

Abstract
Neutrophils from people with poorly controlled diabetes present a primed phenotype and secrete excessive superoxide. Phospholipase A2 (PLA2)-derived arachidonic acid (AA) activates the assembly of NADPH oxidase to generate superoxide anion. There is a gap in the current literature regarding which PLA2 isoform regulates NADPH oxidase activation. The aim of this study was to identify the PLA2 isoform involved in the regulation of superoxide generation in neutrophils and investigate if PLA2 mediates priming in response to pathologic hyperglycemia. Neutrophils were isolated from people with diabetes mellitus and healthy controls, and HL60 neutrophil-like cells were grown in hyperglycemic conditions. Incubating neutrophils with the Ca2+-independent PLA2 (iPLA2) inhibitor bromoenol lactone (BEL) completely suppressed fMLP-induced generation of superoxide. The nonspecific actions of BEL on phosphatidic acid phosphohydrolase-1, p47phox phosphorylation, and apoptosis were ruled out by specific assays. Small interfering RNA knockdown of iPLA2 inhibited superoxide generation by neutrophils. Neutrophils from people with poorly controlled diabetes and in vitro incubation of neutrophils with high glucose and the receptor for advanced glycation end products ligand S100B greatly enhanced superoxide generation compared with controls, and this was significantly inhibited by BEL. A modified iPLA2 assay, Western blotting, and PCR confirmed that there was increased iPLA2 activity and expression in neutrophils from people with diabetes. AA (10 μM) partly rescued the inhibition of superoxide generation mediated by BEL, confirming that NADPH oxidase activity is, in part, regulated by AA. This study provides evidence for the role of iPLA2 in enhanced superoxide generation in neutrophils from people with diabetes mellitus and presents an alternate pathway independent of protein kinase C and phosphatidic acid phosphohydrolase-1 hydrolase signaling.
Neutrophil function has been shown to be altered in diabetes; one of the major neutrophil functional changes in diabetes is increased extracellular superoxide generation (1–5). The chronic hyperglycemia of poorly controlled diabetes can prime neutrophils and monocytes, resulting in an exaggerated inflammatory response and tissue damage (1–5). The neutrophil respiratory burst seems to be related directly to glycemic control with an increase in protein kinase C (PKC) and NADPH oxidase activity (5–7). Mechanistically, hyperglycemia results in increased phosphorylation of p47phox, leading to an increase in the generation of superoxide anion (O2−) (5–7). Recent work has shown that p47phox, a key protein in the assembly of NAPDH oxidase, prematurely translocates to the membrane and associates with p22phox in neutrophils from diabetic subjects (4). This was also observed in cells cultured in high glucose (HG) and the receptor for advanced glycation end products (RAGE) ligand S100B. The premature translocation of p47phox in neutrophils in response to hyperglycemia results in increased superoxide generation. Upon activation, the cytosolic subunits p47phox, p67phox, and p40phox of the NADPH oxidase translocate to the plasma membrane and bind with the cytochrome b558 (gp91phox and p22phox) complex (8). Translocation of the cytoplasmic components to the membrane and their association with cytochrome b558 renders the complex functional; the cytochrome then transfers electrons from NADPH to O2 to create O2− (superoxide anion) (9). The assembly of the subunits of the NADPH oxidase on the membrane is not sufficient; the final activation requires arachidonic acid (AA) (10, 11). Results from several studies suggest that AA release catalyzed by phospholipase A2 (PLA2) is necessary for both the activation and the maintenance of O2− generation by the NADPH oxidase (10–12).
PLA2 comprises a superfamily of enzymes that catalyze the hydrolysis of membrane phospholipid sn-2 ester bonds, generating free fatty acid and a lysophopholipid (13, 14). The PLA2 reaction is the primary pathway through which AA is liberated from membrane phospholipids, providing substrate for enzymatic conversion of the eicosanoids, which include PGs and leukotrienes (LTs) (15). The PLA2 family consists of 15 groups and many subgroups and includes five distinct types of enzymes, namely secreted PLA2 (sPLA2), cytosolic PLA2 (cPLA2), Ca2+-independent PLA2 (iPLA2), platelet-activating factor acetylhydrolases, and lysosomal PLA2 (15). Different isoforms of PLA2 play roles in regulation of inflammation. iPLA2 is associated with the initiation of inflammation, whereas sPLA2 and cPLA2 are involved in the resolution of inflammation (16). The role of PLA2 in the generation of superoxide in neutrophils is not clear. In cPLA2 null mice, it was shown that superoxide generation was not inhibited, suggesting that cPLA2 may not be involved (17). iPLA2, on the other hand, has been shown to be associated with AA mobilization and to be necessary for superoxide generation by neutrophils stimulated with Aroclor 1242, an organochloride compound (18). Because augmentation of an activated neutrophil respiratory burst requires AA generation in response to advanced glycation end products, through which neutrophil NADPH oxidase may be upregulated, enhancing reactive oxygen species output (19), we hypothesize that iPLA2 mediates the hyperglycemia-mediated neutrophil-generated oxidative stress in diabetes. It is also not known if the iPLA2-mediated superoxide generation in neutrophils involves PKC activation. In this study, we examine the role of iPLA2 in the priming of enhanced superoxide generation by neutrophils in diabetes.
Materials and Methods
Reagents
N-fMLP, S100B, cytochrome C, and propranolol hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO). A23187, pyrrolidine-1, and bromoenol lactone (BEL) were purchased from EMD Biosciences (San Diego, CA). LTB4 ELISA kits were purchased from R&D Systems (Minneapolis, MN). AA, rabbit polyclonal antiserum to iPLA2, and a cPLA2 assay kit were purchased from Cayman Chemicals (Ann Arbor, MI). HL60 cells, RPMI medium, and Iscove's medium were obtained from American Type Cell Culture (Manassas, VA). Trizol reagent was purchased from Invitrogen (Carlsbad, CA). High-capacity cDNA reverse transcriptase kits, real-time PCR probes for β-actin, iPLA2, and master mix were obtained from Applied Biosystems (Foster City, CA). Amaxa Nucleofector V solution and human monocyte Nucleofector medium were purchased from Amaxa (Gaithersburg, MD). Small interfering RNA (siRNA) for GAPDH, nontargeting, and iPLA2 were obtained from Dharmacon RNAi technologies (Lafayette, CO). Ab to p47phox was purchased from BD Biosciences (San Jose, CA) and PKC phosphospecific substrate Ab was obtained from Cell Signaling Technology (Danvers, MA).
Subject recruitment, neutrophil isolation, and culture
The study was approved by the Institutional Review Board at Boston University, and written informed consent was obtained from all subjects. Individuals with diabetes mellitus were selected and classified according to the criteria of the American Diabetes Association (20) and were recruited at the General Clinical Research Center of Boston University Medical Center. Systemically healthy individuals with no signs of infection or inflammatory disease were recruited as controls. The glycemic control of diabetes was assessed by glycated hemoglobin (HbA1c). Diabetic subjects were subgrouped according to their glycemic control as poor, moderate, or well-controlled using American Diabetes Association definitions (20). Peripheral venous blood was collected into heparinized tubes, and neutrophils were isolated by Ficoll-hypaque gradient centrifugation as described previously (4). Contaminating RBCs were hypotonically lysed; cell preparations were routinely 99% neutrophils with ≥95% viability, as determined by trypan blue exclusion.
Because primary neutrophils cannot be cultured for extended periods, in some experiments neutrophil-like differentiated HL-60 cells were used for culture or transfection. HL-60 cells (American Type Culture Collection) were cultured in flasks containing RPMI 1640 medium supplemented with 10% heat-inactivated FBS (HIFBS), glucose (5.5 mM), and 1.25% dimethyl-sulfoxide (DMSO) for 6 d. In order to simulate the hyperglycemic conditions of diabetes-induced hyperglycemia, differentiated HL-60 cells (1 × 106/ml) were incubated in RPMI 1640 culture medium (− glucose, + glutamine), 10% HIFBS at 37°C, 5% CO2 atmosphere for a period of 24 h under the following conditions: normal glucose (NG; 5.5 mM), HG (25 mM), S100B (5 μg/ml), S100B plus HG as described previously (4). Cells were then counted and the cell number adjusted accordingly for further experiments.
Superoxide assay
Superoxide (O2−) release was monitored spectrophotometrically at 37°C by measuring superoxide dismutase-inhibitable reduction of ferricyto-chrome C at 550 nm in 96-well plates as described previously (5). Cells (0.5 × 105) were suspended in PBS (200 μl/well) and stimulated by the addition of fMLP (1 μM) and the absorbance (OD) recorded in a Vmax kinetic microplate reader (SpectraMax 340PC, Molecular Devices, Sunnyvale, CA) at 550 nm and 37°C. O2− release was measured under conditions of linearity with respect to time and cell number and is expressed as nanomoles of O2−/min/105 neutrophils (5). For chemical inhibition experiments, neutrophils (25 × 106/ml) were incubated with specific inhibitors for 15 min in prewarmed PBS at 37°C. Control neutrophils received an equivalent volume of the diluent. Cell viability was assessed for all concentrations of the inhibitors by trypan blue exclusion.
Quantitative real-time PCR
Total RNA was extracted from neutrophils or HL60 neutrophil-like cells with Trizol reagent (Invitrogen) as per the manufacturer's protocol. The concentration and purity of RNA was estimated by the A260/A280 ratio spectrophotometrically (Nanodrop, Thermo Fisher Scientific, Waltham, MA). The A260/A280 ratio was routinely above 1.6. A total of 1 μg RNA was converted to cDNA using a high-capacity cDNA reverse transcriptase kit (Applied Biosystems). The reaction mixtures were thoroughly mixed and assayed at 25°C for 10 min, 37°C for 120 min, and 85°C for 0.05 min in a thermal cycler (ABI 9700, Applied Biosystems). Quantitative real-time PCR (QRT-PCR) was performed using primers and TaqMan probes for GAPDH and iPLA2 (group VI) labeled with FAM dye (Applied Bio-systems, TaqMan gene expression assays). β-actin was selected as an internal control and amplified using preformulated VIC-TAMRA-labeled TaqMan probes (Applied Biosystems, Endogenous Control). Quantification was performed in an automated thermal cycler (ABI Prism 7000 Sequence Detector, Applied Biosystems). The reaction mixtures were kept at 50°C for 2 min (one cycle), 95°C for 10 min (one cycle), 95°C for 15 s (45 cycles), and finally 60°C for 1 min. The results were analyzed through a software interface and spreadsheet for the calculation of relative expression (GAPDH/β-actin or iPLA2/β-actin).
iPLA2 activity assay
A modified iPLA2 activity assay was used as described previously (21) to detect the activity of iPLA2. A cPLA2 assay kit was obtained from Cayman Chemicals. To determine the activity of iPLA2, a modified Ca2+-free assay buffer was prepared comprising 300 mM NaCl, 60% glycerol, 10 mM HEPES, 8 mM Triton X-100, 4 mM EGTA, and 2 mg/ml of BSA (pH 7.4) (22). Neutrophils (4 × 108) were lysed in 1 ml cold buffer consisting of 50 mM HEPES (pH 7.4) and 1 mM EDTA. The cell suspension was then sonicated in an ice-cold water bath in two bursts of 15 s each at 1.5 amplitude at a setting of 30 mA. The lysate was then spun at 600 × g for 5 min at 4°C. The pellet consisting of unbroken cells and debris was discarded. The supernatant (whole cell fraction) was further centrifuged at 11,000 × g for 30 min at 4°C. The resulting pellet was the membrane-rich fraction and the supernatant was the cytosol-rich fraction. The protein concentration from each fraction was determined by Bradford protein assay. Ten microliters of the sample was incubated in a cPLA2 assay buffer containing 20 mM CaCl2, modified iPLA2 Ca2+-free buffer, and arachidonylthiophosphatidylcholine for 60 min in a 96-well plate. The reaction was stopped by the addition of 10 μl 5',5'-dithio-bis(2-nitrobenzoic acid)/EGTA, and the colorimetric reaction was read at 414 nm in a plate reader. iPLA2 activity was calculated and expressed as nanomoles thiophosphatidylcholine produced/min/ml.
Cell fractionation
Cell fractionation was performed as described previously (4). Briefly, neutrophils (25 × 106/ml) were lysed in ice-cold extraction buffer [20 mM Tris-HCl (pH 7.4), 10 mM HEPES, 25 mM NaCl, 2 mM EDTA, 10 mM EGTA, 1% (weight for weight) protease inhibitor mixture] and ultrasonicated five times at 30 W for 5 s. The whole cell lysate was centrifuged at 1000 × g for 5 min at 4°C. The resulting pellet was discarded, and the supernatant was further centrifuged at 11,000 × g for 30 min. The pellet was suspended in ice-cold extraction buffer with 0.5% Triton X-100 and 1 mM PMSF by ultrasonication five times at 30 W for 5 s and incubated for 20 min at 4°C. Protein content in each fraction was then estimated using the Bradford assay (4). The authenticity of the cell fractions was verified using the plasma membrane marker Na+/K+ ATPase. In experiments where cells were stimulated with fMLP, the reactions were stopped by ice-cold extraction buffer, and the fractions were prepared as described above.
Western blot
The protein samples for Western blotting were prepared as described in the literature (4). Neutrophils were lysed and fractionated as mentioned above by adding 40 μl of six times SDS sample buffer to 200 μl of the reaction mixture and then boiling the samples for 5 min. The final composition of SDS sample buffer after mixing was 2% (w/v) SDS, 58.3 mM Tris-HC1 (pH 6.8), 6% (v/v) glycerol, 5% (v/v) 2-ME, 0.002% (w/v) bromophenol blue, 1% (v/v) protease inhibitor mixture, and 1 mM PMSF. Aliquots of these samples were separated by SDS-PAGE (10–20 μg/lane) on 7.5% or 10% (v/v) polyacrylamide slab gels. The separated proteins were immediately transferred electrophoretically to polyvinylidene difluoride (PVDF) membranes utilizing transfer buffer [25 mM Tris, 192 mM glycine, and 20% (v/v) methanol (pH 8.4)] at 100 V for 1 h at 4°C. Membranes were blocked for 1 h at room temperature with 5% skim milk in TBS (pH 7.6). The blocking buffer was removed, and the membranes were incubated with the appropriate primary Abs (1:500 dilution for p47phox, 1:1000 in 3% skim milk for iPLA2, and 1:500 for Phospho PKC substrate) overnight at 4°C in 20 mM Tris-HCl (pH 7.6) containing 250 mM NaCl, 0.1% (v/v) Tween 20, 1% (w/v) BSA, and 0.002% (w/v) NaN3. The membranes were subsequently washed three times in TBST [20 mM Tris-HC1 (pH 7.6) containing 150 mM NaCl and 0.1% (v/v) Tween 20] and then incubated with secondary Ab (goat anti-mouse or rabbit IgG-HRP conjugate; 1:5000 dilution) in TBST for 1 h at room temperature. Membranes were washed three times in TBST. The activity of HRP was visualized by incubating the membranes for 5 min at room temperature in an ECL detection system (Pierce Chemical Co., Rockford, IL) followed by autoradiography. Membranes were stripped by incubation with reprobing buffer [62.5 mM Tris-HCl (pH 6.7), 2% SDS, 100 mM 2-ME] for 30 min at 56°C followed by three times washing in TBST. The blots were then stained with an antiactin Ab (1:2000 dilution) to confirm that equal amounts of protein were present in each lane of the gel. The band density was measured using an imaging densitometer (Bio-Rad, Hercules, CA) and normalized to the actin band.
Transfection experiments
HL60 cells (1 × 106/ml) were differentiated for 3 d in RPMI 1640 medium supplemented with 1.25% DMSO, 5.5 mM D-glucose, and 10% HIFBS. On day 3, cells (4 × 106) were resuspended in 100 μl of Nucleofector solution in Amaxa-certified cuvettes (Amaxa, Gaithersburg, MD). The cells received 1 μM GFP small interfering nontargeting (si-NT), small interfering GAPDH, or small interfering iPLA2 (si-iPLA2). The cuvettes were then transferred to an Amaxa Nucleofector device (Amaxa). Electroporation was set at S-11 program as per the manufacturer's recommendations. The device utilizes electroporation to create micropores in the cytoplasmic membrane of the cells and facilitates the entry of siRNA into the cytoplasm. After transfection, cells were resuspended in prewarmed Human Monocyte Nucleofector medium (Amaxa) at a concentration of 1 × 106/ml. Cells transfected with GFP were analyzed in a fluorescent microscope 24 and 48 h posttransfection to establish the transfection efficiency. Cells were counted after 3 d (72 h posttransfection) using a hemocytometer utilizing the trypan blue exclusion method. Total RNA was extracted using Trizol re-agent, and whole cell lysates were collected for Western blotting experiments. Superoxide generation was evaluation for si-NT and si-iPLA2– transfected neutrophils.
FACS
Neutrophils (1 × 106/ml) were incubated with BEL (1, 5, 10, 15, and 20 μM), or vehicle (0.1% DMSO and PBS) for 15 min at 37°C. Incubations were stopped on ice, and neutrophils were immediately centrifuged at 100 × g for 10 min. The supernatant was removed, and neutrophils were resuspended in FACS buffer (PBS with 5% FBS) containing FITC-Annexin V (1:50) and PE-CD16 (mouse, antihuman 1:200) or propidium iodide (PI) to label apoptotic neutrophils. Apoptotic neutrophils were characterized by double-positive staining for Annexin V and PI assessed by CellQuest software (BD Biosciences, San Jose, CA).
Statistical analysis
All data are presented as the average of at least three experiments with SEM unless otherwise stated. For comparison between two groups, the Student t test was applied for significance, and for multiple group analysis, one-way ANOVA was used with post hoc Bonferroni or least significance difference corrections.
Results
Superoxide generation mediated by iPLA2 is increased in neutrophils from people with diabetes
Table I compiles the demographic data of the study population. Neutrophils from people with poorly controlled diabetes demonstrated significantly increased superoxide generation compared with healthy controls (Fig. 1A). This suggests a role for glycemic control in neutrophil priming in the patient cohort studied (4–6). BEL was used to investigate the specific involvement of iPLA2 in the generation of superoxide in neutrophils. Neutrophils were pre-incubated with the inhibitor and stimulated with fMLP (1 μM), and superoxide generation was evaluated. BEL inhibited superoxide generation in a dose-dependent manner (IC50;10 μM), and there was a 100% reduction in the superoxide generation in response to 20 μM BEL (Fig. 1B). Preincubation with pyrrolidine-1 (0.01–20 μM) as a specific inhibitor of cPLA2 and DTT (0.001–1 mM)) for sPLA2 did not inhibit the superoxide generation in healthy neutrophils (data not shown). The experiment was repeated with neutrophils from people with diabetes. BEL completely blocked superoxide generation in neutrophils from healthy and diabetic subjects (Fig. 1C), suggesting that the involvement of iPLA2 in superoxide generation is similar in neutrophils from healthy and diabetic subjects.
Table I.
Demographic data of healthy and diabetic subjects
| Healthy | Diabetic | |
|---|---|---|
| n | 71 | 71 |
| Age (mean in y ± SD) | 36.5 ± 9.3 | 54.6 ± 10.6 |
| Gender (no.) | ||
| Male | 43 | 43 |
| Female | 28 | 28 |
| Ethnicity (no.) | ||
| African-American | 15 | 39 |
| Caucasian | 47 | 27 |
| Hispanic | 4 | 3 |
| Asian | 5 | 2 |
| Smoking status (no.) | ||
| Nonsmokers | 60 | 33 |
| Smokers | 3 | 18 |
| Former smokers | 8 | 20 |
| Type of diabetes (no.) | ||
| Type1 | 12 | |
| Type 2 | 59 | |
| Mean HbA1C % (± SD) | 8.0 ± 1.42 | |
| Glycemic control (no., based on HbA1C %) | ||
| Poorly controlled (>8% HbA1C) | 22 | |
| Moderately controlled (7–8% HbA1C) | 19 | |
| Well controlled (<7% HbA1C) | 20 | |
| Duration of diabetes (mean in y ± SD) | 8.84 ± 6.93 | |
| Periodontal condition (no.) | ||
| Healthy | 19 | |
| Mild | 20 | |
| Moderate | 9 | |
| Severe | 23 |
There were no significant differences between the age groups.
HbA1C, glycated hemoglobin.
FIGURE 1.

Neutrophils from poorly controlled diabetic subjects generate significantly increased superoxide regulated by iPLA2. A, Neutrophils (25 × 106/ml) isolated from healthy and diabetic subjects were resuspended in PBS and incubated at room temperature for a period of 15 min. Cells were then stimulated with 1 μM fMLP, and superoxide generation was evaluated using a superoxide dismutase-inhibitable cytochrome C assay. The intensity of the colorimetric reaction was measured in a microplate reader at 550 nm. Data is presented as the mean ± SEM in nanomoles of O2− for healthy (n = 27), well-controlled diabetes (n = 8), moderately controlled diabetes (n = 6), and poorly controlled diabetes (n = 13). B, Neutrophils (25 × 106/ml) were incubated with the indicated concentrations of BEL for 15 min at 37°C and then washed once with PBS. Neutrophils were resuspended in PBS and incubated at room temperature for a period of 15 min. Cells were then stimulated with 1 μM fMLP, and superoxide generation was evaluated using a superoxide dismutase-inhibitable cytochrome C assay. The intensity of the colorimetric reaction was measured in a microplate reader at 550 nm. Data are represented as percent inhibition and are expressed as mean ± SEM for three different donors. C, Neutrophils (25 × 106/ml) from healthy and diabetic subjects were incubated with BEL (20 μM) for 15 min at 37°C and washed once with PBS. Neutrophils were resuspended in PBS and incubated at room temperature for a period of 15 min and were then stimulated with 1 μM fMLP and superoxide generation evaluated. Data are shown as nanomoles of O2− expressed as mean ± SEM for six different donors.
Hyperglycemia-mediated increase in superoxide generation is mediated by iPLA2
To understand the mechanism of increased superoxide generation and the role of iPLA2 in diabetes, an in vitro system modeling the neutrophil response to hyperglycemia was established. For these experiments, HL60 cells were differentiated into neutrophil like cells over a period of 6 d and were then cultured for 24 h with NG (5.5 mM), HG (25 mM), RAGE ligand (S100B; 5 μg/ml), RAGE ligand and HG (S100B+HG) or mannitol (25 mM). The mannitol group served as an osmotic control for the hyperglycemia experiments. The superoxide response from these experimental groups show that compared with NG alone, S100B (p < 0.01) and S100B+HG (p < 0.001) demonstrate significantly increased superoxide generation (Fig. 2A). HG alone did not significantly increase superoxide generation. The combination of HG with S100B demonstrated significantly increased superoxide generation compared with HG alone (p < 0.001). Mannitol also demonstrated significantly increased superoxide generation compared with NG alone, demonstrating the impact of osmotic pressure changes on neutrophil superoxide generation; however, the receptor-mediated stimulation of neutrophil function by the RAGE ligand S100B was more robust. When BEL was added, there was a significant reduction in superoxide generation in all groups, suggesting that the increase in superoxide generation is mediated by iPLA2 (Fig. 2A).
FIGURE 2.

Neutrophils cultured in hyperglycemic medium generate significantly increased superoxide, which is regulated by iPLA2. A, HL-60 cells were differentiated into neutrophils by incubation with 1.25% DMSO for 6 d. The cells were then cultured under NG (5 mM), HG (25 mM), S100B (5 μg/ml), HG + S100B, and 25 mM mannose (MANN) as an osmotic control. After 24 h, the cells were washed and resuspended in PBS and were incubated with BEL 20 μM for 15 min at 37°C. The cells were stimulated with 1 μM fMLP, and superoxide generation was evaluated in nanomoles of O2−. Results are expressed as mean ± SEM for n=4. B, Neutrophils (25 × 106/ml) were incubated with either BEL (20 μM) or propranolol hydrochloride (150 μM) for 15 min at 37°C. Cells were washed once and resuspended in PBS and were stimulated with 1 μM fMLP. Superoxide release was evaluated by the reduction of cytochrome C read at 550 nM. Results are expressed as mean ± SEM for n= 3. *p < 0.05; **p < 0.01 (one-way ANOVA between groups NG, S100B, S100B+HG, and mannitol). #p<0.05; ##p<0.01 (one-way ANOVA between stimulated and BEL-treated groups).
Previous studies have suggested that BEL is also a potent inhibitor of phosphatidic acid phosphohydrolase-1 (PAP-1) (23). PAP-1 catalyzes the conversion of phosphatidic acid into diacylglycerol via the Kennedy pathway. Hence, if BEL-mediated inhibition of PAP-1 results in the suppression of superoxide generation, then the direct inhibition of PAP-1 would give the same results. To investigate the role of PAP-1 in superoxide generation in neutrophils, propranolol hydrochloride, a specific inhibitor of PAP-1, was used. Results from this experiment suggested that propranolol hydrochloride (150 μM) did not significantly inhibit superoxide reduction in neutrophils compared with BEL (Fig. 2B), further confirming that the BEL-mediated inhibition of iPLA2 and not PAP-1 regulates superoxide generation in neutrophils.
iPLA2 knockdown inhibits superoxide generation in neutrophils
In order to confirm the specificity of iPLA2 in neutrophil super-oxide generation, HL-60 cells were used for the knockdown of iPLA2 expression. HL-60 cells were differentiated with 1.25% DMSO for 6 d to create neutrophil-like cells. After 3 d, cells were transfected with si-iPLA2 using the Amaxa Nucleofector device (Amaxa). The transfected reagents were GFP, si-NT, si-GAPDH, and si-iPLA2. The cells were further incubated for 3 d. Transfection efficiency was confirmed by the uptake of GFP and by the knockdown of GAPDH RNA and protein and was found to be 25%, 80%, and 57%, respectively. Transfection with iPLA2 siRNA resulted in ~50% reduction in mRNA levels of iPLA2 (Fig. 3A). The iPLA2 mRNAwas comparable between the control and nontargeted groups. Fig. 3B shows the typical amplicon associated with the reduction of iPLA2 mRNA in the targeted group. This knockdown resulted in 30% inhibition of fMLP-stimulated superoxide generation (Fig. 3C). The data from iPLA2 knockdown experiments confirm the chemical inhibition of iPLA2 by BEL, suggesting that iPLA2 is the key PLA2 isoform involved in superoxide generation by neutrophils.
FIGURE 3.

iPLA2 knockdown inhibits super-oxide generation in neutrophils A, HL60 cells (1 × 106 /ml) were differentiated with 1.25% DMSO in RPMI 1640 media supplemented with 10% HIFBS and D-glucose (5.5 mM) for 3 d. On day 3, cells were transfected with nontargeting siRNA (1 μM) and si-iPLA2 (1 μM) by nucleofection and re-suspended in the differentiation media. Nontransfected differentiated cells served as controls. After 72 h, cells were collected, and RNA was extracted by Trizol reagent and reverse transcribed to cDNA. Real-time PCR using probes for actin and iPLA2 was set to 45 cycles to estimate the fold change of iPLA2 normalized to actin. B, Figure shows a typical amplicon of real-time PCR. C, Cells transfected with si-iPLA2 and nontargeting siRNA were counted and resuspended in PBS at a concentration of 25 × 106/ml and stimulated with 1 μM fMLP. Cytochrome C reduction was read in a spectrophotometer at 550 nm for triplicate samples. Data are presented as nanomoles of O2− and expressed as mean ± SEM. p < 0.05.
iPLA2 expression is increased in neutrophils from people with diabetes and mediates superoxide generation
The hypothesis to be tested is that superoxide generation is increased in neutrophils from people with diabetes, which is mediated, at least in part, by an increase in iPLA2 activity. To investigate possible increased expression of iPLA2 in neutrophils from diabetic subjects, QRT-PCR and Western blotting experiments were undertaken. The QRT-PCR data shows that neutrophils from people with poorly controlled diabetes (Hb1Ac >8) had significantly increased iPLA2 mRNA compared with healthy controls (Fig. 4A). Western blotting reveals that iPLA2 was exclusively expressed in the membrane fraction of the neutrophils with no expression in the cytosol. The iPLA2 protein migrates at 85 kDa on a 10% SDS-PAGE gel (data not shown). Neutrophils from people with diabetes showed significantly increased expression of membrane-bound iPLA2 compared with healthy controls (Fig. 4B). In Fig. 4B, we show a representative image of increased iPLA2 protein in the whole cell fraction of neutrophils from people with diabetes.
FIGURE 4.

Neutrophils from people with poorly controlled diabetes mellitus exhibit increased expression of iPLA2. A, Total RNA was extracted from resting neutrophils from healthy and diabetic subjects, and 1 μg of RNA was used to prepare cDNA using RTPCR. One hundred nanograms of cDNA was used for QRT-PCR using probes for actin and iPLA2 to estimate the fold change of iPLA2 normalized to actin for healthy (n = 23), well-controlled diabetes (n = 6), moderately controlled diabetes (n = 6), and poorly controlled diabetes (n = 10). B, Twenty micrograms of whole cell and membrane fraction was run on 10% SDS-PAGE gel and transferred to a PVDF membrane. The membranes were probed overnight with iPLA2 Ab (in 3% skim milk). iPLA2 membrane fractions and one representative whole cell image was quantified by densitometry and normalized to actin. Results are expressed as mean ± SEM. n = 6. p < 0.05.
iPLA2 activity is increased in neutrophils from people with diabetes
To investigate whether increased superoxide generation in neutrophils from people with diabetes is directly related to increased iPLA2 activity, a modified iPLA2 assay was used. The results of experiments using normal neutrophils show that the basal PLA2 activity in neutrophil cytoplasmic membrane preparations is significantly increased in the presence of EDTA (Fig. 5A), suggesting that membrane-bound basal PLA2 activity is calcium-independent and confirming the specific involvement of iPLA2. The next experiment was undertaken in the presence of EDTA and without calcium to specifically investigate iPLA2 activity in the membrane fractions of neutrophils from healthy and diabetes donors. The membrane-bound iPLA2 activity is significantly increased in neutrophils from people with diabetes compared with healthy controls (Fig. 5B).
FIGURE 5.

Calcium-independent PLA2 activity is increased in membrane fractions of neutrophils from diabetic subjects. A, Neutrophils (4 × 108/ml) were sonicated in a homogenization buffer supplemented with protease inhibitor. Cells were then fractionated to isolate the membrane components. The reaction was initiated by the addition of sample to the substrate arachidonyl thiophosphatidyl choline. The reactions were stopped by the addition of 5',5'-dithio-bis(2-nitrobenzoic acid)/EGTA, and the plate was read at 415 nm. The buffers for this assay were calcium free and contained EDTA to chelate all calcium, thus activating calcium-independent PLA2. Results are expressed as mean ± SEM for n = 3. B, The membrane fraction of neutrophils from diabetic subjects demonstrated a significant increase in calcium-independent PLA2 activity. Data represent percent increase of iPLA2 activity over healthy controls and are expressed as mean ± SEM for n = 4. p, 0.05.
iPLA2 does not regulate cell apoptosis or cell death in neutrophils
Because iPLA2 is associated with multiple, diverse processes in neutrophils (14), we wanted to examine the involvement of iPLA2 in apoptosis of neutrophils. FACS analysis was used to determine Annexin V expression (apoptotic marker) and PI binding to DNA (necrotic marker). FACS analysis confirmed that >95% of cells isolated were CD16+ neutrophils. Results also revealed that inhibition of iPLA2 did not induce significant apoptotic or necrotic cell death compared with the vehicle control (Fig. 6A). Neutrophils treated with BEL (20 μM) were also counted in a hemocytometer using the trypan blue exclusion method to assess viability. The viability of the neutrophils was not impacted by the inhibition of iPLA2. There was no significant difference in the Annexin V-positive neutrophils incubated in PBS, DMSO (0.1%), and 20 μM BEL (Fig. 6B).
FIGURE 6.

BEL does not stimulate apoptosis in neutrophils. A, Neutrophils (10 ± 106/ml) were incubated with PBS, DMSO (0.1%), and BEL (1– 20 μM) for 15 min at 37°C. Cells were labeled with Annexin V or PI to detect apoptotic and necrotic neutrophils. The upper right quadrant of the dot blot represents Annexin V- and PI-positive cells. The histogram represents Annexin V-positive cells at a threshold setting of 102 FL1-H. B, Annexin V-positive cells were counted and are represented on a bar graph, and data are represented as percent cells positive and as mean ± SEM for n = 3.
iPLA2 regulates superoxide generation independently of p47phox or pleckstrin phosphorylation
The phosphorylation and translocation of p47phox to the cytoplasmic membrane is a key event in superoxide generation catalyzed by NADPH oxidase. To investigate whether the inhibition of iPLA2 impacts PKC-dependent p47phox phosphorylation, normal neutrophils were stimulated with fMLP (1 μM) in the presence or absence of BEL (20 μM) for a period of 180 s because studies have shown that neutrophils stimulated for 180 s with fMLP induced p47phox phosphorylation and translocation (24, 25). In un-stimulated cells, p47phox was not phosphorylated. Maximum phosphorylation of p47phox was evident at 30 s with a gradual reduction at 180 s (Fig. 7). There was no significant difference in the phosphorylation of p47phox in cells treated with or without the iPLA2 inhibitor at 30, 60, and 180 s. The Ab used for these experiments detects phosphorylated PKC substrates (26, 27). Therefore, in addition to p47phox, pleckstrin is also labeled (27). Pleckstrin phosphorylation was also not changed with the inhibition of iPLA2, suggesting that the iPLA2 regulation of superoxide generation in neutrophils is independent of the phosphorylation of these PKC substrates.
FIGURE 7.

BEL does not inhibit the phosphorylation of p47phox. Neutrophils (25 × 106/ml) were incubated with or without BEL (20 μM) and then stimulated with 1 μM fMLP for 30, 60, or 180 s. The reaction was stopped by adding ice-cold lysis buffer, and samples were prepared for Western blotting. Equal amounts of protein (10 μg/lane) were loaded on 10% SDS-PAGE gels and transferred to PVDF membranes. Membranes were then probed with anti-PKC phosphoserine substrate Ab (1:500). The membranes were stripped and reprobed with anti-p47phox (1:500). Data were obtained by a densitometry, and the results are presented as phospho-p47phox/total p47phox ratio. Results are expressed as mean ± SEM for n = 3.
AA rescues neutrophil superoxide inhibition by BEL
Our data suggests that inhibition of iPLA2 by BEL abolishes superoxide generation in neutrophils. To prove our hypothesis that iPLA2-derived AA activates NAPDH oxidase, we added exogeneous AA to neutrophils treated with BEL (20 μM). The data shows that AA (10 μM) partly restores NADPH oxidase activity, resulting in superoxide generation (Fig. 8). AA alone did not stimulate the superoxide generation by neutrophils (data not shown).
FIGURE 8.
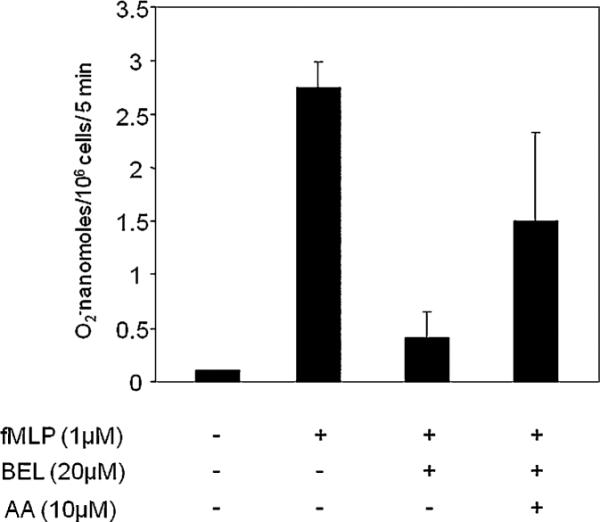
AA partly restores the activity of iPLA2 in superoxide generation in neutrophils. Neutrophils (25 × 106/ml) were incubated with BEL (20 μM) for 15 min at 37°C and then washed at 1000 rpm for 10 min at room temperature with PBS. Cells were then incubated with AA (10 μM) at 37°C for 30 min before being stimulated by 1 μM fMLP. Superoxide generation released was assessed by cytochrome C reduction and was read in a spectrophotometer at 550 nm for triplicate samples. Data are presented as nanomoles of O2−. Results are expressed as mean ± SEM for n = 3.
Discussion
Hyperglycemia-mediated priming of neutrophils results in an enhanced oxidative burst in diabetes mellitus (1–5). This study demonstrates a role for iPLA2 in superoxide generation in neutrophils and shows that iPLA2 might at least, in part, contribute to neutrophil priming in diabetes. Findings from this study suggest that the fMLP-induced iPLA2-dependent superoxide generation is independent of the PKC and p47phox phosphorylation. Increased superoxide generation by neutrophils from diabetes is blocked by specific inhibitors of iPLA2, and in normal neutrophils cultured under hyperglycemic conditions, priming and superoxide generation is prevented. This is the first study to demonstrate a role for iPLA2 in superoxide generation in neutrophils from people with diabetes mellitus.
Neutrophils from poorly controlled diabetic subjects generate significant amounts of superoxide compared with healthy controls (4, 5), exhibit increased PKC activity, diglyceride formation, and p47phox phosphorylation (5), and are characterized by a premature translocation of p47phox to the plasma membrane, which was also consistent with findings from in vitro hyperglycemia experiments (4). These findings strongly support a primed neutrophil phenotype in poorly controlled diabetes, resulting in an increased oxidative stress. The data presented in this study extend and further clarify these findings. To identify the role of iPLA2 in superoxide generation, we used BEL, a chemical inhibitor known to potently inhibit iPLA2 (28). Because BEL has been shown to have nonspecific actions (23), we performed experiments to rule out BEL-mediated inhibition of PAP-1, stimulation of apoptosis, and p47phox phosphorylation. The data reveals that BEL completely inhibits superoxide generation by normal neutrophils, which is consistent with previous reports (18). Taken together, it is apparent that BEL is only effective at high concentrations in the range of 10 μM for inhibition of superoxide generation in neutrophils (18). Conversely, the cPLA2 inhibitor pyrrolidine-1 did not have activity on superoxide generation by neutrophils, confirming previous findings (19). A study on a human myeloid cell line PLB-985 expressing antisense cPLA2 mRNA failed to activate NAPDH oxidase activity, and addition of exogenous AA fully restored this activity (29). Another study in mice reported that disruption of the cPLA2-α gene suppressed neutrophil AA release but had no effect on NAPDH oxidase activity (17). Our data suggest that superoxide generation is regulated by the iPLA2 rather than the cPLA2. We used specific cPLA2-α inhibitor pyrrolidine-1 and DTT to rule out the involvement of cPLA2-α and sPLA2 in neutrophil superoxide generation. We confirmed these findings with suppression of iPLA2 expression by RNA silencing. The resulting data confirm the specificity of the involvement of iPLA2 in superoxide generation by the human neutrophils and further suggest that increased iPLA2 expression and function, in part, contribute to the neutro-phil priming in people with diabetes.
Because hyperglycemia primes neutrophils for enhanced superoxide generation and iPLA2 directly regulates this process, we wanted to investigate whether increased expression and activity of iPLA2 was involved in priming. We report that an increase in superoxide generation in neutrophils from people with diabetes is associated with an increase in iPLA2 activity, the expression of iPLA2 at the message, and protein level. To this end, to separate the activity of iPLA2 and cPLA2, an assay developed previously was used to assess cPLA2 and iPLA2 activity independently. iPLA2 activity is enhanced by depleting calcium levels (21), and when neutrophil membrane fractions were treated with 4 mM EGTA in the absence of any exogenous calcium, this group shows a significant increase in basal PLA2 activity compared with the activity in the presence of 20 mM of exogenous calcium, suggesting that the predominant PLA2 activity from neutrophil membrane fractions is calcium independent and therefore reflects iPLA2 activity. The results are in agreement with data from another study, which showed that iPLA2 activity was increased in the presence of BAPTA-AM or thapsigargin (calcium chelators) (22). In the presence of intracellular calcium, binding of calmodulin to the iPLA2 renders the enzyme inactive and release of calmodulin in the absence of calcium activates iPLA2 (30, 31). The data from our PLA2 activity experiments suggest that in the presence of EGTA, chelation of intracellular calcium releases calmodulin from its binding site on iPLA2 and renders it active. This calcium-independent PLA2 activity was significantly elevated in neutrophils from diabetes. Another PLA2 isoform has been shown to exhibit similar calcium-independent activity (32); a novel 60.9-kDa membrane-bound calcium-independent cPLA2-γ, also known as group cPLA2-IVC. To date however, the expression of cPLA2-γ in neutrophils is not known and warrants further investigation.
iPLA2 has also been shown to drive the onset of acute pleurisy-induced inflammation in a male Wistar rat model through the generation of PGE2, LTB4, platelet-activating factor, and the β form of pro-IL-1 (16). During the resolution phase, there was induction of sPLA2 (type II and V) and cPLA2. Because the complications of diabetes are associated with inflammation, it would be interesting to investigate if the increase in the activity and expression of iPLA2 in neutrophils is associated with a reduction of activity and expression of cPLA2 and sPLA2.
Our findings (Fig. 8) show that the suppression of O2− by BEL-mediated iPLA2 inhibition can be partly restored by AA, suggesting the activation of NADPH oxidase by AA. Hence, one of the mechanisms by which iPLA2 induces O2− release will be AA liberation from membrane phospholipids. Studies have shown that AA is necessary for the activation of O2− production and the associated H+ channel-mediated efflux (33, 34). AA can act by increasing the affinity of NADPH to the oxidase, regulation of a proton channel, by facilitating the translocation of cytosolic components to the membrane, increasing the affinity of GTP binding sites in the membrane and enhancing the dissociation of Rac G protein from its regulatory molecule, GDI (34). AA mediates priming of NADPH oxidase and enhances the production of superoxide generation when cells are challenged with a secondary stimulus like fMLP (34). There is also evidence that PGE2, a metabolite of AA, suppresses bacterial killing by inhibiting NAPDH oxidase in alveolar macrophages (35). It was shown that PGE2 increases cAMP-1 levels via activation of G protein coupled E prostanoid (EP) receptors EP2 and EP4. The increased cAMP-1 levels inhibited the phosphorylation and translocation p47phox to the cytoplasmic membrane (35). The data reported in this study suggest novel pathways of host immune response in the regulation of NADPH oxidase activity by iPLA2, AA, and PGE2.
Neutrophils prelabeled with [3H] AA display increased [3H] arachidonate release on exposure to advanced glycation endproduct-albumin over exposure to albumin alone (19). Advanced glycation endproduct augmentation of the activated neutrophil respiratory burst required AA generation, through which neutrophil NADPH oxidase may be upregulated, enhancing reactive oxygen species output (19). Neutrophils cultured under hyperglycemia-like conditions demonstrated significantly elevated superoxide generation compared with NG alone (Fig. 2A). This was pronounced in neutrophils cultured with S100B, which is known to bind the RAGE receptor and induces significant phosphorylation of p47phox and its premature translocation to the cytoplasmic membrane (4, 7). BEL significantly blocked the hyperglycemia-mediated priming of iPLA2 under these conditions, suggesting a mechanism that involves iPLA2 activity. Because AA partly rescued the superoxide inhibition by BEL, it is possible that excess release of AA by iPLA2 in part, at least, contributes to the priming mechanism in hyperglycemic conditions. It may be speculated that different pools of AA exist that serve different functions; in the case of iPLA2, AA is directed to superoxide generation and cPLA2-produced AA is primarily routed toward eicosanoid generation (18). In this study, we were able to provide evidence that iPLA2-mediated AA release, in part, contributes to NADPH oxidase activity. Studies have demonstrated that two parallel pathways regulate NADPH oxidase activation: p47phox phosphorylation and PLA2 activation (36, 37). Our findings confirm the existence of these two routes where inhibition of iPLA2 by BEL inhibited fMLP-induced superoxide generation, and this was not due to suppression of p47phox phosphorylation. However, simultaneous activation of both of these pathways is critical in the generation of superoxide anion in neutrophils. We also confirmed the role of iPLA2 in superoxide generation in a differentiated neutrophil-like HL60 model by suppressing iPLA2 mRNA expression. The inhibition of superoxide by BEL in differentiated HL60 cells was 75–80% (Fig. 2A). We also determined the transfection efficiency using GFP as a marker. The data reveals that the transfection efficiency in HL-60 cells was 25%, which is similar to findings of Magalhães et al. (38). Like other investigators, we are unable to achieve 100% transfection efficiency with HL-60 cells. The interpretation of a 30% knockdown in activity with 25% transfection efficiency as significant is consistent with the literature (38).
In conclusion, this study demonstrates that increased iPLA2 expression and activity in people with uncontrolled diabetes might, at least in part, contribute to the priming of neutrophils for enhanced superoxide generation upon secondary stimulus and confirms the regulatory role of iPLA2 in neutrophil superoxide generation. The regulation of superoxide generation in neutrophils by iPLA2 involves a mechanism that is independent of PKC and PAP-1 hydrolase signaling.
Acknowledgments
We thank Ms. Amanda Blackwood for laboratory assistance.
This work was supported in part by United States Public Health Service Commissioned Corps Grants DE-15566, DE-16933, DE-18917, and RR-00533.
Abbreviations used in this paper
- AA
arachidonic acid
- BEL
bromoenol lactone
- cPLA2
cytosolic PLA2
- EP
E prostanoid
- HbA1C
glycated hemoglobin
- HG
high glucose
- HIFBS
heat-inactivated FBS
- iPLA2
Ca2+-independent PLA2
- LT
leukotriene
- NG
normal glucose
- PAP-1
phosphatidic acid phosphohydrolase-1
- PI
propidium iodide
- PKC
protein kinase C
- PLA2
phospholipase A2
- PVDF
polyvinylidene difluoride
- RAGE
receptor for advanced glycation end products
- si-iPLA2
small interfering iPLA2
- si-NT
small interfering nontargeting
- siRNA
small interfering RNA
- sPLA2
secreted PLA2.
Footnotes
Disclosures The authors have no financial conflicts of interest.
References
- 1.Wierusz-Wysocka B, Wysocki H, Siekierka H, Wykretowicz A, Szczepanik A, Klimas R. Evidence of polymorphonuclear neutrophils (PMN) activation in patients with insulin-dependent diabetes mellitus. J. Leukoc. Biol. 1987;42:519–523. doi: 10.1002/jlb.42.5.519. [DOI] [PubMed] [Google Scholar]
- 2.Hand WL, Hand DL, Vasquez Y. Increased polymorphonuclear leukocyte respiratory burst function in type 2 diabetes. Diabetes Res. Clin. Pract. 2007;76:44–50. doi: 10.1016/j.diabres.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 3.Gupta A, Tripathi AK, Tripathi RL, Madhu SV, Banerjee BD. Advanced glycosylated end products-mediated activation of polymorphonuclear neutrophils in diabetes mellitus and associated oxidative stress. Indian J. Biochem. Biophys. 2007;44:373–378. [PubMed] [Google Scholar]
- 4.Omori K, Ohira T, Uchida Y, Ayilavarapu S, Batista EL, Jr., Yagi M, Iwata T, Liu H, Hasturk H, Kantarci A, Van Dyke TE. Priming of neutrophil oxidative burst in diabetes requires preassembly of the NADPH oxidase. J. Leukoc. Biol. 2008;84:292–301. doi: 10.1189/jlb.1207832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karima M, Kantarci A, Ohira T, Hasturk H, Jones VL, Nam BH, Malabanan A, Trackman PC, Badwey JA, Van Dyke TE. Enhanced superoxide release and elevated protein kinase C activity in neutrophils from diabetic patients: association with periodontitis. J. Leukoc. Biol. 2005;78:862–870. doi: 10.1189/jlb.1004583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gyurko R, Siqueira CC, Caldon N, Gao L, Kantarci A, Van Dyke TE. Chronic hyperglycemia predisposes to exaggerated inflammatory response and leukocyte dysfunction in Akita mice. J. Immunol. 2006;177:7250–7256. doi: 10.4049/jimmunol.177.10.7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding Y, Kantarci A, Hasturk H, Trackman PC, Malabanan A, Van Dyke TE. Activation of RAGE induces elevated O2- generation by mononuclear phagocytes in diabetes. J. Leukoc. Biol. 2007;81:520–527. doi: 10.1189/jlb.0406262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheppard FR, Kelher MR, Moore EE, McLaughlin NJ, Banerjee A, Silliman CC. Structural organization of the neutrophil NADPH oxidase: phosphorylation and translocation during priming and activation. J. Leukoc. Biol. 2005;78:1025–1042. doi: 10.1189/jlb.0804442. [DOI] [PubMed] [Google Scholar]
- 9.Burg ND, Pillinger MH. The neutrophil: function and regulation in innate and humoral immunity. Clin. Immunol. 2001;99:7–17. doi: 10.1006/clim.2001.5007. [DOI] [PubMed] [Google Scholar]
- 10.Dana R, Malech HL, Levy R. The requirement for phospholipase A2 for activation of the assembled NADPH oxidase in human neutrophils. Biochem. J. 1994;297:217–223. doi: 10.1042/bj2970217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shmelzer Z, Haddad N, Admon E, Pessach I, Leto TL, Eitan-Hazan Z, Hershfinkel M, Levy R. Unique targeting of cytosolic phospholipase A2 to plasma membranes mediated by the NADPH oxidase in phagocytes. J. Cell Biol. 2003;162:683–692. doi: 10.1083/jcb.200211056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henderson LM, Chappell JB, Jones OT. Superoxide generation is inhibited by phospholipase A2 inhibitors. Role for phospholipase A2 in the activation of the NADPH oxidase. Biochem. J. 1989;264:249–255. doi: 10.1042/bj2640249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balsinde J, Winstead MV, Dennis EA. Phospholipase A(2) regulation of arachidonic acid mobilization. FEBS Lett. 2002;531:2–6. doi: 10.1016/s0014-5793(02)03413-0. [DOI] [PubMed] [Google Scholar]
- 14.Balsinde J, Balboa MA. Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A2 in activated cells. Cell. Signal. 2005;17:1052–1062. doi: 10.1016/j.cellsig.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 16.Gilroy DW, Newson J, Sawmynaden P, Willoughby DA, Croxtall JD. A novel role for phospholipase A2 isoforms in the checkpoint control of acute inflammation. FASEB J. 2004;18:489–498. doi: 10.1096/fj.03-0837com. [DOI] [PubMed] [Google Scholar]
- 17.Rubin BB, Downey GP, Koh A, Degousee N, Ghomashchi F, Nallan L, Stefanski E, Harkin DW, Sun C, Smart BP, et al. Cytosolic phospholipase A2-α is necessary for platelet-activating factor biosynthesis, efficient neutrophil-mediated bacterial killing, and the innate immune response to pulmonary infection: cPLA2-α does not regulate neutrophil NADPH oxidase activity. J. Biol. Chem. 2005;280:7519–7529. doi: 10.1074/jbc.M407438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tithof PK, Peters-Golden M, Ganey PE. Distinct phospholipases A2 regulate the release of arachidonic acid for eicosanoid production and su-peroxide anion generation in neutrophils. J. Immunol. 1998;160:953–960. [PubMed] [Google Scholar]
- 19.Wong RKM, Pettit AI, Quinn PA, Jennings SC, Davies JE, Ng LL. Advanced glycation end products stimulate an enhanced neutrophil respiratory burst mediated through the activation of cytosolic phospholipase A2 and generation of arachidonic Acid. Circulation. 2003;108:1858–1864. doi: 10.1161/01.CIR.0000089372.64585.3B. [DOI] [PubMed] [Google Scholar]
- 20.Expert Committee on the Diagnosis and Classification of Diabetes Mellitus Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 2000;23(Suppl 1):S4–S19. [PubMed] [Google Scholar]
- 21.Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nat. Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- 22.Csutora P, Zarayskiy V, Peter K, Monje F, Smani T, Zakharov SI, Litvinov D, Bolotina VM. Activation mechanism for CRAC current and store-operated Ca2+ entry: calcium influx factor and Ca2+-independent phospholipase A2beta-mediated pathway. J. Biol. Chem. 2006;281:34926–34935. doi: 10.1074/jbc.M606504200. [DOI] [PubMed] [Google Scholar]
- 23.Fuentes L, Pérez R, Nieto ML, Balsinde J, Balboa MA. Bromoenol lactone promotes cell death by a mechanism involving phosphatidate phosphohydrolase-1 rather than calcium-independent phospholipase A2. J. Biol. Chem. 2003;278:44683–44690. doi: 10.1074/jbc.M307209200. [DOI] [PubMed] [Google Scholar]
- 24.Dewas C, Fay M, Gougerot-Pocidalo MA, El-Benna J. The mitogen-activated protein kinase extracellular signal-regulated kinase 1/2 pathway is involved in formyl-methionyl-leucyl-phenylalanine-induced p47phox phosphorylation in human neutrophils. J. Immunol. 2000;165:5238–5244. doi: 10.4049/jimmunol.165.9.5238. [DOI] [PubMed] [Google Scholar]
- 25.Dang PM, Fontayne A, Hakim J, El Benna J, Périanin A. Protein kinase C zeta phosphorylates a subset of selective sites of the NADPH oxidase component p47phox and participates in formyl peptide-mediated neutrophil respiratory burst. J. Immunol. 2001;166:1206–1213. doi: 10.4049/jimmunol.166.2.1206. [DOI] [PubMed] [Google Scholar]
- 26.Ohira T, Zhan Q, Ge Q, VanDyke T, Badwey JA. Protein phosphorylation in neutrophils monitored with phosphospecific antibodies. J. Immunol. Methods. 2003;281:79–94. doi: 10.1016/s0022-1759(03)00278-3. [DOI] [PubMed] [Google Scholar]
- 27.Ding Y, Kantarci A, Badwey JA, Hasturk H, Malabanan A, Van Dyke TE. Phosphorylation of pleckstrin increases proinflammatory cytokine secretion by mononuclear phagocytes in diabetes mellitus. J. Immunol. 2007;179:647–654. doi: 10.4049/jimmunol.179.1.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. Suicide inhibition of canine myocardial cytosolic calcium-independent phospholipase A2. Mechanism-based discrimination between calcium-dependent and -independent phospholipases A2. J. Biol. Chem. 1991;266:7227–7232. [PubMed] [Google Scholar]
- 29.Dana R, Leto TL, Malech HL, Levy R. Essential requirement of cytosolic phospholipase A2 for activation of the phagocyte NADPH oxidase. J. Biol. Chem. 1998;273:441–445. doi: 10.1074/jbc.273.1.441. [DOI] [PubMed] [Google Scholar]
- 30.Jenkins CM, Wolf MJ, Mancuso DJ, Gross RW. Identification of the calmodulin-binding domain of recombinant calcium-independent phospholipase A2beta. implications for structure and function. J. Biol. Chem. 2001;276:7129–7135. doi: 10.1074/jbc.M010439200. [DOI] [PubMed] [Google Scholar]
- 31.Balsinde J, Dennis EA. Distinct roles in signal transduction for each of the phospholipase A2 enzymes present in P388D1 macrophages. J. Biol. Chem. 1996;271:6758–6765. doi: 10.1074/jbc.271.12.6758. [DOI] [PubMed] [Google Scholar]
- 32.Underwood KW, Song C, Kriz RW, Chang XJ, Knopf JL, Lin L-L. A novel calcium-independent phospholipase A2, cPLA2-γ, that is prenylated and contains homology to cPLA2. J. Biol. Chem. 1998;273:21926–21932. doi: 10.1074/jbc.273.34.21926. [DOI] [PubMed] [Google Scholar]
- 33.Mollapour E, Linch DC, Roberts PJ. Activation and priming of neutrophil nicotinamide adenine dinucleotide phosphate oxidase and phospholipase A(2) are dissociated by inhibitors of the kinases p42(ERK2) and p38(SAPK) and by methyl arachidonyl fluorophosphonate, the dual inhibitor of cytosolic and calcium-independent phospholipase A(2) Blood. 2001;97:2469–2477. doi: 10.1182/blood.v97.8.2469. [DOI] [PubMed] [Google Scholar]
- 34.Henderson LM, Chappell JB. The NADPH-oxidase-associated H+ channel is opened by arachidonate. Biochem. J. 1992;283:171–175. doi: 10.1042/bj2830171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Serezani CH, Chung J, Ballinger MN, Moore BB, Aronoff DM, Peters-Golden M. Prostaglandin E2 suppresses bacterial killing in alveolar macrophages by inhibiting NADPH oxidase. Am. J. Respir. Cell Mol. Biol. 2007;37:562–570. doi: 10.1165/rcmb.2007-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Dowd YM, El-Benna J, Perianin A, Newsholme P. Inhibition of formyl-methionyl-leucyl-phenylalanine-stimulated respiratory burst in human neutrophils by adrenaline: inhibition of Phospholipase A2 activity but not p47phox phosphorylation and translocation. Biochem. Pharmacol. 2004;67:183–190. doi: 10.1016/j.bcp.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 37.Bauldry SA, Wooten RE. Induction of cytosolic phospholipase A2 activity by phosphatidic acid and diglycerides in permeabilized human neutrophils: interrelationship between phospholipases D and A2. Biochem. J. 1997;322:353–363. doi: 10.1042/bj3220353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magalhães MA, Zhu F, Sarantis H, Gray-Owen SD, Ellen RP, Glogauer M. Expression and translocation of fluorescent-tagged p21-activated kinase-binding domain and PH domain of protein kinase B during murine neutrophil chemotaxis. J. Leukoc. Biol. 2007;82:559–566. doi: 10.1189/jlb.0207126. [DOI] [PubMed] [Google Scholar]