

Arterial smooth muscle cells (SMCs) normally reside in the arterial wall in a differentiated contractile state. Following injury, such as that caused by angioplasty or stenting, or in more chronic conditions, such as atherosclerosis, some SMCs in the arterial wall convert to a less differentiated state, often termed de-differentiated, synthetic, or proliferative state.1 These de-differentiated or synthetic SMCs gain the ability to respond to growth factors, and to migrate and proliferate. At the same time, they lose several cytoskeletal markers of differentiation and most likely their ability to respond to contractile stimuli and to contribute to vessel contraction. It has long been known that there is an inverse correlation between SMC proliferation and their expression of cytoskeletal markers of differentiation.2 Understanding of the factors regulating SMC differentiation is important, because maintaining SMC differentiation in conditions normally associated with increased SMC de-differentiation would most likely prevent restenosis following angioplasty and development of irreversible atherosclerotic lesions characterized by SMC proliferation and migration.
What are the extracellular cues that determine the differentiation state of SMCs? It has been known for decades that the extracellular matrix (ECM) surrounding SMCs is one important factor, and perhaps the principal factor, for maintaining SMCs in a differentiated state. Thus, SMCs isolated from the arterial wall and plated onto laminin or type IV collagen retain their differentiated phenotype to a greater extent than do SMCs plated onto fibronectin or monomeric type I collagen.3-4 Interestingly, the ECM surrounding the SMC not only regulates the SMC's differentiation state, but also determines whether the cell is able to respond to the action of growth factors, such as platelet-derived growth factor (PDGF). In addition, the three-dimensional structure of the ECM is known to be of major importance in regulating SMC responsiveness to growth factors and their proliferative capacity. For example, monomeric type I collagen supports SMC proliferation and PDGF mitogenic signaling, whereas the same collagen in a fibrillar state efficiently prevents PDGF signaling and subsequent proliferation.5 The ECM of the arterial wall is dramatically altered following angioplasty and in atherosclerosis, and it has been hypothesized that changes in ECM precede de-differentiation and proliferation and migration of SMCs.6
The study by Wang and colleagues, published in this issue of Circulation Research now adds another ECM protein, cartilage oligomeric matrix protein (COMP; also known as thrombospondin 5), to the group of ECM components that regulates SMC differentiation state.7 COMP is a pentameric glycoprotein expressed at high levels in cartilage, but also in other tissues. Wang and colleagues demonstrate that COMP levels are reduced following balloon-injury of rat carotid arteries, and by PDGF-stimulation of SMCs in culture, concomitant with decreased expression of the SMC differentiation markers smooth muscle α-actin, SM22α, and calponin.7 Furthermore, reduction in endogenous COMP expression by siRNA resulted in reduced expression of the above SMC differentiation markers, suggesting that SMC binding to COMP favors maintenance of a differentiated phenotype. Reciprocally, overexpression of COMP promoted a differentiated and contractile phenotype of isolated SMCs, and inhibited PDGF-induced signaling. This group showed last year that forced overexpression of COMP suppressed neointima formation after arterial injury.8 In the present study,7 this observation has been extended to demonstrate that overexpression of COMP prevents downregulation of calponin and SM22α in the rat carotid balloon injury model, and that it also increases arterial contraction in response to phenylephrine.
The ECM controls SMC phenotype through a large number of integrins and other adhesion molecules. ECM proteins that help maintain SMCs in a differentiated phenotype, such as fibrillar type I collagen and type IV collagen, are expressed in normal uninjured blood vessels, as are their integrins/receptors (Figure 1). Integrin α8β1 also appears to belong to the group of integrins that maintains SMC differentiation,9 but the ligand responsible for mediating this effect has not yet been identified. In the study by Wang et al.,7 integrin α7β1, but not α8β1, was shown to mediate the SMC differentiating effect of COMP. The α7β1 integrin is a laminin receptor, and an α7-integrin-deficient mouse,10 mimics the effects of COMP-deficiency on neointimal formation, suggesting that this integrin is particularly important in maintaining the SMC in a differentiated state.
Figure 1. Interactions of extracellular matrix proteins and smooth muscle cell integrins regulate their differentiation state.
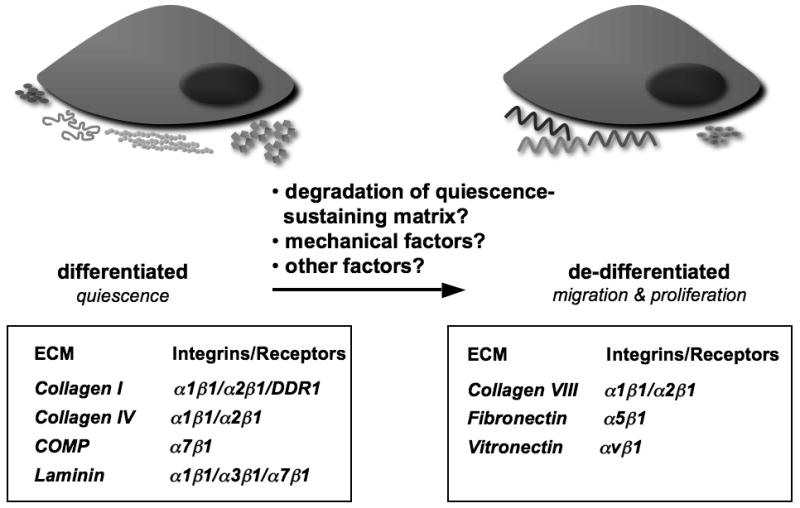
The work of Wang and colleagues7 adds COMP to the repertoire of extracellular matrix (ECM) proteins whose binding to their adhesion receptors, primarily integrins, contributes to the maintenance of the quiescent state of differentiated SMC. Following injury and in atherosclerosis, new ECM proteins are detected that in vitro promote SMC migration and proliferation associated with the de-differentiated SMC phenotype. It is less clear what triggers the transition from the differentiated to de-differentiated phenotype. Future studies of distinct regulatory mechanisms controlling COMP expression and turnover may help determine key components of this transition. DDR1, discoidin domain receptor 1
On the other hand, integrins whose ECM ligands promote SMC de-differentiation are often not expressed under normal conditions. For example, the fibronectin receptor, α5β1, is not expressed in quiescent vessels in vivo, nor are fibronectin fibrils detected. However, following injury, α5β1 is upregulated in less differentiated SMCs, and fibronectin fibril assembly is localized to these same cells.11 Similarly, collagen VIII is not a constituent of the normal vessel wall, but is expressed in remodeling tissues and stimulates SMC migration and matrix metalloproteinase synthesis.12 Vitronectin is a component of plasma that infiltrates the vessel wall following injury or endothelial dysfunction, and in vitro induces loss of contractility through integrin α5β1.13 Thus, integrins through interactions with specific ECM components, some of which are dynamically altered following injury, can control the SMC differentiation phenotype.
In vivo transition of SMC to the de-differentiated phenotype is associated with major remodeling of the vascular tissue. Proteases capable of degrading the ECM, particularly the matrix metalloproteinases, have essential roles in structurally digesting and altering matrix proteins, including their conversion to signaling molecules.14 It has been hypothesized that degraded matrix, such a type I collagen, may be required to release SMC from quiescence-sustaining matrices in which they are embedded.6 COMP is cleaved by proteases of the closely related ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) family, ADAMTS-7 and ADAMTS-12. 8,15 Thus, the reduced levels of COMP following vascular injury might be due to a combination of proteolysis and reduced transcription. In addition to increased proteolysis of quiescence-sustaining ECM components, SMCs and other cells in the vascular wall synthesize ECM components that support de-differentiation.
Although the studies of Wang and colleagues7 demonstrate a clear role for COMP-integrin α7β1 interactions in the maintenance of rat SMC differentiation in vivo in the rat carotid artery balloon injury model, what is the function of COMP in human vascular disease and other models of vascular injury, such as atherosclerosis? It is possible that regional differences in integrin α7β1 expression, such as those reported in humans for integrin α5β1,16 may restrict COMP's contribution to maintenance of SMC quiescence to specific anatomic sites.
The study by Wang and colleagues7 has increased our understanding of the ECM-integrin interactions that determine the SMC differentiation state, but a number of important questions remain to be answered. For example, is COMP responsible for the effects of other ECM proteins on SMC differentiation? It is possible that e.g. fibronectin promotes SMC de-differentiation by altering the ability of SMCs to produce and secrete COMP, whereas laminin or type IV collagen might have the opposite effect. Alternatively, does COMP act in part through its catalysis of collagen fibrillogenesis?17 To what extent do cells other than SMCs promote de-differentiation, for example macrophages as a source of proteases in atherosclerosis? Do lipid mediators,18-19 or other systemic factors help tip the balance? Furthermore, what strategies could be used to inhibit de-differentiation of SMCs in animal models and in human subjects? Answers to these questions are likely to bring us closer to strategies for the treatment of several vascular diseases. We are following this field with interest as we enter a new decade of research.
Acknowledgments
Sources of Funding: Research in the authors' laboratories is supported by grants from the NIH (HL018645, HL067267, and HL081795 to E.W.R. and HL062887, HL092969, and HL097365 to K.E.B).
Non-standard Abbreviations and Acronyms
- ADAMTS
a disintegrin and metalloproteinase with thrombospondin motifs
- COMP
cartilage oligomeric matrix protein
- ECM
extracellular matrix
- PDGF
platelet-derived growth factor
- SMC
smooth muscle cell
Footnotes
Disclosures: None
References
- 1.Campbell GR, Campbell JH. The phenotypes of smooth muscle expressed in human atheroma. Ann NY Acad Sci. 1990;598:143–158. doi: 10.1111/j.1749-6632.1990.tb42286.x. [DOI] [PubMed] [Google Scholar]
- 2.Fager G, Hansson GK, Gown AM, Larson DM, Skalli O, Bondjers G. Human arterial smooth muscle cells in culture: inverse relationship between proliferation and expression of contractile proteins. In Vitro Cell Dev Biol. 1989;25:511–520. doi: 10.1007/BF02623563. [DOI] [PubMed] [Google Scholar]
- 3.Hedin U, Bottger BA, Forsberg E, Johansson S, Thyberg J. Diverse effects of fibronectin and laminin on phenotypic properties of cultured arterial smooth muscle cells. J Cell Biol. 1988;107:307–319. doi: 10.1083/jcb.107.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orr AW, Lee MY, Lemmon JA, Yurdagul A, Jr, Gomez MF, Bortz PD, Wamhoff BR. Molecular mechanisms of collagen isotype-specific modulation of smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2009;29:225–231. doi: 10.1161/ATVBAHA.108.178749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koyama H, Raines EW, Bornfeldt KE, Roberts JM, Ross R. Fibrillar collagen inhibits arterial smooth muscle proliferation through regulation of cdk2 inhibitors. Cell. 1996;87:1069–1078. doi: 10.1016/s0092-8674(00)81801-2. [DOI] [PubMed] [Google Scholar]
- 6.Raines EW, Koyama H, Carragher NO. The extracellular matrix dynamically regulates smooth muscle cell responsiveness to PDGF. Ann NY Acad Sci. 2000;902:39–51. doi: 10.1111/j.1749-6632.2000.tb06299.x. [DOI] [PubMed] [Google Scholar]
- 7.Wang L, Zheng J, Du Y, Huang Y, Li J, Liu B, Liu C, Zhu Y, Gao Y, Xu Q, Kong W, Wang X. Cartilage oligomeric matrix protein maintains the contractile phenotype of vascular smooth muscle cells by interacting with α7β1 integrin. Circ Res. 2010;106:XXX–XXX. doi: 10.1161/CIRCRESAHA.109.202762. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Zheng J, Bai X, Liu B, Liu CJ, Xu Q, Zhu Y, Wang N, Kong W, Wang X. ADAMTS-7 mediates vascular smooth muscle cell migration and neointima formation in balloon-injured rat arteries. Circ Res. 2009;104:688–698. doi: 10.1161/CIRCRESAHA.108.188425. [DOI] [PubMed] [Google Scholar]
- 9.Zargham R, Touyz RM, Thibault G. α8 integrin overexpression in de-differentiated vascular smooth muscle cells attenuates migratory activity and restores the characteristics of the differentiated phenotype. Atherosclerosis. 2007;195:303–312. doi: 10.1016/j.atherosclerosis.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Welser JV, Lange N, Singer CA, Elorza M, Scowen P, Keef KD, Gerthoffer WT, Burkin DJ. Loss of the α7 integrin promotes extracellular signal-regulated kinase activation and altered vascular remodeling. Circ Res. 2007;101:672–681. doi: 10.1161/CIRCRESAHA.107.151415. [DOI] [PubMed] [Google Scholar]
- 11.Pickering JG, Chow LH, Li S, Rogers KA, Rocnik EF, Zhong R, Chan BM. α5β1 integrin expression and luminal edge fibronectin matrix assembly by smooth muscle cells after arterial injury. Am J Pathol. 2000;156:453–465. doi: 10.1016/s0002-9440(10)64750-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou G, Mulholland D, Gronska MA, Bendeck MP. Type VIII collagen stimulates smooth muscle cell migration and matrix metalloproteinase synthesis after arterial injury. Am J Path. 2000;156:467–476. doi: 10.1016/S0002-9440(10)64751-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dahm LM, Bowers CW. Vitronectin regulates smooth muscle contractility via αv and β1 integrin. J Cell Sci. 1998;111:1175–1183. doi: 10.1242/jcs.111.9.1175. [DOI] [PubMed] [Google Scholar]
- 14.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Bio. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu CJ, Kong W, Xu K, Luan Y, Ilalov K, Sehgal B, Yu S, Howell RD, Di Cesare PE. ADAMTS-12 associates with and degrades cartilage oligomeric matrix protein. J Biol Chem. 2006;281:15800–15808. doi: 10.1074/jbc.M513433200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davenpeck KL, Marcinkiewicz C, Wang D, Niculescu R, Shi Y, Martin JL, Zalewski A. Regional differences in integrin expression: role of α5β1 in regulating smooth muscle cell functions. Circ Res. 2001;88:352–358. doi: 10.1161/01.res.88.3.352. [DOI] [PubMed] [Google Scholar]
- 17.Halász K, Kassner A, Mörgelin M, Heinegård D. COMP acts as a catalyst in collagen fibrillogenesis. J Biol Chem. 2007;282:31166–31173. doi: 10.1074/jbc.M705735200. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi K, Takahashi M, Nishida W, Yoshida K, Ohkawa Y, Kitabatake A, Aoki J, Arai H, Sobue K. Phenotypic modulation of vascular smooth muscle cells induced by unsaturated lysophosphatidic acids. Circ Res. 2001;89:251–258. doi: 10.1161/hh1501.094265. [DOI] [PubMed] [Google Scholar]
- 19.Frontini MJ, O'Neil C, Sawyez C, Chan BM, Huff MW, Pickering JG. Lipid incorporation inhibits Src-dependent assembly of fibronectin and type I collagen by vascular smooth muscle cells. Circ Res. 2009;104:832–841. doi: 10.1161/CIRCRESAHA.108.187302. [DOI] [PubMed] [Google Scholar]