

SUMMARY
FANCM remodels branched DNA structures and plays essential roles in the cellular response to DNA replication stress. Here we show that FANCM forms a conserved DNA remodeling complex with a histone-fold heterodimer, MHF. We find that MHF stimulates DNA binding and replication fork remodeling by FANCM. In the cell, FANCM and MHF are rapidly recruited to forks stalled by DNA interstrand crosslinks, and both are required for cellular resistance to such lesions. In vertebrates, FANCM-MHF associates with the Fanconi anemia (FA) core complex, promotes FANCD2 monoubiquitination in response to DNA damage, and suppresses sister-chromatid exchanges. Yeast orthologs of these proteins function together to resist MMS-induced DNA damage and promote gene conversion at blocked replication forks. Thus, FANCM-MHF is an essential DNA remodeling complex that protects replication forks from yeast to human.
Keywords: FANCM, Fanconi Anemia, DNA repair, Histone fold, CENP-S, CENP-X
INTRODUCTION
Fanconi anemia (FA) is a genetically heterogeneous syndrome characterized by genomic instability, congenital abnormalities, bone marrow failure, and cancer predisposition. FA can be caused by mutation in any of 13 different FANC genes. The hallmark of FA cells is their hypersensitivity to drugs that induce DNA interstrand crosslinks (ICLs), which prevent separation of the complementary strands of DNA and completely block replication and other essential processes. Increasing evidence suggests that FANC proteins act both as signal transducers and as DNA processing molecules in a DNA damage response network, the FA-BRCA network, which is essential to repair or bypass ICLs during DNA replication (Thompson and Hinz, 2009; Wang, 2007).
The FANC proteins can be divided into three groups, based on their participation at different stages of the FA-BRCA network (Wang, 2007). Group I acts at the early stage of the pathway and consists of eight FANC proteins, FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, and FANCM. These proteins, together with FAAP100 and FAAP24, constitute the FA core complex. The FA core complex has two main functions. First, it monoubiquitinates group II FA proteins, FANCD2 and FANCI, in response to DNA damage or during the S phase of the cell cycle; and second, it directly participates in DNA repair through a DNA remodeling enzyme, FANCM (Xue et al., 2008). Group II FANC proteins include FANCD2 and FANCI, which form the FANCI-FANCD2 complex. Once ubiquitinated, this complex associates with chromatin and colocalizes with the Group III FANC proteins, FANCD1/BRCA2, FANCN/PALB2 and FANCJ/BRIP1. The group III proteins all interact with BRCA1 and are thought to mediate HR-dependent DNA repair.
FANC proteins and FAAPs usually have orthologs in vertebrates but not in yeast. A single exception is FANCM, which not only has an ortholog in yeast, but also in archea (Wang, 2007). This implies that the DNA remodeling function of FANCM is important enough to be conserved through evolution. The yeast orthologs of FANCM, MPH1 in S. cerevisiae and Fml1 in S. pombe, possess a helicase motif and are capable of dissociating D-loops (an HR intermediate) and suppressing chromosome crossover recombination induced by double-strand breaks (Banerjee et al., 2008; Prakash et al., 2009; Sun et al., 2008). Fml1 can also stimulate replication fork reversal in vitro, and promote gene conversion at stalled replication forks in vivo (Sun et al., 2008).
Human FANCM contains an ATP-dependent branch-point translocase activity which promotes migration of Holliday junctions, replication fork reversal, and dissociation of D-loops (Gari et al., 2008a; Gari et al., 2008b; Xue et al., 2008). The ATP-dependent activity of FANCM is required for cellular resistance to DNA crosslinking agents, but is dispensable for the monoubiquitination function of the FA core complex (Rosado et al., 2009; Singh et al., 2009; Xue et al., 2008). FANCM binds specifically and with high affinity to Holliday junctions and replication forks (Gari et al., 2008b; Xue et al., 2008), and this DNA binding activity seems to be required for efficient monoubiquitination of FANCD2 (Xue et al., 2008). FANCM has a DNA-binding partner, FAAP24, which can target FANCM to single-strand DNA, an intermediate of both DNA replication and repair (Ciccia et al., 2007). The FANCM/FAAP24 dimer is needed to tolerate damage induced by UV and camptothecin, and for suppression of crossover recombination (Rosado et al., 2009; Singh et al., 2009).
Here, we describe a histone-fold protein complex, named MHF, as a new component of the FA core complex and an essential co-factor of FANCM. We present evidence that MHF and FANCM form a conserved DNA remodeling complex that maintains genomic stability from yeast to human.
RESULTS
Identification of MHF1 and MHF2 as integral components of the FA core complex
To search for additional components of the FA core complex, we fractionated HeLa nuclear extract by gel-filtration chromatography, collected peak fractions containing FANCM (peak 1 in Figure 1A), and immunoprecipitated with a FANCM antibody. Silver-staining (Figure 1B), mass spectrometry analyses (data not shown), and immunoblotting (Figure 1C) revealed the presence of all known components of the FA core complex (FANC-A,-B, -C, -E, -F, -G -L, -M, FAAP100 and FAAP24) in the FANCM immunoprecipitate. Most of the Bloom syndrome complex components (BLM, TOPIIIα, RMI1/BLAP75, RPA70, and RPA32) were also identified by mass spectrometry (data not shown), supporting our previous findings that the FA core and BLM complexes associate in a super-complex, BRAFT (Wang, 2007).
Figure 1. Identification of MHF1 and MHF2 as integral components of the FA core complex.

(A) Immunoblotting showing Superose 6 gel-filtration profiles of FANCM, MHF1 and MHF2 in HeLa nuclear extract. The peak fractions and those corresponding to marker proteins are indicated at the bottom. (B) A silver-stained gel showing that the complex purified by a FANCM antibody from the pooled peak 1 fractions in (A) contained MHF1, MHF2 and other components of FA core and BLM complexes. IP indicates immunoprecipitation. (C) Immunoblotting shows that MHF1, MHF2 and other FA core complex components were present in the immunoprecipitates isolated from peak 1 fractions by MHF1, FANCM or FANCA antibodies. Nuclear extract (NE) was used as a loading control. (D) As described in (B), except a MHF1 antibody was used in IP. (E) Immunoblotting shows that MHF2 co-immunoprecipitated with MHF1 and other FA core complex components from HeLa cells stably expressing Flag-tagged MHF2, but not from control HeLa cells. A Flag antibody was used in IP. (F) As described in (B), except that peak 2 fractions in (A) were used for IP by a MHF1 antibody. (see also Figures S1 and S2).
Silver-staining detected two polypeptides of about 16 and 10 kDa that were not previously identified (Figure 1B). Mass spectrometry revealed the 16 kDa protein as CENP-S or APITD1 (gene accession ID: NP_954988) (Foltz et al., 2006; Krona et al., 2004), and the 10 kDa polypeptide as a protein with accession ID of A8MT69 or CENP-X (Amano et al., 2009). A8MT69 has a confusing name of STRA13, which refers to two distinct proteins: one is A8MT69, and the other is an unrelated helix-loop-helix transcription factor. Of 56 published articles on STRA13, only one is on A8MT69, which shows that deletion of the A8MT69 ortholog in fission yeast may result in sensitivity to several genotoxins (Deshpande et al., 2009). To avoid the confusion, we renamed the 16 and 10 kDa polypeptides as MHF1 and MHF2, respectively (for FANCM (or MPH1)-associated Histone Fold protein 1 or 2).
The following evidence indicates that MHF1 and MHF2 are components of the FA core complex. First, immunoblotting showed that antibody against MHF1 or MHF2 recognized the corresponding polypeptide in the immunoprecipitate obtained with either FANCM or FANCA antibody (Figure 1C). Second, one peak of MHF1 or MHF2 on Superose 6 column is coincident with that of FANCM, supporting the notion that they are present in the same FA core complex (Figure 1A, peak 1). Third, reciprocal immunoprecipitation using a MHF1 antibody from the pooled Superose fractions of peak 1 obtained the same set of polypeptides isolated by the FANCM antibody, including components of both FA core and BLM complexes, as evidenced by silver-staining (Figure 1D), mass spectrometry (data not shown), and immunoblotting (Figure 1C). Finally, MHF2-associated polypeptides isolated by a Flag antibody from the extract of HeLa cells stably expressing Flag-tagged MHF2 also contained MHF1, FANCM and other FA core complex components (Figure 1E).
We noticed that MHF1 and MHF2 co-fractionate in two peaks on a Superose 6 column (Figure 1A). While peak 1 corresponds to the FA core complex of about 1 MDa, peak 2 corresponds to a complex of much smaller size. When peak 2 fractions were immunoprecipitated with a MHF1 antibody, only MHF1 and MHF2 were isolated (Figure 1F), as revealed by mass spectrometry (data not shown), indicating that these two proteins could comprise a complex distinct from the FA core complex. The two MHF proteins appear to be approximately equimolar amounts on the silver-stained gel, suggesting that they are likely obligate partners. Direct interaction between MHF1 and MHF2 was observed by mammalian two-hybrid analyses (Figure S1).
MHF1 and MHF2 are conserved from human to yeast and form a new histone-fold complex
Bioinformatics revealed that both MHF proteins contain a histone-fold, which can mediate both protein-protein and protein-DNA interactions (Figure S2A and B) (Arents and Moudrianakis, 1993). Proteins containing this motif often associate to form heterodimeric or heterotetrameric complexes that bind to bent DNA. Our purification of MHF1 and MHF2 as a stoichiometric complex from HeLa extract (Figure 1F) fits well with the bioinformatic prediction that they form a histone-fold complex.
We co-expressed MHF1 with HIS-tagged MHF2 in E. coli, and purified the complex using Talon metal affinity chromatography. Coomassie-staining revealed that the two proteins were present in approximately equimolar amounts (Figure 2A, lane 3), indicating that they indeed form a heterodimeric complex. We named this complex MHF.
Figure 2. MHF binds double-strand and branch-structured DNAs, but not single-strand DNA.
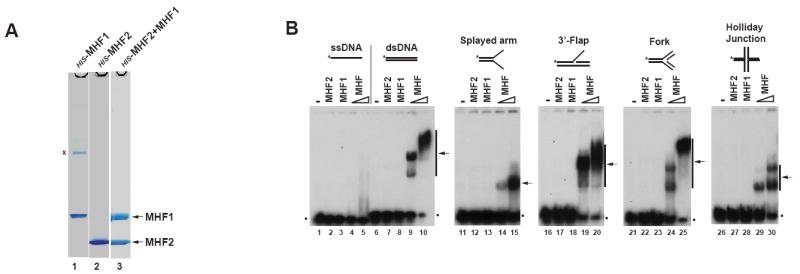
(A) Coomassie blue-stained SDS-PAGE gels showing HIS-tagged recombinant proteins MHF1, MHF2 and MHF complex purified from E.coli. A nonspecific band was marked with “x”. (B) Electrophoretic mobility shift assay (EMSA) for testing DNA binding activity of MHF1, MHF2 and MHF complex. A variety of DNA substrates (0.5nM) illustrated at the top were incubated with 0.6 μM MHF1, 0.6 μM MHF2 and increasing amounts (0.3 and 0.6μM) of MHF complex, respectively. Asterisks denote 32P label at the DNA 5’ end. The arrows indicate the shift bands of MHF-DNA complex. The dots represent free DNA probe. (see also Figure S3).
MHF possesses DNA binding properties distinct from FANCM and FAAP24
Like other histone-fold complexes, MHF was found to bind double-strand DNA (dsDNA), but not single-strand DNA (ssDNA) (Figure 2B, lanes 1-10). This activity requires the MHF complex because individual MHF subunit lacked the activity.
MHF was also found to bind several structured DNAs containing different branch points (Figure 2B, lanes 11-30). Notably, the affinity of MHF to these structures was either similar or somewhat reduced compared to that of dsDNA of the same length (compare lanes 10, 15, 20, 25 and 30), indicating that MHF has no increased affinity for branched DNA, and its binding may even be precluded by such structures. The binding of MHF to dsDNA was further visualized by electron microscopy (Figure S3A and B). MHF appeared to form clusters which result in compaction of DNA, suggesting self-association between MHF proteins.
The DNA binding characteristics of MHF differ from those of FANCM and FAAP24, which specifically recognize branched and ssDNA, respectively (Ciccia et al., 2007; Gari et al., 2008b; Xue et al., 2008). We propose that these proteins constitute a molecular machine that binds cooperatively to different parts of a stalled replication fork: FANCM binds the branch point, whereas MHF and FAAP24 associate with dsDNA and ssDNA regions, respectively (see Discussion). Moreover, we have also shown that MHF can bind chromatin (Figure S3C) and cooperate with histone octamers to assemble into nucleosome structures in vitro (Figure S3D and E), which is consistent with MHF aiding FANCM association with DNA in vivo. Furthermore, MHF can efficiently anneal complementary single-stranded DNAs (albeit not when they are pre-bound by RPA) (Figure S3F and G), which could assist the catalysis of branch point migration by FANCM.
MHF and FANCM co-evolved and form an independent complex
Bioinformatics analyses revealed that MHF1 and MHF2 orthologs are present in all eukaryotes, including yeast (Figure S2C and D). This feature is shared by FANCM but not by other FANC proteins or FAAPs, most of which have orthologs only in vertebrates (Wang, 2007). The data suggest that FANCM, MHF1 and MHF2 may perform functions important to all eukaryotes that favor their co-evolution.
The co-evolution of FANCM and the two MHF proteins implies that they may constitute a complex that lacks other FANC proteins. To distinguish this complex from the FA core complex in HeLa cells, we omitted the Superose 6 fractionation step that was used for enrichment and purification of the FA core complex (Figure 1D). We performed immunoprecipitation directly from HeLa extract with the same MHF1 antibody, and obtained FANCM, MHF1 and MHF2 as the only three major polypeptides (Figure 3A). Other FANC proteins can only be detected by immunoblotting due to their lower levels (data not shown). The data suggest that significant amounts of MHF and FANCM are present in a complex largely free of other FANC proteins. We named this complex FANCM-MHF.
Figure 3. MHF stimulates DNA binding and fork reversal activities of FANCM.
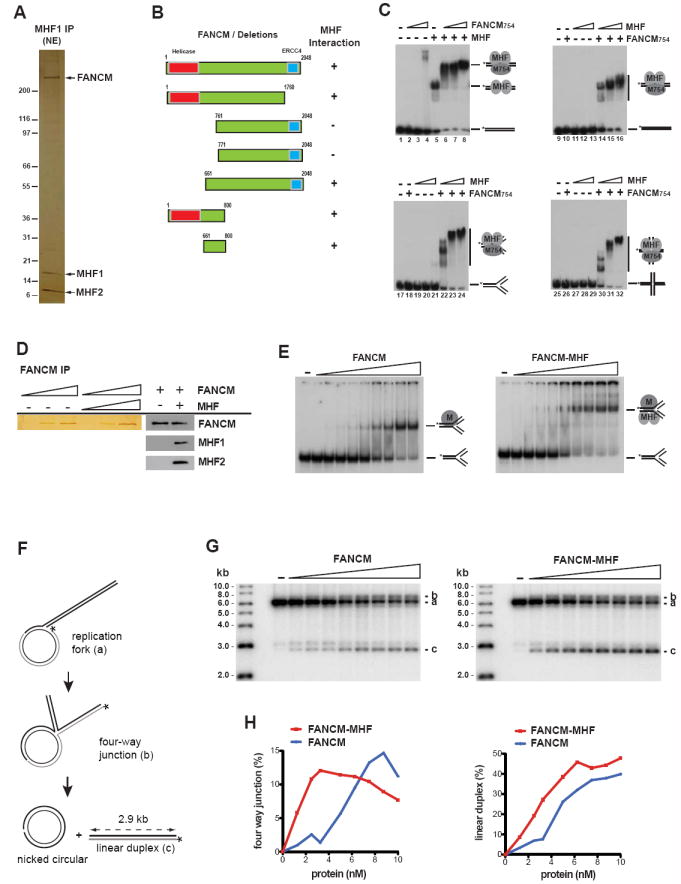
(A) A silver-stained gel showing FANCM-MHF complex immunoprecipitated directly by a MHF1 antibody from HeLa nuclear extract (NE). Three major polypeptides were identified as FANCM, MHF1 and MHF2 by mass spectrometric analyses. (B) A diagram shows the mapping of MHF-interaction domain within FANCM. Left panels show FLAG-tagged wild type and various deletion mutants of FANCM used in Figure S4D. Right panels show the presence or absence of interactions between various FANCM constructs and MHF. (C) EMSA showing the DNA binding activity of MHF and FANCM754. The reaction contained the DNA substrate (0.5nM) with or without recombinant FANCM754 protein or MHF complex as indicated. The protein concentrations are: FANCM754: 22.5, 45 and 90 nM in lanes 2 to 4 and 6 to 8, respectively; MHF: 300 nM in lanes 5 to 8; FANCM754: 15nM in lanes 10, 14, 15, 16, 18, 22, 23, 24, 26, 30, 31, 32; MHF: 50, 100 and 150nM in lanes 11 to 13, 14 to 16, 19 to 21, 22 to 24, 27 to 29, and 30 to 32, respectively. The shifted bands of indicated protein-DNA complex and free DNA probe are illustrated. (D) A silver-stained gel showing Flag-FANCM purified from baculovirs-infected insect cells (Sf21), either alone or in association with the MHF complex (left panels). At these protein concentrations, MHF proteins were hardly detectable by silver- staining due to their low molecular mass. Western blots confirmed that MHF1 and MHF2 were co-immunoprecipitated with Flag-FANCM (right panels). (E) Comparison of the DNA binding activity of purified FANCM and FANCM-MHF complex on fork DNA substrate by EMSA. 5’-32P-labeled synthetic replication forks (0.5 nM) were incubated for 30 min at room temperature with increasing concentrations of FANCM or FANCM-MHF (0, 0.125, 0.25, 0.5, 1, 2, 4, 6, 8, 10 nM). MHF does not bind this substrate at 10nM. (F) Experimental scheme. Fork reversal converts the replication fork into a four-way junction (b). Complete fork reversal dismantles the joint molecules into nicked circular and labeled linear duplex (c). (G) Increasing concentrations of Flag-FANCM or Flag-FANCM-MHF (0, 0.125, 0.25, 0.5, 1, 2, 4, 6, 8, 10 nM) were incubated with the replication fork substrate (0.5 nM) for one hour at 37°C. The different labeled species are (top down) the four-way junction intermediate (b), the original replication fork substrate (a), and the linear duplex end product of fork reversal (c). (H) Product formation shown in (G) was quantified by PhosphorImaging and expressed as percentage of total DNA. (see also Figure S4).
Majority of MHF and FANCM do not associate with the FA core complex
We quantitatively immunodepleted the FA core complex from HeLa extract with the FANCA antibody, and found that less than 30% of FANCM, MHF, and FAAP24 were co-depleted (Figure S4A), Similarly, depletion of FAAP24 or MHF from the extract co-depleted FANCA by less than 25% (Figure S4B and C). These data suggest that majority of FANCM and its two partners do not associate with the FA core complex. Conversely, immunodepletion of either FAAP24 or MHF co-depleted FANCM by about 70% and 85% (Figure S4B and C), indicating that most of FANCM in cells associates with either or both of its partners.
MHF and FAAP24 can form separate complexes with FANCM
We immunoprecipitated MHF from the FAAP24-depleted extract and obtained FANCM but no FAAP24 (Figure S4B, lane 4), suggesting that MHF and FANCM can form a complex without FAAP24. Similarly, we immunoprecipitated FAAP24 from the MHF-depleted extract and obtained FANCM but no MHF (Figure S4C, lane 4), indicating FAAP24 and FANCM can also form a complex without MHF. Together with the results in Figure 1 which showed that FANCM and both partners co-IP, our data suggest that FANCM can associate with its partners in a combinatorial manner to form distinct complexes: FANCM-MHF, FANCM-FAAP24, and FANCM-MHF-FAAP24.
MHF interacts with FANCM and promotes its DNA binding activity
We mapped the MHF-interaction domain within FANCM to a region near the helicase domain of FANCM (aa. 661-800) by IP-Western analyses of a series of FANCM deletion mutants (Figures 3B and S4D). We have previously shown that both the full-length FANCM protein and its N-terminal fragment (1-754 aa.) encompassing the helicase domain (FANCM754) have high affinity for branched DNA structures, but not for dsDNA (Gari et al., 2008b; Xue et al., 2008). The current findings that MHF interacts with both FANCM and dsDNA predict that MHF may recruit FANCM to dsDNA. Consistent with this prediction, FANCM754 alone exhibited little dsDNA binding activity (Figure 3C, lanes 2-4), while MHF and FANCM754 together displayed strong binding activity (see the supershifted band in Figure 3C, lanes 6 to 8 vs. 5).
FANCM and MHF bind DNA synergistically
The fact that FANCM and the two MHF proteins can form a complex raised a possibility that they may bind DNA cooperatively. Indeed, not only did MHF enhance the DNA binding of FANCM754, FANCM754 also stimulated the DNA binding activity of MHF (Figure 3C; the unbound DNA was reduced in lane 6 compared to lane 5). Moreover, FANCM754 and MHF bound DNA synergistically at low protein concentrations: while either protein alone showed little binding activity, both proteins together displayed an activity much higher than the sum of individuals (Figure 3C; less than 1% of dsDNA was shifted in lanes 10-13, whereas 50-90% of DNA was shifted in lanes 14-16). This synergy was also observed for fork and Holliday junction (HJ) substrates (Figure 3C, lanes 17 to 32).
We reconstituted the FANCM-MHF complex by co-expressing full-length recombinant FANCM, MHF1 and MHF2 proteins in insect cells, and purified the trimeric complex (Figure 3D). Incubation of FANCM-MHF with synthetic replication forks led to formation of a defined protein-DNA complex whose mobility was reduced compared to that of FANCM-fork complex, suggesting that FANCM, MHF1 and MHF2 bind together to branched DNA molecules (Figure 3E). Moreover, the FANCM-MHF complex had a stronger fork binding activity than FANCM alone (Figure 3E), providing further evidence that FANCM-MHF binds DNA cooperatively.
MHF stimulates replication fork reversal activity of FANCM
FANCM exhibits an ATP-dependent replication fork reversal activity, which may stabilize stalled forks and facilitate assembly of DNA damage signaling and repair complexes (Gari et al., 2008a). We found that recombinant FANCM-MHF had a fork reversal activity stronger than that of FANCM (Figure 3F, G and H). Both FANCM and FANCM-MHF catalyzed reversal of a model replication fork into a four-way junction intermediate, and led to the formation of a labeled linear DNA duplex, the end product of complete fork reversal (Figure 3G and H). At low protein concentrations, however, FANCM-MHF produced higher levels of four-way junction intermediate (about 5 to 7 -fold) and linear regression product (about 2 to 3 -fold) than FANCM alone (Figure 3G and H). These data establish FANCM-MHF as a DNA remodeling machine, and suggest that MHF is a crucial co-factor for FANCM in both binding and ATP-dependent remodeling of DNA.
MHF is required for stability of FANCM and for activation of the FA pathway
We next examined whether MHF is required for FANCM function in vivo. SiRNA depletion of either MHF1 or MHF2 in HeLa cells reduced the level of FANCM in whole cell lysates (Figure 4A) and the chromatin fractions (Figure 4B), suggesting that MHF is required for stability of FANCM and may also be important for chromatin association of FANCM. Depletion of one MHF protein also reduced the protein level of the other (Figure 4A), providing additional evidence that MHF1 and MHF2 are direct interacting partners and depend on each other for stability.
Figure 4. MHF is required for stability of FANCM, activation of the FA pathway and cellular resistance to DNA damaging agents.

(A and B) Immunoblotting shows that depletion of MHF1 or MHF2 reduces the level of FANCM in whole cell lysates (A) and chromatin fractions (B) of HeLa cells. Nontargeting siRNA oligos were used as a control. α-Tubulin, Histone H3 and Actin were included as loading controls. (C and D) Immunoblotting shows that HeLa cells depleted of MHF1 (C) or MHF2 (D) have reduced levels of monoubiquitinated FANCD2 and FANCI in the presence of MMC (60 ng/ml) or cisplatin (5 μM)(Cis). “L” (long) and “S” (short) represent ubiquitinated and non-ubiquitinated forms, respectively. The ratio between long and short forms was obtained by using KODAK Molecular Imaging Software and shown below the blots. (E, F and G) Clonogenic survival assays of HeLa cells depleted of MHF1, MHF2 or FANCM by siRNA following the treatment with MMC, MMS or CPT at the indicated concentrations. Three independent experiments were performed. The results were reproducible, and a representative data of mean surviving percentage with S.E.M. from triplicate cultures are shown. (see also Figure S5).
HeLa cells depleted of either MHF protein displayed reduced levels of monoubiquitinated FANCD2 and FANCI in response to DNA crosslinking drugs, mitomycin C (MMC) and cisplatin (Figure 4C and D). The cells also exhibited hypersensitivity to these drugs (Figures 4E and S5A), and increased chromosomal breaks in response to MMC (Figure S5B). All these phenotypes are characteristics for cells defective in FA core complex components, indicating that MHF is important for normal functions of the core complex and the FA pathway.
We noticed that HeLa cells depleted of FANCM or MHF were sensitive to methyl methanesulfonate (MMS), a DNA alkylating agent (Figure 4F). This feature has been found in hamster cells mutated of FANCG (Tebbs et al., 2005), and in yeast FANCM (Banerjee et al., 2008; Sun et al., 2008) and MHF mutants (see below), suggesting that some DNA repair functions of FANCM-MHF are conserved in lower eukaryotes.
Cells depleted of MHF also exhibited sensitivity to camptothecin (CPT), a topoisomerase I inhibitor (Figure 4G). This feature was observed in cells depleted of FANCM but not in those lacking other components of the FA core complex (Rosado et al., 2009; Singh et al., 2009). The results suggest that FANCM-MHF may have functions independent of the FA core complex.
MHF and FANCM are rapidly recruited to DNA interstrand crosslinks (ICLs) that block replication
Because cells depleted of FANCM or MHF are hypersensitive to drugs that induce ICLs, we examined whether FANCM-MHF localizes at ICLs using two independent techniques. First, we utilized laser-activated psoralen conjugates to generate ICLs within a localized region in nucleus, and then visualize proteins recruited to this region by indirect immunofluorescence (Thazhathveetil et al., 2007). Both FANCM and MHF1 were recruited to the ICLs within 15 min after photoactivation, in a subgroup of cells in random culture (about 15%) (Figure 5A and data not shown), while they were not recruited to DNA monoadducts generated by laser-activated angelicin (Thazhathveetil et al., 2007). Co-staining with a cell cycle marker (NPAT) revealed that the recruitment occurred only in S phase cells (Figure S6A, B, C, and data not shown). When cells were synchronized in S phase, the recruitment was increased to approximately 50% of the cell population (Figure S6D). The fact that the recruitment of FANCM and MHF to ICLs occurs only during S phase provides in vivo evidence for FANCM-MHF to remodel replication forks blocked by crosslinked DNA. Notably, the recruitment of FANCM (but not γ-H2AX) was strongly diminished in cells depleted of either MHF1 or MHF2 (Figure 5B and C). This may be due to reduced stability (Figure 4A) and/or impaired targeting of FANCM in the absence of MHF.
Figure 5. MHF is rapidly recruited to ICL sites in S phage cells and required for FANCM recruitment.

(A) Images showing that MHF1 and FANCM were recruited to psoralen-induced ICLs beginning 15 mins after laser photoactivation in S phase cells. The yellow arrow indicates the position of the laser-targeted region. (B) Images showing that HeLa cells depleted of MHF1 or MHF2 by siRNA have deficient recruitment of FANCM to psoralen-induced ICLs. Nontargeting siRNA oligos and γ-H2AX were used as controls. Images represent 15 mins post treatment time point. The recruitment of FANCM and γ-H2AX to ICL sites was indicated by yellow and white arrows, respectively. (C) Graph showing the percentages of cells in which FANCM was recruited to psoralen-induced ICLs at various time points after photoactivation. No recruitment of FANCM to ICLs was observed in MHF2-depleted cells at 30 mins post treatment. HeLa cells were either transfected with siRNA against MHF1, MHF2 or control siRNA and synchronized in S phase prior to the experiment. Values are averages from two independent experiments with error bars representing standard error of the mean. (D) Schematic representation of the plasmid substrates used in the eCHIP assay. The presence of psoralen-ICL is indicated. Cells expressing EBNA (+) support replication of the plasmid, whereas those lacking it (-) do not. (E) Recruitment of MHF1 to the site of ICL was detected by eChIP. The enrichment of MHF1 and FAAP24 at the ICL in replicating (293EBNA) and nonreplicating (HEK293) DNA substrates was reflected by their relative recruitment ratio compared to the control substrate without ICL. The relative recruitment was derived by normalizing the comparative concentration (from real-time PCR) of crosslinked substrate to that of the unmodified control substrate. Error bars represent standard deviation from three independent experiments. (see also Figure S6).
We also used eChIP, a chromatin-IP based method that detects proteins at a site-specific psoralen ICL on an episomal plasmid transfected into cells (Figure 5D)(Shen et al., 2009). This plasmid contains the replication origin of Epstein-bar virus (EBV), so that the plasmid without the ICL can undergo replication unidirectionally in EBNA-293 cells that express the Epstein-bar nuclear antigen 1 (EBNA). For the plasmid carrying the ICL (which was positioned 488 bp downstream of the replication origin), the replication fork is stalled by the crosslink, and proteins accumulated at the stalled fork can be detected by ChIP-PCR using a primer set that amplifies a DNA fragment near the crosslink. The same plasmid can be introduced into standard 293 cells that lack EBNA for detection of proteins recruited to ICL under non-replicating conditions. We found that MHF1 was enriched about 5-fold at the ICL when the episomal vector was allowed to replicate in EBNA-293 cells (Figure 5E). However, the enrichment was reduced to less than 2-fold in the 293 cells that do not support vector replication. In comparison, FAAP24 was enriched at the ICL under both replicating and non-replicating conditions (Figure 5E), in agreement with previous findings (Shen et al., 2009). These data are congruent with the notion that MHF functions at the replication forks blocked by ICLs, most likely in the form of the FANCM-MHF complex.
MHF and FANCM act in the same pathway for FANCD2 monoubiquitination and suppression of sister-chromatid exchange (SCE)
We have screened FA patients for mutations in MHF1 and MHF2, but have failed to identify such individuals (data not shown). To study the functions of MHF genetically, we inactivated MHF1 in chicken DT40 cells (Figure S7A, B and C). Compared to wildtype cells, MHF1-/- cells exhibited a lower level of FANCM and MHF2, a reduced level of monoubiquitinated FANCD2 (Figure 6A, lanes 1-2), and a decreased number of FANCD2 nuclear foci (Figure S7D and E). Introduction of human MHF1 into MHF1-/- cells restored FANCD2 monoubiquitination, and also resulted in over expression of FANCM (2-fold) and MHF2 (11-fold) compared to wildtype cells (Figure 6A, lanes 1-3). These findings are consistent with siRNA data from HeLa cells that MHF1 is required for normal FANCD2 monoubiquitination and for stability of FANCM and MHF2.
Figure 6. MHF and FANCM act in the same pathway for FANCD2 monoubiquitination and suppression of SCE in chicken DT40 Cells.
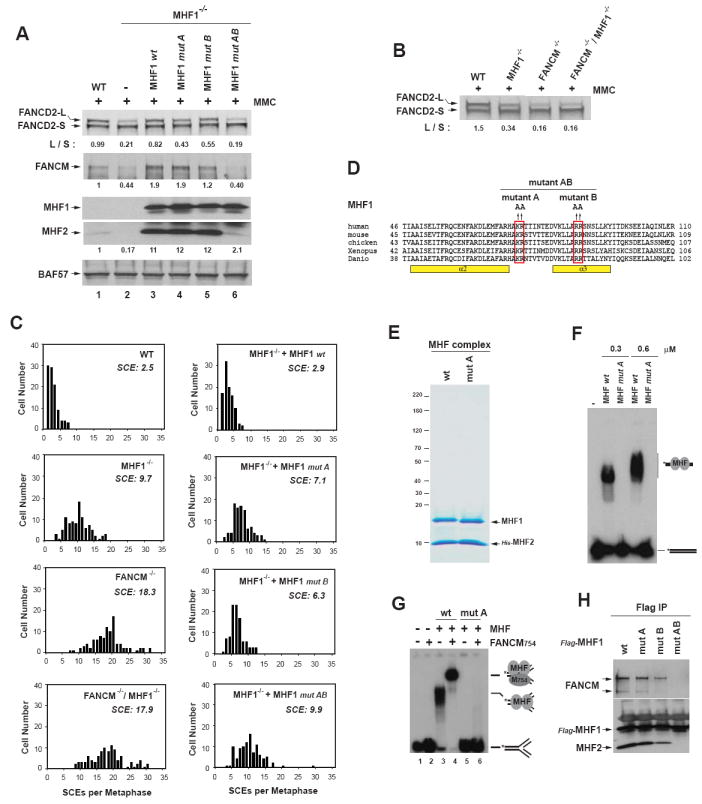
(A) Immunoblotting shows levels of FANCD2, FANCM, and MHF2 in whole cell lysates from various DT40 cells (wildtype (WT), MHF1-/- cells, and MHF1-/- cells complemented with human wildtype (wt) or mutant MHF1 as described in (D)). The ratio between the monoubiquitinated and unubiquitinated FANCD2 (L/S) was shown. Cells were treated with MMC (50 ng/ml) for 18 hr. BAF57 was used as a loading control. (B) Immunoblotting shows levels of monoubiquitinated and unubiquitinated FANCD2 in whole cell lysates from various DT40 cells as indicated on the top. (C) Histograms showing spontaneous SCE levels of various DT40 cells as indicated within each graph. MHF1-/- cells transfected with human MHF1 wildtype (wt) or mutants (described in D) were also shown. The mean number of SCEs per metaphase is listed. (D) Illustration of three human MHF1 point mutants generated by substituting 2 clusters of positively-charged amino acid residues (KR and RR) to alanine as indicated. (E) A Coomassie blue-stained SDS-PAGE gel shows the purified recombinant MHF complex containing MHF1 of either wildtype (wt) or mutant A (mut A). (F) Comparison of the DNA binding activity of MHF complexes containing either wildtype or mutant A of MHF1 by EMSA. Reactions contain 32P-labeled dsDNA substrate (0.5nM) and 0.3 or 0.6μM of the MHF complex as indicated. (G) Comparison of the DNA binding activity of FANCM754 with either wildtype (wt) or mutant A MHF by EMSA. The reaction contained the 32P-labeled fork DNA substrate (0.5nM) with or without FANCM754 protein or MHF complex (wt or mut A). The protein concentrations are: FANCM754: 22.5nM (lanes 2, 4 and 6); MHF: 450 nM (lanes 3 and 5) and 300 nM (lanes 4 and 6). (H) IP-Western analyses show association between FANCM and various Flag-tagged MHF1 point mutants in HEK293 cells. (see also Figure S7).
We generated FANCM-/-/MHF1-/--double mutant cells (data not shown), and found that they contained the levels of monoubiquitinated FANCD2 comparable to that of FANCM-/- cells in the presence of MMC (Figure 6B). These results suggest that MHF and FANCM act in a common pathway to promote efficient monoubiquitination of FANCD2.
DT40 cells inactivated of FANC genes exhibit higher levels of SCEs (Rosado et al., 2009). The level of SCEs in MHF1-/- cells was about 3 to 4-fold higher than that of wildtype cells (9.7 vs. 2.5), and this elevated SCE level could be corrected by expression of human MHF1 (Figure 6C). The data suggest that MHF participates in suppression of SCEs in DT40 cells. The SCE level in MHF1-/- cells is lower than that of FANCM-/- cells (9.7 vs. 18.3), suggesting that without MHF, FANCM remains partially active in maintaining genome integrity (Figure 6C). The SCE level of FANCM-/-/MHF1-/- cells is comparable to that of FANCM-/- cells (17.9 vs. 18.3), indicating that MHF and FANCM act through the same pathway to suppress SCEs.
MHF1-/- DT40 cells lacked cellular sensitivity and chromosomal breakage in response to DNA ICL drugs (data not shown), which are phenotypes of DT40 cells inactivated of FANC genes. These results differ from those of siRNA studies in HeLa cells. Possibly, the balance between DNA repair and cell death pathways may be different between these cells.
The DNA binding activity of MHF is required for normal FANCD2 monoubiquitination and full suppression of SCE
To study whether the DNA binding activity of MHF is required for its function in vivo, we generated three MHF1 point mutants by substituting 2 clusters of conserved positively charged amino acid residues with alanine: mutant A (K73A/R74A), B (R87A/R88A), and AB (K73A/R74A/R87A/R88A) (Figure 6D). Mutagenesis of similar residues in other histone-fold proteins can disrupt protein-DNA interactions (Hori et al., 2008). We co-expressed these mutants with MHF2 in E.coli, and found that only mutant A can be co-purified with MHF2 in a stable complex (Figure 6E and data not shown). Notably, the recombinant complex containing mutant A lacked dsDNA binding activity (Figure 6F), and also failed to recruit FANCM to fork DNA (Figure 6G, compare lanes 4 and 6), indicating that MHF requires its DNA binding activity to recruit FANCM to DNA. Co-IP analyses in HEK293 cells showed that mutant A retained normal association with MHF2 and FANCM (Figure 6H). Importantly, MHF1-/- DT40 cells stably expressing mutant A had a lower level of monoubiquitinated FANCD2 (Figure 6A, lanes 3-4), and a higher SCE frequency (Figure 6C) than cells transfected with wildtype MHF1, suggesting that the DNA binding activity of MHF is required for normal FANCD2 monoubiquitination and full suppression of SCE. Compared to MHF1-/- null cells, the mutant A-expressing MHF1-/- cells reproducibly exhibited a higher level of monoubiquitinated FANCD2 and a lower SCE frequency (Figure 6A, lanes 2 and 4; 6C), indicating that this mutant remains partially functional even though it lacks the ability to bind DNA. This partial function might be due to the ability of mutant A to stabilize FANCM and MHF2, as the latter two proteins were recovered to levels higher than not only those of MHF1-/- null cells, but also those of wildtype cells (Figure 6A, lanes 1, 2 and 4). The findings that mutant A-complemented cells had a higher than normal amount of FANCM but still exhibited abnormal FANCD2 monoubiquitination and SCE frequency suggest that the stabilization and over expression of FANCM cannot substitute the function of MHF in vivo.
The interaction between MHF and FANCM is essential for FANCM stability
We also analyzed MHF1 mutant B and AB using the same assays. These mutants cannot be co-purified with MHF2 in a stable complex from E.coli (data not shown), indicating that protein-interactions between MHF1-MHF2 were impaired. Consistent with such an impairment, co-IP in HEK293 cells showed that mutant B was partially defective in association with MHF2 and FANCM, whereas mutant AB was completely deficient (Figure 6H). Notably, the levels of FANCM and MHF2 in the mutant B-complemented MHF1-/- DT40 cells were restored to near those of cells expressing wildtype MHF1 or mutant A (Figure 6A). This is in contrast to mutant AB-complemented MHF1-/- cells, in which the levels of FANCM and MHF2 were not restored and similar to those of MHF1-/- null cells, suggesting that the stability of these two proteins strongly depends on their interactions with MHF1.
The levels of monoubiquitinated FANCD2 and SCE in mutant B-complemented MHF1-/- cells were found to be intermediate between MHF1-/- null cells and those expressing wildtype MHF1 (Figure 6A and C). In contrast, the levels in the mutant AB-complemented MHF1-/- cells were indistinguishable to those of the MHF1-/- null cells. The degree of defects in FANCD2 monoubiquitination and SCE by the two mutants appears to correlate with their ability to stabilize FANCM. Together with the data of mutant A, the results suggest that MHF has two important activities: binding to DNA and stabilizing FANCM by protein-protein interactions. Only when both activities are inactivated (as in mutant AB), MHF completely loses its ability to promote FANCD2 monoubiquitination and suppress SCEs.
Budding yeast MHF and FANCM orthologs work in the same pathway in resistance to MMS-induced DNA damage
We investigated whether yeast orthologs of MHF (Figure S2C and D) and FANCM work together in DNA repair like their vertebrate counterparts. We disrupted budding yeast orthologs of MHF1 and MHF2 in a srs2 mutant background, and found that both mutants displayed hypersensitivity to MMS (Figure 7A, middle panels, compare mhfΔ srs2Δ doubles with srs2Δ single mutant). This feature resembles that of a mph1Δ mutant, which was previously shown to have MMS hypersensitivity in combination with a srs2Δ mutant (Banerjee et al., 2008) (Figure 7A). The mhf1Δ mph1Δ or mhf2Δ mph1Δ double mutants displayed MMS hypersensitivity similar to that of the mph1Δ single mutant in the srs2Δ background (Figure 7A, bottom panels). Furthermore, the survival frequencies of the triple mutant strains at multiple MMS concentrations were statistically indistinguishable from srs2Δ mph1Δ (Figure 7B). Collectively, these results indicate that both MHF proteins are epistatic with Mph1 in tolerating MMS-induced DNA damage. In addition, a mhf1Δmhf2Δ double mutant exhibited MMS hypersensitivity indistinguishable from each mhfΔ single mutant in the srs2Δ background (Figure 7A, middle panels), consistent with the evidence in mammalian cells that the two proteins work in the same complex. The MMS resistance of the mhf1Δ or mhf2Δ mutant is weaker than that of the mph1Δ mutant, reminiscent of results in DT40 cells where the SCE level of the MHF1-/- mutant is lower than that of the FANCM-/- mutant (Figure 6C). FANCM or MPH1 may therefore be partially functional without MHF. Taken together, these data support the findings in mammalian cells and indicate that the MHF proteins play an evolutionarily conserved role--cooperating with MPH1/FANCM to protect genome integrity.
Figure 7. Yeast MHF and FANCM orthologs work in the same pathway to resist MMS-induced DNA damage and promote gene conversion at stalled replication forks.
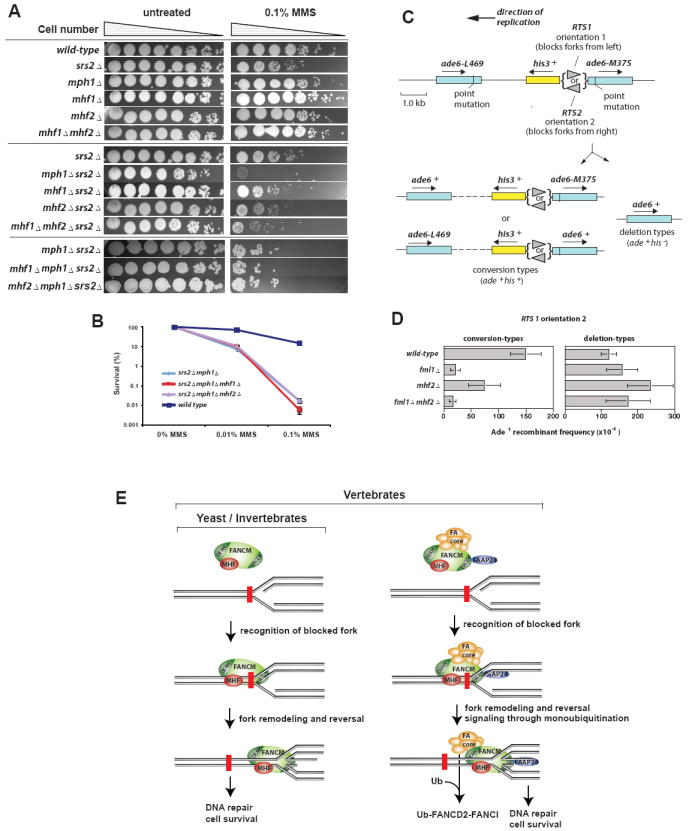
(A) Qualitative spot tests for strain sensitivities to MMS reveal a common role and shared pathway for Mph1, Mhf1, and Mhf2 in DNA damage resistance. (B) Quantitative analysis of MMS sensitivities shows that the triple mutants are not statistically distinguishable from srs2Δ mph1Δ at two different MMS concentrations. P values for the mhf1Δ and mhf2Δ triple mutants, respectively, versus srs2Δ mph1Δ are as follows: 0.35, 0.23 (0.01% MMS); 0.36, 0.97 (0.1% MMS). (C) Schematic showing the direct repeat recombination substrate on chromosome 3 of S. pombe plus the two types of Ade+ recombinants (Sun et al., 2008). (D) Ade+ recombinant frequencies for various strains as indicated. Error bars are the standard deviations. (E) A model describes common and unique functions of FANCM-MHF and FA core complex in signaling and repairing of blocked replication forks. The former acts in yeast, invertebrates and vertebrates, whereas the latter functions only in vertebrates. While both complexes can promote reversal of blocked forks (indicated by the red brick) to allow subsequent repair, only the FA core complex has a signaling role in monoubiquitinating FANCD2 and FANCI.
MHF2 and FANCM act in the same pathway to promote gene conversion at stalled replication forks in fission yeast
The FANCM ortholog in S. pombe, Fml1, promotes Rad51-dependent gene conversion at blocked replication forks (Sun et al., 2008). To investigate if MHF is also required, we deleted the mhf2 gene and assessed what impact this had on the frequency of Ade+ recombinants induced by a polar replication fork barrier (RTS1) positioned between a direct repeat of ade6- heteroalleles on chromosome 3 of S. pombe (Figure 7C). This region is replicated unidirectionally, and consequently only orientation 2 of RTS1 blocks replication between the repeats. The block of replication results in a strong induction of Ade+ recombinants, which arise both from Rad51-dependent gene conversion, and Rad51-dependent and independent deletions (Sun et al., 2008). A His3+ gene positioned between the repeats enables these two types of recombinants to be distinguished: Ade+His3+ are conversion-types, whereas Ade+His3- are deletion-types. In a mhf2Δ mutant, the frequency of conversion-types in the absence of replication fork blockage at RTS1 (i.e. when RTS1 is in orientation 1) is not significantly different from wild-type (data not shown). However, when RTS1 is in orientation 2 and replication forks are blocked, the frequency of conversion-types is reduced approximately two-fold in a mhf2Δ mutant compared to wild-type (Figure 7D, left panel). This reduction is statistically significant (P = <0.0001), but is less than with the fml1Δ mutant, which shows a 7-fold reduction. Possibly, Fml1 retains some ability to promote gene conversion without MHF. Importantly the frequency of conversion-types in a fml1Δ mhf2Δ double mutant is the same as in a fml1Δ single mutant, indicating that Mhf2 acts in the same pathway to promote gene conversion at blocked replication forks as Fml1 does.
Intriguingly, the loss of conversion-types in the mhf2Δ mutant is accompanied by an approximately two-fold increase in deletion-types (Figure 7D, right panel), which again is statistically significant (P = <0.0001). This increase is dependent on Fml1 as it is suppressed in a fml1Δ mhf2Δ double mutant (P = <0.01). These data suggest that MHF may act to prevent Fml1 mediated Rad51-dependent strand invasion events from giving rise to deletions.
DISCUSSION
A network of DNA caretaker proteins keeps replication forks under surveillance to prevent gross chromosomal rearrangements in S phase. Here we have identified a conserved DNA remodeling complex that protects replication forks in eukaryotes. This complex contains FANCM, a branched DNA binding and remodeling enzyme, as well as two histone fold proteins, MHF1 and MHF2. In vertebrates, MHF1 and MHF2 are bona fide components of the FA core complex, and are required for normal activation of the FA pathway, including optimal monoubiquitination of the FANCI-FANCD2 complex in response to DNA damage, cellular resistance to DNA crosslinking drugs, and prevention of chromosomal breakage. MHF and FANCM also constitute a FANC-independent complex, FANCM-MHF, which is rapidly recruited to blocked forks in vivo. Our genetic analyses in yeast suggest that FANCM-MHF is a conserved complex that promotes gene conversion at blocked replication forks, in contrast to the FA core complex, which exists only in vertebrates (Figure 7E).
MHF is a histone fold complex that functions in DNA damage response and at centromeres
Histone fold proteins can facilitate transcription (TAF octamer in TFIID), chromatin-remodeling (Chrac14/Chrac16 in the CHRAC complex), and stabilization of centromeres (CENP-T/CENP-W). Our finding that MHF functions with FANCM extends the participation of histone-fold complexes to the DNA damage response. While our manuscript was under review, Amano and colleagues described a CENP-S complex that is identical to MHF, with MHF1 and MHF2 being CENP-S and CENP-X, respectively (Amano et al., 2009). They showed that this complex is essential for the stable assembly of kinetochore structure and normal mitosis. Centromeric DNA is intrinsically difficult to replicate and rich in replication pause sites. MHF may target FANCM at centromeres to forestall the disastrous encounter of replication forks with secondary DNA structures and/or tightly DNA bound protein complexes.
FANCM and MHF constitute a conserved DNA remodeling complex
MHF1 and MHF2 have yeast orthologs that are genetically epistatic with the FANCM orthologs in resistance to MMS and promotion of gene conversion at stalled forks. This leads us to propose that FANCM and the two MHF proteins constitute a core complex that remodels DNA structure in yeast and higher eukaryotes. In accord with this hypothesis, human MHF and FANCM can be co-immunoprecipitated as a complex that is largely free of the other FA core complex components. The FANCM-MHF complex can be reconstituted by baculovirus-expressed proteins, and the resulting complex possesses DNA binding and fork-reversal activities stronger than FANCM alone. Both FANCM and MHF are rapidly recruited to ICLs specifically in S phase, and the recruitment of MHF1 to ICLs is stimulated by DNA replication. Moreover, depletion of either FANCM or MHF in HeLa cells results in cellular sensitivity to DNA crosslinking drugs, which blocks progression of replication forks. Furthermore, FML1 and MHF mutants in S. pombe are defective in promoting gene conversion at replication fork barriers. These findings suggest that FANCM-MHF may stabilize and remodel blocked replication forks to facilitate DNA damage signaling and repair.
How might MHF contribute to the remodeling activity of FANCM-MHF? MHF may anchor the complex to DNA damage sites in chromatin. Consistent with this, MHF can bind in vitro assembled chromatin template and its depletion reduces the level of FANCM in the chromatin fraction and abrogates recruitment of FANCM to laser-induced ICLs. A MHF point mutant unable to bind dsDNA is defective in recruiting FANCM to DNA in vitro, and fails to promote normal FANCD2 monoubiquitination and fully suppress SCE in DT40 cells. Our biochemical studies suggest that at stalled forks, FANCM most likely binds the DNA branch point of replication intermediates (Gari et al., 2008b; Xue et al., 2008), whereas MHF would associate with surrounding dsDNA. Binding of MHF to dsDNA may help to recruit and orient FANCM at the branch point. In the FA core complex, FAAP24 may further facilitate this process by anchoring FANCM to the surrounding ssDNA region of stalled forks. After FANCM is recruited to stalled forks, MHF may facilitate repair or bypass of the stalled forks by stimulating fork reversal activity of FANCM. This stimulation may be independent of the role of MHF in FANCD2 monoubiquitination, which has been shown not to require remodeling activity of FANCM (Rosado et al., 2009; Singh et al., 2009; Xue et al., 2008).
In a cellular context, MHF may play an additional role in stabilizing FANCM via protein-protein interactions. This was revealed because the MHF mutant A is only partially defective in FANCD2 monoubiquitination and SCE suppression despite inactivation of its DNA binding activity. We found a strong correlation between FANCM stability and its ability to associate with various MHF point mutants. MHF mutant A has normal association with FANCM and can fully stabilize FANCM as the wildtype protein. Mutant B has reduced association with FANCM and cannot restore FANCM to the level by the wildtype protein. Mutant AB is completely defective in FANCM association so that it fails to stabilize FANCM. Notably, mutant AB which lacks both DNA binding activity and FANCM association is completely deficient in promoting normal FANCD2 monoubiquitination and suppressing SCE. These data suggest that MHF requires both DNA-binding and FANCM association to function in vivo.
FANCM-MHF and FA core complex may have independent functions
In vertebrates, MHF also plays a signaling role as part of the FA core complex, as its depletion or inactivation of its DNA binding activity reduces FANCD2 monoubiquitination in response to DNA damage. A recent report shows that FANCD2 monoubiquitination in vitro requires only FANCL but not other subunits of the FA core complex (Alpi et al., 2008), suggesting that the role of MHF in this reaction is indirect. A previous study suggested that the DNA binding activity of FANCM is required for FANCD2 monoubiquitination (Xue et al., 2008), possibly by anchoring FA core complex to chromatin (Kim et al., 2008). Our findings that MHF stabilizes FANCM and cooperates with FANCM to bind DNA suggest that MHF indirectly contributes to this process by stimulating recruitment of FANCM and associated FA core complex to chromatin.
In addition to their differential roles in FANCD2 monoubiquitination, FA core complex and FANCM-MHF appear to have other independent functions. For example, HeLa cells lacking MHF or FANCM display MMS and camptothecin hypersensitivity, a cellular feature absent in human cells deficient of other FA core complex components. Moreover, FANCM and MHF are epistatic in suppression of SCEs in DT40 cells, whereas FANCM and FANCC are not (Rosado et al., 2009).
In vertebrates, FANCM-MHF associates with not only components of the FA core complex, but also subunits of the BLM complex (BLM, Topo IIIa, RMI and RPA). FANCM and BLM suppress SCE through the same pathway in DT40 cells (Rosado et al., 2009). These results reinforce our earlier findings that FANC and BLM complexes work together in the BRAFT super-complex to protect genome integrity (Wang, 2007). Elucidation of the structure and function of this large complex should shed new light on the mechanism of replication fork surveillance.
EXPERIMENTAL PROCEDURES
Gel filtration, immunoprecipitation and protein identification
Superose 6 gel filtration and immunoprecipitation were performed as described (Ciccia et al., 2007). The eluted immunoprecipitates were resolved by 8-16%Tris-Glycine gel (InVitrogen), visualized by silver-staining, and were subjected to mass spectrometry analyses.
Fork regression assay
The plasmid-based replication fork reversal assay was described in (Gari et al., 2008a).
Protein Recruitment to laser-induced localized ICLs
We followed the previous protocol to detect proteins recruited at laser-induced localized ICLs (Thazhathveetil et al., 2007).
Detecting proteins recruited to a site-specific psoralen-ICL by eChIP
The eChIP was carried out as described (Shen et al., 2009).
Budding yeast strains and MMS sensitivity assay
We followed the previous protocols used for Mph1 study to generate Mhf mutants and performed MMS sensitivity assay (Banerjee et al., 2008).
S. pombe strains and recombination assay
Strain construction and the direct repeat recombination assay has followed published protocols (Sun et al., 2008).
Detailed Experimental materials and methods can be found in Supplemental Information.
Supplementary Material
Acknowledgments
We thank Y. Xue for providing recombinant FANCM754 protein, Drs. KJ Patel for FANCM-/- cells and antibody, M. Takata for chicken FANCD2 antibody, Y. Li for FAAP24 antibody, and D. Schlessinger for critical reading of the manuscript. This work was supported in part by the Intramural Research Program of the National Institute on Aging (AG000688-07), National Institute of Health (HL007781 and CA112775), the Fanconi Anemia Research Foundation and Swiss National Science Foundation (3100A0-116275). A.C. was supported by a Swiss National Science Foundation Professorship (PP00A-118991). MCW and FO were supported by a Wellcome Trust Senior Research Fellowship to MCW.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alpi AF, Pace PE, Babu MM, Patel KJ. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol Cell. 2008;32:767–777. doi: 10.1016/j.molcel.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Amano M, Suzuki A, Hori T, Backer C, Okawa K, Cheeseman IM, Fukagawa T. The CENP-S complex is essential for the stable assembly of outer kinetochore structure. J Cell Biol. 2009;186:173–182. doi: 10.1083/jcb.200903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arents G, Moudrianakis EN. Topography of the histone octamer surface: repeating structural motifs utilized in the docking of nucleosomal DNA. Proc Natl Acad Sci U S A. 1993;90:10489–10493. doi: 10.1073/pnas.90.22.10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Smith S, Oum JH, Liaw HJ, Hwang JY, Sikdar N, Motegi A, Lee SE, Myung K. Mph1p promotes gross chromosomal rearrangement through partial inhibition of homologous recombination. J Cell Biol. 2008;181:1083–1093. doi: 10.1083/jcb.200711146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, Laghmani el H, Joenje H, McDonald N, de Winter JP, et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol Cell. 2007;25:331–343. doi: 10.1016/j.molcel.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Deshpande GP, Hayles J, Hoe KL, Kim DU, Park HO, Hartsuiker E. Screening a genome-wide S. pombe deletion library identifies novel genes and pathways involved in genome stability maintenance. DNA Repair (Amst) 2009;8:672–679. doi: 10.1016/j.dnarep.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Black BE, Bailey AO, Yates JR, 3rd, Cleveland DW. The human CENP-A centromeric nucleosome-associated complex. Nat Cell Biol. 2006;8:458–469. doi: 10.1038/ncb1397. [DOI] [PubMed] [Google Scholar]
- Gari K, Decaillet C, Delannoy M, Wu L, Constantinou A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci U S A. 2008a;105:16107–16112. doi: 10.1073/pnas.0804777105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari K, Decaillet C, Stasiak AZ, Stasiak A, Constantinou A. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol Cell. 2008b;29:141–148. doi: 10.1016/j.molcel.2007.11.032. [DOI] [PubMed] [Google Scholar]
- Hori T, Amano M, Suzuki A, Backer CB, Welburn JP, Dong Y, McEwen BF, Shang WH, Suzuki E, Okawa K, et al. CCAN makes multiple contacts with centromeric DNA to provide distinct pathways to the outer kinetochore. Cell. 2008;135:1039–1052. doi: 10.1016/j.cell.2008.10.019. [DOI] [PubMed] [Google Scholar]
- Kim JM, Kee Y, Gurtan A, D’Andrea AD. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood. 2008;111:5215–5222. doi: 10.1182/blood-2007-09-113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krona C, Ejeskar K, Caren H, Abel F, Sjoberg RM, Martinsson T. A novel 1p36.2 located gene, APITD1, with tumour-suppressive properties and a putative p53-binding domain, shows low expression in neuroblastoma tumours. Br J Cancer. 2004;91:1119–1130. doi: 10.1038/sj.bjc.6602083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, Satory D, Dray E, Papusha A, Scheller J, Kramer W, Krejci L, Klein H, Haber JE, Sung P, Ira G. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 2009;23:67–79. doi: 10.1101/gad.1737809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado IV, Niedzwiedz W, Alpi AF, Patel KJ. The Walker B motif in avian FANCM is required to limit sister chromatid exchanges but is dispensable for DNA crosslink repair. Nucleic Acids Res. 2009;37:4360–4370. doi: 10.1093/nar/gkp365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Do H, Li Y, Chung W-Y, Tomasz M, de Winter JP, Xia B, Elledge SJ, Wang W, Li L. Recruitment of Fanconi Anemia and breast cancer proteins to DNA damage sites is differentially governed by replication. Mol Cell. 2009;35:716–723. doi: 10.1016/j.molcel.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh TR, Bakker ST, Agarwal S, Jansen M, Grassman E, Godthelp BC, Ali AM, Du CH, Rooimans MA, Fan Q, et al. Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia complementation group M. Blood. 2009;114:174–180. doi: 10.1182/blood-2009-02-207811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Nandi S, Osman F, Ahn JS, Jakovleska J, Lorenz A, Whitby MC. The FANCM ortholog Fml1 promotes recombination at stalled replication forks and limits crossing over during DNA double-strand break repair. Mol Cell. 2008;32:118–128. doi: 10.1016/j.molcel.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebbs RS, Hinz JM, Yamada NA, Wilson JB, Salazar EP, Thomas CB, Jones IM, Jones NJ, Thompson LH. New insights into the Fanconi anemia pathway from an isogenic FancG hamster CHO mutant. DNA Repair (Amst) 2005;4:11–22. doi: 10.1016/j.dnarep.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Thazhathveetil AK, Liu ST, Indig FE, Seidman MM. Psoralen conjugates for visualization of genomic interstrand cross-links localized by laser photoactivation. Bioconjug Chem. 2007;18:431–437. doi: 10.1021/bc060309t. [DOI] [PubMed] [Google Scholar]
- Thompson LH, Hinz JM. Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: Mechanistic insights. Mutat Res. 2009;668:54–72. doi: 10.1016/j.mrfmmm.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- Xue Y, Li Y, Guo R, Ling C, Wang W. FANCM of the Fanconi anemia core complex is required for both monoubiquitination and DNA repair. Hum Mol Genet. 2008;17:1641–1652. doi: 10.1093/hmg/ddn054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.