
Table 2.
The confusion matrix for the full system described in the text. The order of classes is from the largest to the smallest.
| Cytoplasm | Nucleus | Mitochondirion | ER | Vacuole | Nucleolus | Cell periphery |
Vacuolar membrane |
Nuclear periphery |
Spindle pole |
Endosome | Late Golgi |
Actin | Peroxisome | Lipid particle |
Golgi | Bud neck |
Early Golgi |
Microtubule | ER to Golgi |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cytoplasm | 97.0 | 0.0 | 0.9 | 1.1 | 0.2 | 0.4 | 0.5 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Nucleus | 0.4 | 93.5 | 1.6 | 0.0 | 1.8 | 1.6 | 0.0 | 0.0 | 0.4 | 0.0 | 0.2 | 0.0 | 0.2 | 0.0 | 0.0 | 0.0 | 0.2 | 0.0 | 0.0 | 0.0 |
| Mitochondrion | 1.6 | 0.8 | 94.2 | 1.0 | 0.0 | 0.4 | 0.0 | 0.0 | 0.0 | 0.4 | 0.6 | 0.4 | 0.0 | 0.0 | 0.2 | 0.0 | 0.2 | 0.0 | 0.0 | 0.0 |
| ER | 8.2 | 0.0 | 0.4 | 87.3 | 1.1 | 0.0 | 1.5 | 0.0 | 1.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.4 | 0.0 | 0.0 | 0.0 | 0.0 |
| Vacuole | 8.3 | 4.1 | 0.8 | 5.0 | 75.2 | 0.0 | 1.7 | 5.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Nucleolus | 0.0 | 13.0 | 1.4 | 0.0 | 1.4 | 82.6 | 0.0 | 0.0 | 0.0 | 0.0 | 1.4 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Cell periphery |
1.8 | 0.0 | 3.5 | 10.5 | 0.0 | 3.5 | 80.7 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Vacuolar membrane |
0.0 | 0.0 | 5.6 | 3.7 | 11.1 | 0.0 | 0.0 | 66.7 | 7.4 | 1.9 | 3.7 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Nuclear periphery |
1.9 | 0.0 | 0.0 | 3.8 | 3.8 | 1.9 | 0.0 | 3.8 | 81.1 | 0.0 | 3.8 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Spindle pole |
0.0 | 0.0 | 7.7 | 0.0 | 0.0 | 12.8 | 0.0 | 0.0 | 0.0 | 64.1 | 12.8 | 0.0 | 0.0 | 2.6 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Endosome | 0.0 | 2.9 | 11.8 | 5.9 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 8.8 | 61.8 | 2.9 | 0.0 | 2.9 | 0.0 | 2.9 | 0.0 | 0.0 | 0.0 | 0.0 |
| Late Golgi |
3.0 | 3.0 | 15.2 | 3.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 9.1 | 57.6 | 0.0 | 0.0 | 3.0 | 3.0 | 0.0 | 3.0 | 0.0 | 0.0 |
| Actin | 0.0 | 0.0 | 7.4 | 22.2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 3.7 | 55.6 | 7.4 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 3.7 |
| Peroxisome | 0.0 | 0.0 | 19.0 | 4.8 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 9.5 | 14.3 | 0.0 | 4.8 | 33.3 | 14.3 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Lipid particle |
0.0 | 0.0 | 21.1 | 10.5 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 10.5 | 15.8 | 5.3 | 5.3 | 31.6 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Golgi | 0.0 | 0.0 | 0.0 | 13.3 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 6.7 | 20.0 | 0.0 | 0.0 | 13.3 | 26.7 | 0.0 | 20.0 | 0.0 | 0.0 |
| Bud neck |
6.7 | 0.0 | 33.3 | 13.3 | 0.0 | 6.7 | 0.0 | 0.0 | 0.0 | 0.0 | 6.7 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 33.3 | 0.0 | 0.0 | 0.0 |
| Early Golgi |
0.0 | 0.0 | 9.1 | 18.2 | 0.0 | 0.0 | 0.0 | 9.1 | 0.0 | 0.0 | 0.0 | 27.3 | 0.0 | 0.0 | 0.0 | 36.4 | 0.0 | 0.0 | 0.0 | 0.0 |
| Microtubule | 0.0 | 20.0 | 60.0 | 0.0 | 0.0 | 20.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| ER to Golgi |
0.0 | 0.0 | 0.0 | 50.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 50.0 |
Table 3.
Comparison of accuracy with previous results.
| Accuracy Chen et al. 2007 (%) | Accuracy this work (%) | |
|---|---|---|
| Cytoplasm | 94.4 | 97 |
| Nucleus | 88.6 | 93.5 |
| Mitochondrion | 90.9 | 94.2 |
| ER | 70.2 | 87.3 |
| Vacuole | 73.6 | 75.2 |
| Nucleolus | 73.5 | 82.6 |
| Cell periphery | 75.4 | 80.7 |
| Vacuolar membrane | 51.9 | 66.7 |
| Nuclear periphery | 77.4 | 81.1 |
| Spindle pole | 64.1 | 64.1 |
| Endosome | 47.1 | 61.8 |
| Late Golgi | 12.1 | 57.6 |
| Actin | 22.2 | 55.6 |
| Peroxisome | 14.3 | 33.3 |
| Lipid particle | 0 | 31.6 |
| Golgi | 6.7 | 26.7 |
| Bud neck | 0 | 33.3 |
| Early Golgi | 0 | 0 |
| Microtubule | 0 | 0 |
| ER to Golgi | 0 | 50 |
| Average by class for 20 classes | 43.1 | 58.6 |
| Average by image for 20 classes | 81.0 | 87.8 |
| Average by image for 4 major classes | 92.7 | 95.3 |
We also report the accuracies of classifying just the four major classes in Table 3. For the four-major-class task, both our approach and the previous approach were configured slightly differently compared with the 20-class task. In our approach, we did not use SRDA because it induces over-reduction for such a small number of classes. Our approach achieves a higher accuracy, approximately halving the error rate.
Given that our approach does not involve segmentation, it was of interest to determine whether classification accuracy showed dependence on cell density. The number of cells per image (as determined using the graphical model segmentation approach used previously) was as high as 51, with a mean of 18.7 and a standard deviation of 7.8. Images of all densities were correctly classified, and the few incorrectly classified images showed a similar distribution of cell density as the whole collection.
Computational Efficiency
Table 4 reports the running time of each component on a computer with a 2.66 GHz Intel CPU and 2 GB of RAM. As can be seen in the table, our approach takes about one hour to perform the entire process. This is a great improvement over the previous system, which takes several days. The improved speed is achieved primarily because segmentation of each image to find cell boundaries is avoided and feature calculation is more rapid. In addition, SRDA significantly reduces classification time through finding only 19 combinations of features among 640 features in the 20-class task. For example, after SRDA feature selection, SVM classification time was reducesd to approximately 1/25 of the time required for all features. This computational efficiency allows investing more effort to find proper parameters in the SVM classification so that the classification accuracy can be improved.
Table 4.
Running time of each component.
| Component | Running time |
|---|---|
| Image preprocessing | 646 sec |
| Rotation-invariant feature extraction | 49 min |
| Feature selection using SRDA | 15 sec |
| Classification using an SVM with the RBF kernel | 230 sec |
Contributions of System Components to Overall Accuracy
Table 5 shows the performance drops in terms of overall accuracy in our method when each of three processes that characterize our approach is disabled. Note that in this analysis only one process is disabled, leaving the other two functional. When the intensity adjustment process is disabled, the overall accuracy drops from 87.8% to 80.1%, showing a performance drop of 7.7%. In the cases that disable orientation adjustment and linear discriminant analysis, 5.3% and 2.8% performance drops are observed respectively.
Table 5.
Contributions of system components to accuracy. Performance drop is calculated as the difference between the original accuracy (87.8%) and the accuracy when each part of the approach is not applied.
| Process disabled | Overall accuracy (%) | Performance drop (% points) |
|---|---|---|
| Intensity adjustment | 80.1 | 7.7 |
| Orientation adjustment | 82.5 | 5.3 |
| Linear discriminant analysis | 85.0 | 2.8 |
Rotation Invariance Comparison
The comparison of three different methods to achieve rotation-invariance is show in Table 6. As can be seen, our method shows the best performance in terms of overall accuracy. The maximum response method may negatively impact the performance because important spatial information is lost in the process of achieving rotation invariance. In addition, the number of features may be too small to distinguish different classes effectively. The reason that the dominant gradient orientation method performs poorly may be due to the incompatibility of this method with Gabor features. The dominant gradient orientation method is originally devised for the SIFT descriptor which produces the gradient orientation histogram. The resulting major orientations from the histogram are not likely to correlate perfectly with the major orientations from the Gabor filter responses. Also, using the gradient orientation histogram to determine the major orientations does not reflect relative spatial information found among the patches from the same yeast cell. Therefore, rotations based on the dominant gradient orientation method may jumble up the Gabor features patch by patch.
Table 6.
Comparison of rotation invariance methods
| Rotation invariance method | Accuracy (%) |
|---|---|
| Local orientation | 87.8 |
| Maximum response | 82.1 |
| Dominant gradient orientation | 82.4 |
| No orientation adjustment | 82.5 |
Analysis of Prediction Confidence
It is possible to return the predictions sorted in terms of confidence. Figure 3 shows the behavior of prediction accuracy as predictions are made with varying threshold on that confidence. Note that an accuracy of approximately 92% can be achieved when only the most confident 50% of predictions are considered.
Fig. 3.
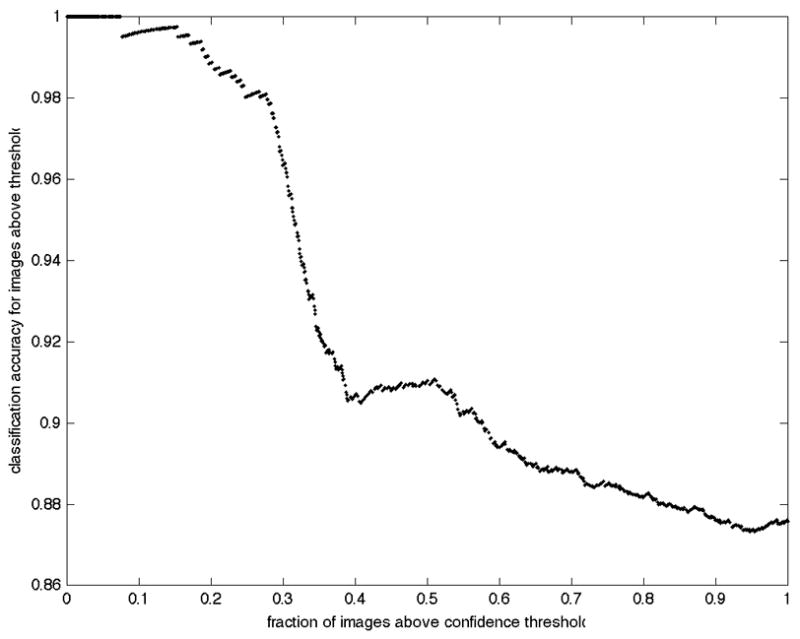
Improved accuracy for high confidence predictions. The overall accuracy of only those predictions with confidence above a given threshold is displayed as a function of the fraction of images whose confidence was greater than that threshold.
Conclusion
In this paper, we have introduced a framework for yeast image classification that outperforms previously reported results. We anticipate that this framework can also be successfully applied to other fluorescence microscope images depicting subcellular patterns. In addition, utilizing more recent and efficient descriptors such as SIFT might be expected to improve classification accuracy. Future work will be required to test these expectations.
The automated classification framework described here is computationally efficient and reduces potential human biases in making assignments. Therefore, we anticipate that its natural applications include proteome-scale high-throughput analysis of subcellular location in which computational efficiency may be an important consideration.
Fig. 1.
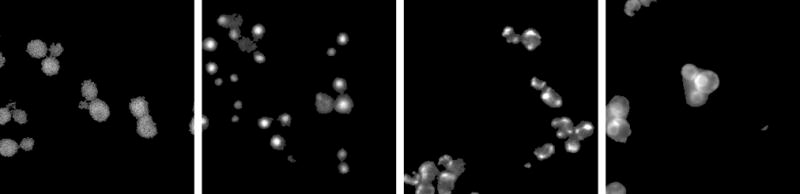
Example GFP images from the four major classes in the UCSF yeast GFP fusion database. Each panel shows a 256 × 256 pixel region in the center of the original image for a randomly chosen protein from a given class. From left to right, the proteins shown (and their subcellular locations) are YNL267W (Cytoplasm), YPL011C (Nucleus), YDL120W (Mitochondrion), and YOR254C (Endoplasmic Reticulum). The images were scaled and background-corrected as described in the methods.
Acknowledgments
This work was supported in part by National Science Foundation grants EEEC-0540865 (to Takeo Kanade) and EF-0331657 (to R.F.M.). Facilities and infrastructure support were provided by NIH National Technology Center for Networks and Pathways grant U54 RR022241 (to Alan Waggoner).
LITERATURE CITED
- 1.Boland MV, Murphy RF. A Neural Network Classifier Capable of Recognizing the Patterns of all Major Subcellular Structures in Fluorescence Microscope Images of HeLa Cells. Bioinformatics. 2001;17(12):1213–1223. doi: 10.1093/bioinformatics/17.12.1213. [DOI] [PubMed] [Google Scholar]
- 2.Conrad C, Erfle H, Warnat P, Daigle N, Lorch T, Ellenberg J, Pepperkok R, Eils R. Automatic Identification of Subcellular Phenotypes on Human Cell Arrays. Genome Research. 2004;14:1130–1136. doi: 10.1101/gr.2383804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamilton NA, Pantelic RS, Hanson K, Teasdale RD. Fast automated cell phenotype image classification. BMC Bioinformatics. 2007;8:110. doi: 10.1186/1471-2105-8-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425(6959):686–91. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 5.Starkuviene V, Liebel U, Simpson JC, Erfle H, Poustka A, Wiemann S, Pepperkok R. High-content screening microscopy identifies novel proteins with a putative role in secretory membrane traffic. Genome Res. 2004;14(10A):1948–56. doi: 10.1101/gr.2658304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aturaliya RN, Fink JL, Davis MJ, Teasdale MS, Hanson KA, Miranda KC, Forrest AR, Grimmond SM, Suzuki H, Kanamori M, Kai C, Kawai J, Carninci P, Hayashizaki Y, Teasdale RD. Subcellular localization of mammalian type II membrane proteins. Traffic. 2006;7(5):613–25. doi: 10.1111/j.1600-0854.2006.00407.x. [DOI] [PubMed] [Google Scholar]
- 7.Garcia Osuna E, Hua J, Bateman NW, Zhao T, Berget PB, Murphy RF. Large-scale automated analysis of location patterns in randomly tagged 3T3 cells. Ann Biomed Eng. 2007;35(6):1081–7. doi: 10.1007/s10439-007-9254-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayashi A, Da-Qiao D, Tsutsumi C, Chikashige Y, Masuda H, Haraguchi T, Hiraoka Y. Localization of gene products using a chromosomally tagged GFP-fusion library in the fission yeast Schizosaccharomyces pombe. Genes Cells. 2009;14(2):217–25. doi: 10.1111/j.1365-2443.2008.01264.x. [DOI] [PubMed] [Google Scholar]
- 9.Uhlen M, Bjorling E, Agaton C, Szigyarto CA, Amini B, Andersen E, Andersson AC, Angelidou P, Asplund A, Asplund C, Berglund L, Bergstrom K, Brumer H, Cerjan D, Ekstrom M, Elobeid A, Eriksson C, Fagerberg L, Falk R, Fall J, Forsberg M, Bjorklund MG, Gumbel K, Halimi A, Hallin I, Hamsten C, Hansson M, Hedhammar M, Hercules G, Kampf C, Larsson K, Lindskog M, Lodewyckx W, Lund J, Lundeberg J, Magnusson K, Malm E, Nilsson P, Odling J, Oksvold P, Olsson I, Oster E, Ottosson J, Paavilainen L, Persson A, Rimini R, Rockberg J, Runeson M, Sivertsson A, Skollermo A, Steen J, Stenvall M, Sterky F, Stromberg S, Sundberg M, Tegel H, Tourle S, Wahlund E, Walden A, Wan J, Wernerus H, Westberg J, Wester K, Wrethagen U, Xu LL, Hober S, Ponten F. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics. 2005;4(12):1920–32. doi: 10.1074/mcp.M500279-MCP200. [DOI] [PubMed] [Google Scholar]
- 10.Chen SC, Zhao T, Gordon GJ, Murphy RF. Automated image analysis of protein localization in budding yeast. Bioinformatics. 2007;23(13):i66–71. doi: 10.1093/bioinformatics/btm206. [DOI] [PubMed] [Google Scholar]
- 11.Chen S-C, Zhao T, Gordon GJ, Murphy RF. A Novel Graphical Model Approach to Segmenting Cell Images. Proc CIBCB. 2006:1–8. [Google Scholar]
- 12.de Carvalho MAG, Lotufo RdA, Couprie M. Morphological segmentation of yeast by image analysis. Image and Vision Computing. 2007;25(1):34–39. [Google Scholar]
- 13.Kvarnstrom M, Logg K, Diez A, Bodvard K, Kall M. Image analysis algorithms for cell contour recognition in budding yeast. Opt Express. 2008;16(17):12943–57. doi: 10.1364/oe.16.012943. [DOI] [PubMed] [Google Scholar]
- 14.Saito TL, Sese J, Nakatani Y, Sano F, Yukawa M, Ohya Y, Morishita S. Data mining tools for the Saccharomyces cerevisiae morphological database. Nucleic Acids Res. 2005;33:W753–7. doi: 10.1093/nar/gki451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niemisto A, Selinummi J, Saleem R, Shmulevich I, Aitchison J, Yli-Harja O. Extraction of the Number of Peroxisomes in Yeast Cells by Automated Image Analysis. Proc EMBS. 2006:2353–2356. doi: 10.1109/IEMBS.2006.259890. [DOI] [PubMed] [Google Scholar]
- 16.Gordon A, Colman-Lerner A, Chin TE, Benjamin KR, Yu RC, Brent R. Single-cell quantification of molecules and rates using open-source microscope-based cytometry. Nat Methods. 2007;4(2):175–81. doi: 10.1038/nmeth1008. [DOI] [PubMed] [Google Scholar]
- 17.Narayanaswamy R, Moradi EK, Niu W, Hart GT, Davis M, McGary KL, Ellington AD, Marcotte EM. Systematic definition of protein constituents along the major polarization axis reveals an adaptive reuse of the polarization machinery in pheromone-treated budding yeast. J Proteome Res. 2009;8(1):6–19. doi: 10.1021/pr800524g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolinski H, Petrovic U, Mattiazzi M, Petschnigg J, Heise B, Natter K, Kohlwein SD. Imaging-based live cell yeast screen identifies novel factors involved in peroxisome assembly. J Proteome Res. 2009;8(1):20–7. doi: 10.1021/pr800782n. [DOI] [PubMed] [Google Scholar]
- 19.Huang K, Murphy RF. Automated classification of subcellular patterns in multicell images without segmentation into single cells. Proc ISBI. 2004:1139–1142. [Google Scholar]
- 20.Maree R, Geurts P, Wehenkel L. Random subwindows and extremely randomized trees for image classification in cell biology. BMC Cell Biol. 2007;8 (Suppl 1):S2. doi: 10.1186/1471-2121-8-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varma M, Zisserman A. Classifying Images of Materials: Achieving Viewpoint and Illumination Independence. Proc ECCV. 2002;3:255–271. [Google Scholar]
- 22.Dana KJ, Ginneken Bv, Nayar SK, Koenderink JJ. Reflectance and texture of real-world surfaces. ACM Trans Graph. 1999;18(1):1–34. [Google Scholar]
- 23.Schmid C. Constructing models for content-based image retrieval. Proc CVPR. 2001;2:II-39–II-45. [Google Scholar]
- 24.Lazebnik S, Schmid C, Ponce J. A sparse texture representation using local affine regions. IEEE Trans Pattern Analysis and Machine Intelligence. 2005;27(8):1265–1278. doi: 10.1109/TPAMI.2005.151. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Marsza ek M, Lazebnik S, Schmid C. Local Features and Kernels for Classification of Texture and Object Categories: A Comprehensive Study. International Journal of Computer Vision. 2007;73(2):213–238. [Google Scholar]
- 26.Lowe DG. Distinctive Image Features from Scale-Invariant Keypoints. International Journal of Computer Vision. 2004;60:91–110. [Google Scholar]
- 27.Manjunath BS, Ma WY. Texture features for browsing and retrieval of image data. IEEE Trans Pattern Analysis and Machine Intelligence. 1996;8(18):837–842. [Google Scholar]
- 28.Cai D, He X, Han J. SRDA: An Efficient Algorithm for Large-Scale Discriminant Analysis. IEEE Trans Knowledge and Data Engineering. 2008;20(1):1–12. [Google Scholar]
- 29.Hsu C-W, Lin C-J. A comparison of methods for multi-class support vector machines. IEEE Trans Neural Networks. 2002;13:415–425. doi: 10.1109/72.991427. [DOI] [PubMed] [Google Scholar]