

Abstract
The microtubule-actin crosslinking factor 1 (MACF1) is a ubiquitous cytoskeletal linker protein with multiple spliced isoforms expressed in different tissues. The MACF1a isoform contains microtubule and actin binding regions and is expressed at high levels in the nervous system. Macf1−/− mice are early embryonic lethal and hence the role of MACF1 in the nervous system could not be determined. We have specifically knocked out MACF1a in the developing mouse nervous system using Cre/loxP technology. Mutant mice died within 24–36 hrs after birth of apparent respiratory distress. Their brains displayed a disorganized cerebral cortex with a mixed layer structure, heterotopia in the pyramidal layer of the hippocampus, disorganized thalamocortical and corticofugal fibers, and aplastic anterior and hippocampal commissures. Embryonic neurons showed a defect in traversing the cortical plate. Our data suggest a critical role for MACF1 in neuronal migration that is dependent on its ability to interact with both microfilaments and microtubules.
INTRODUCTION
Plakins are multi-domain proteins implicated in cytoskeletal regulation, intercellular interactions, and cell signaling (Ruhrberg and Watt, 1997; Sonnenberg and Liem, 2007). The Microtubule-Actin Crosslinking Factor 1 (Macf1) gene encodes large proteins capable of bridging microfilaments and microtubules (MTs) (Leung et al., 1999; Sun et al., 1999). The two major MACF1 isoforms characterized to date, MACF1a and MACF1b, result from alternative splicing (Leung et al., 1999; Lin et al., 2005). Both proteins contain N-terminal actin-binding domains (ABD), followed by plakin domains, extended spectrin repeat domains, EF-hand motifs, and C-terminal microtubule-binding domains (MTBDs) (Sonnenberg and Liem, 2007). The MACF1 ABD consists of two calponin homology (CH) domains and can bind actin filaments in mammalian cells, whereas the MTBD region can bind to and bundle MTs in transfected cells (Sun et al., 2001). MACF1b contains additional plakin repeats and associates with the Golgi apparatus (Lin et al., 2005). Alternative initiation of both MACF1a and MACF1b can result in additional isoforms (Jefferson et al., 2006).
MACF1 is expressed ubiquitously, with high levels detected in lung and nervous tissues (Leung et al., 1999). Standard gene knock-outs of MACF1 in mice have resulted in mutants that are embryonic lethal, with all embryos dead by E10. The early developmental defect was shown to be related to Wnt signaling. MACF1 associated with a complex containing several classic Wnt signaling molecules including axin, APC, β-catenin, and GSK3β. MACF1 may be necessary for the translocation of axin, β–catenin, and GSK3β to the plasma membrane in response to Wnt activation of frizzled/LRP5/6 and for the subsequent degradation of axin (Chen et al., 2006). MTs of primary Macf1-null fibroblasts showed skewed cytoplasmic trajectories and altered dynamic instability, resulting in uncoordinated migration in wounding assays. Rescue of these defects required both the ABD and MTBD domains (Kodama et al., 2003). In addition, MACF1 has been shown to regulate MT binding of the plus-end-tracking protein CLASP2, suggesting a role in MT stabilization and cell motility (Drabek et al., 2006). Consistent with such a role, a recent report shows that MACF1 coordinates MT, actin, and focal adhesion dynamics in migrating epithelial cells (Wu et al., 2008).
MACF1 homologs have been identified in Drosophila (shortstop, shot) and C. elegans (Vab10). Vab-10 mutants exhibit body morphology defects (Bosher et al., 2003). Mutations in shot are particularly interesting, since they result in multiple defects including axonal extension, dendrite morphology, epidermal muscle attachment, and tendon cell differentiation (Subramanian et al., 2003). Shot recruits EB1/APC to promote microtubule assembly at the muscle-tendon junction. The name shortstop was coined because the mutant phenotype showed a failure to extend motor and sensory neurons to their correct length and reach their target. The shot mutant axons were able to initiate extension and the morphology of the growth cone appeared normal. Shot is also required for the extension and elaboration of dendritic branches. The axonal extension defect can be rescued by a construct that consists only of the actin and microtubule binding domains. These studies suggest that the link between actin and microtubules is important in axon extension in Drosophila.
A plakin related to MACF1 is BPAG1 (bullous pemphigoid antigen 1). BPAG1 also has multiple isoforms, and BPAG1a has a similar domain structure to MACF1a. The dystonia musculorum (dt) mouse is a naturally occurring BPAG1 knock-out mouse that shows sensory neuron degeneration, apparently due to the absence of BPAG1a in these neurons (Brown et al., 1995; Goryunov et al., 2007). However, unlike the Drosophila shot mutants, there are no widespread abnormalities in the rest of the nervous system of the dt animals. MACF1 might be compensating for BPAG1a in other parts of the nervous system and it is therefore of interest to determine the specific role(s) that MACF1 might play in the nervous system.
Cortical development is a highly coordinated process of neuronal migration and differentiation (Hatten, 1999; Rakic, 1990). During embryonic development, neuronal progenitors give rise to newborn neurons in the ventricular zone. The immature neurons migrate along the processes of radial glia towards the pial surface and form a six-layer cortex in an inside-out fashion, with later-born neurons terminally differentiating in more outer layers (Kawauchi and Hoshino, 2008). Mutations in cytoskeletal elements have been described that perturb radial neuronal migration in various human disorders, including lissencephaly, subcortical band heterotopia (double cortex syndrome), and periventricular heterotopia (Gressens, 2006; Kerjan and Gleeson, 2007). A role for cytoskeletal linker proteins in these defects has not been previously reported.
Because of the early lethality of Macf1 knockout embryos, tissue-specific functions of MACF1 at later developmental stages remain unknown. In order to study MACF1 functions in the mouse nervous system, we generated a nervous-system-specific Macf1 knockout using the loxP/Cre technology. The mutant mice die shortly after birth and display multiple brain defects associated with impaired neuronal migration and axonal extension.
RESULTS
Macf1 inactivation in the nervous system
To generate a conditional Macf1 knockout allele, we floxed exons 6 and 7 of the Macf1 gene (Fig. 1A). These exons encode the second CH domain of the ABD region of the MACF1 proteins. Upon successfully targeting the gene with a neo-cassette-containing vector in ES cells (Fig. 1B), we eliminated the neo cassette in vivo by crossing Macf1-targeted females with E2a-Cre males (Fig. 1C). Floxed (F) allele-carrying progeny were backcrossed to WT mice to ensure that the F allele had been incorporated into the germline. From these crosses, we also obtained mice that were heterozygous for a completely recombined (R) allele. In mating these heterozygous mice, Macf1F/F and Macf1F/R mice were born at Mendelian frequencies and were apparently indistinguishable from WT littermates (data not shown). However, no Macf1R/R mice were born from Macf1+/R heterozygote crosses. The phenotype of Macf1R/R embryos was similar to the previously reported Macf1 knockout, with all homozygous embryos apparently dead by E10.5 (Fig. 1D). These data confirm that the deletion of exons 6 and 7 of the Macf1 gene in the entire embryo results in a phenotype similar to the previously described Macf1 knockout (Chen et al., 2006)
Figure 1.
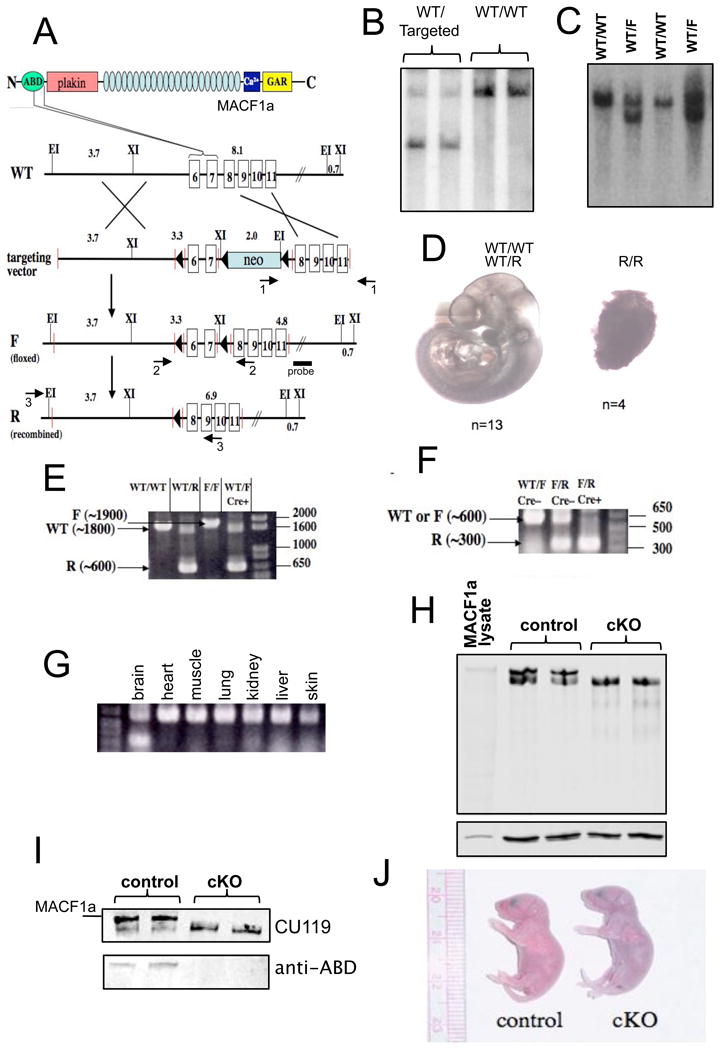
Generation of mutant Macf1 mice. (A) Schematic of the targeting strategy. Exons 6 and 7, encoding the C-terminal half of the MACF1 ABD, were floxed in the targeting construct. Recombination of this construct into the wild-type Macf1 locus resulted in a targeted Macf1 allele. The neo cassette was eliminated in vivo, resulting in a floxed allele (F). Tissue-specific Cre recombination gives rise to a fully recombined (R) allele that lacks the two exons. Primer pairs 1 and 2 were used for genotyping; pair 3, for RT-PCR. Position of the Southern blotting probe is indicated. Targeted ES cells (B) and floxed heterozygous animals (C) were genotyped by Southern blotting. Two mutant samples and 2 wild-type controls are shown in each panel. For fragment lengths see Materials and Methods. (D) Mice carrying two copies of the R allele die early in embryonic development. Two litters of E10.5 embryos resulting from Macf1+/R heterozygote matings were dissected. (E) F-to-R allele conversion in the brains of Macf1 mutant mice carrying a Cre transgene under the control of the nestin promoter was confirmed by genomic PCR. The WT, F, and R products are indicated with arrows. Note the R band, and the apparent absence of an F band, in the Macf1+/F;nes-Cre lane. RT-PCR confirmed the elimination of mRNA containing the ABD region in the brains of Macf1F/R;nes-Cre animals (F) but not in other tissues (G). (H) Western blotting of Macf1F/R (control) brain extracts with CU119 antibody revealed a high molecular weight doublet, with the top band co-migrating with plasmid-derived MACF1a (MACF1 lysate). The top band was absent in Macf1F/R;nes-Cre (cKO) brains. Lower panel shows tubulin as a loading control. (I) Re-probing the control and cKO brain extracts with a polyclonal antibody against the ABD (Antolik et al., 2007) showed that only the top band contains this domain, indicating that cKO brains no longer express MACF1a but still contain the ABD-less MACF1 protein, MACF1c. Position of plasmid-derived MACF1a is also indicated. (J) cKO mice did not have any gross abnormalities compared with control mice but died within 24–36 hrs after birth, apparently of respiratory distress. Note the cyanotic appearance of the mutant pup.
To inactivate the Macf1 gene in the nervous system, we used a mouse line that expresses Cre recombinase throughout the nervous system under the control of a nestin promoter (nestin-Cre) (Tronche et al., 1999). Macf1F/F females were crossed with Macf1+/R;nes-Cre males. Approximately 25% of the offspring from these crosses died at P0. The genotypes of these animals were invariably found to be Macf1F/R;nes-Cre. In the remainder of this article, we will refer to animals and tissues with this genotype as cKO (conditional knockout). Unless indicated otherwise, the genotype of control mice is Macf1F/R (Cre-negative).
The efficiency and specificity of the Cre-mediated gene alteration in the brain was confirmed by genomic PCR (Fig. 1E) and RT-PCR (Fig. 1F, G). Western blotting of WT and control brains with CU119, an antibody directed against the plakin domain of MACF1 (Lin et al., 2005), revealed a high-molecular weight doublet. The top band was the same size as MACF1a expressed ectopically in transfected cells (Leung et al., 1999) (Fig. 1H), corresponding to a protein of ~630 kDa, whereas the faster migrating band still had a molecular weight >500 kDa (the 200 kDa marker is at the bottom of the gel). Only the top band was absent in the cKO brains, whereas the lower band remained. Since our knock-out construct targeted exons in the ABD, the lower band could represent a previously unreported MACF1 isoform lacking the ABD that had an initiation site 5′ to the plakin domain and was otherwise identical to MACF1a. To test this possibility, we used a polyclonal antibody against the MACF1 ABD (Antolik et al., 2007). As expected, only the top band was detected in the control brains and neither band was observed in the cKO lysates (Fig. 1I). Mass spectrometry confirmed that the more rapidly migrating isoform did not contain any peptides corresponding to the ABD region, but contained peptides corresponding to other regions of MACF1a. We were not able to obtain an N-terminal sequence of this faster migrating isoform to determine the start site for this isoform. We will refer to this new isoform as MACF1c.
Most cKO newborns died in the first 24 hrs after birth, and all cKO mice died within 36 hrs of an apparent respiratory insufficiency. The animals started gasping, became cyanotic, and expired shortly while characteristically stretching backwards (Fig. 1J). They were able to nurse, as their stomachs contained milk. At birth, they exhibited no gross morphological defects and could not readily be distinguished from their wild-type littermates.
A common cause for an inability to breathe in newborn mice is a defect in the innervation of the diaphragm (Blanchi et al., 2003). We examined the phrenic nerves and acetylcholine receptor endplates of cKO diaphragms from newborn mice (P0) by staining them with a polyclonal neurofilament heavy chain antibody and α-bungarotoxin (Berg et al., 1972). We found no differences in the extent of phrenic nerve branching and endplate density between control and mutant animals (Supplemental Fig. 1), suggesting that diaphragm innervation and endplate formation were unaffected in the cKO mice.
Multiple neuronal developmental deficits in cKO brains
We examined the general structural anatomy of the cKO brain. For these and the following experiments, we analyzed a total of 8 pairs of mice from different litters. As noted before the control mice were Macf1F/R (Cre-negative). No differences were seen between wild-type and Macf1F/R (Cre-negative) mice. Hematoxylin/eosin staining of sagittal sections of E17.5 brains revealed a pronounced disorganization of the normal stratified structure in cKO cortices compared with control cortices (Fig. 2). The distinctive tri-laminar (cortical plate – subplate/intermediate zone – ventricular zone) appearance of the control cortex was perturbed in the cKO cortex, with both the cortical plate and ventricular zone appearing much less compact and the subplate region poorly defined (Fig. 2A, B). Abnormal accumulations of cells were present in the intermediate zone of both dorsal and ventral cerebrum in the cKO brain, suggestive of perturbations in the normal migration of cortical projection neurons and interneurons, respectively. The marginal and ventricular zones in the cKO cortices were thinner than in the control cortices (Fig. 2B, D). The overall thickness of the cortical well in the control and cKO cortices were similar but the cellular distribution across the cortex was more even in the cKO brain resulting in a blurring of compartmental boundaries (Supplemental Fig. 2).
Figure 2.
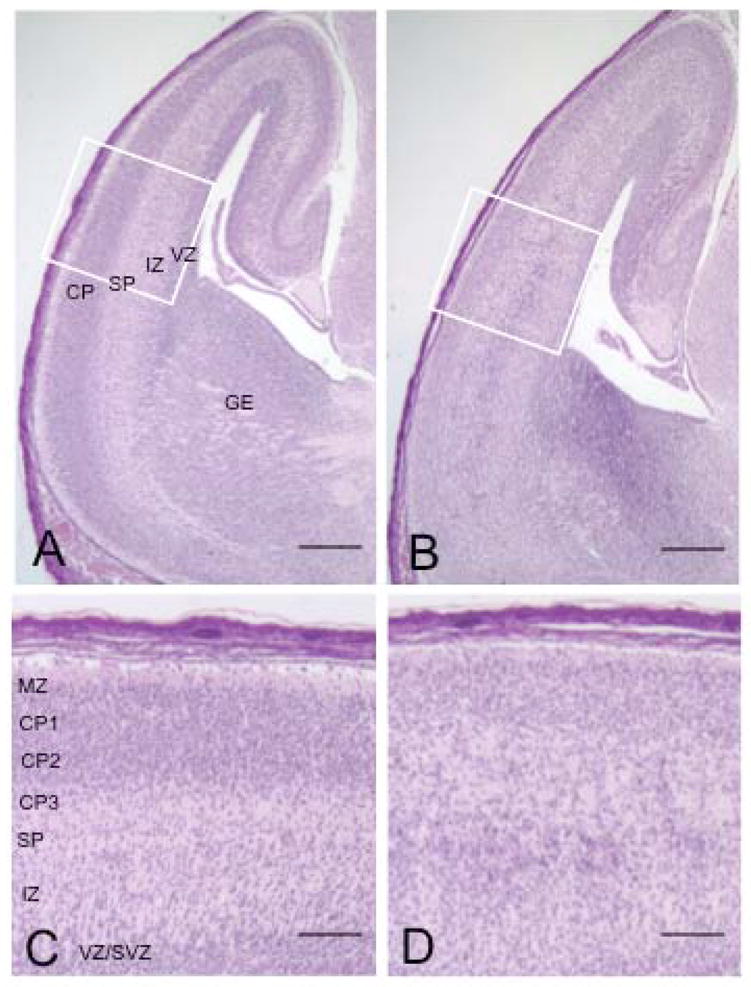
Disorganization of the cerebral cortex in Macf1 nestin-Cre animals. Sagittal sections of control (A, C) or mutant (B, D) E17.5 brains were stained with hematoxylin/eosin. The control cortex (A) contained a sharply defined cortical plate, a thin but distinct subplate, a low-cell-density intermediate zone, and a robust ventricular/subventricular zone. In contrast, the mutant cortex displayed a hypotrophic cortical plate, a reduced ventricular zone, and a loose band of cells positioned below the subplate in the normally clear intermediate zone (B). Higher magnifications also revealed a thinner marginal zone and a more even cell distribution across the cKO cortex compared with the control (C, D). CP, cortical plate; CP1–3, upper, middle, and lower tiers of the cortical plate; IZ, intermediate zone; MZ, marginal zone; SP, subplate; VZ/SVZ, ventricular/subventricular zones. Similar results were obtained from six different pairs of animals from different litters. Scale bars: A, B=250μm; C, D=50μm
Silver and Nissl staining of coronal P0 brain sections at different levels revealed additional morphological changes in cKO brains (Fig. 3). The lateral ventricles were smaller and shaped differently in the cKO animals (Fig. 3A, B) and the anterior commissure, a major route of axonal crossing between the hemispheres, was severely aplastic in the cKO brain compared to control brain (Fig. 3C, D). There appeared to be significantly fewer fibers in the corpus callosum of the cKO brain as compared to the control brain (Fig. 3E–H). The hippocampal commissure was also aplastic in cKO animals compared to controls (Fig. 4A, B, arrows). The thalamocortical fibers appeared much thinner in the cKO animals (Fig. 4A, B, arrowheads, and 4C, D). In the control brains, these fibers formed a distinct tract that extended along the ventral surface of the cortex (Fig. 4E), whereas the fibers in the cKO brains were less compact than in the control brains, with many axons extending tangentially towards the cortical surface (Fig. 4F). Staining with antibodies against α-internexin, a neuronal intermediate filament expressed in thalamocortical fibers (Fliegner et al., 1994; Kaplan et al., 1990) and TAG-1, a cell adhesion molecule expressed in corticofugal fibers (Denaxa et al., 2001) revealed a disorganization of both types of fibers and a more diffuse distribution in the cKO brains compared to the control brains (Fig. 4G, H). Taken together, these data indicate a possible defect in axonal extension and/or guidance in Macf1 cKO animals.
Figure 3.
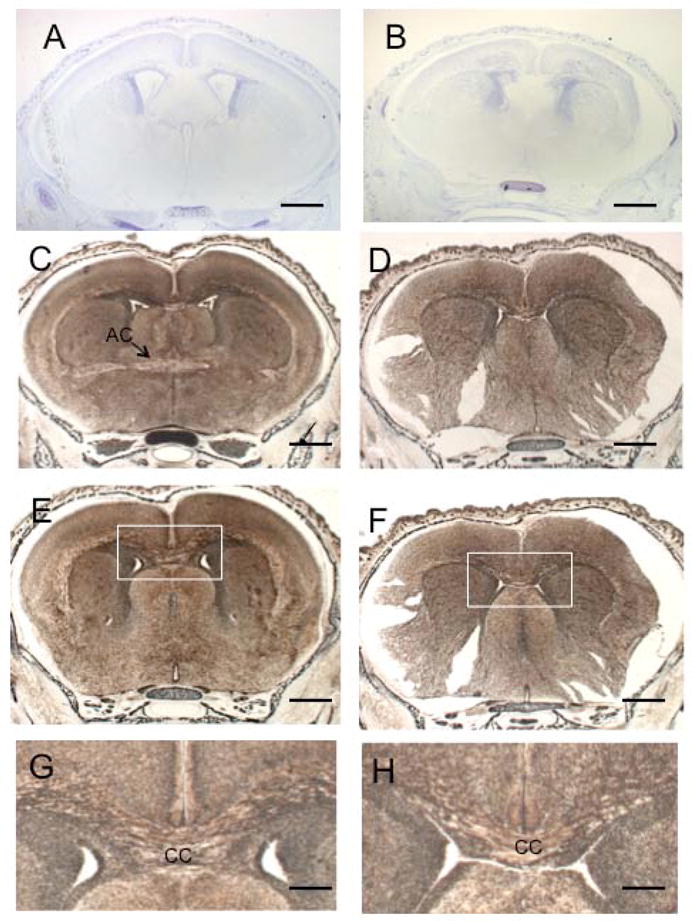
Multiple developmental defects in Macf1 nestin-cKO brains. (A, B) Control (A) or mutant (B) P0 coronal sections were stained with Nissl. The anterior commissure (AC) in the cKO brain was severely aplastic. The shapes of the lateral ventricles (LV) were altered. (C–H) Control (C, E, G) or mutant (D, F, H) P0 coronal sections were silver-stained. Note the anterior commissure in the control brain (C, arrow) and its apparent absence in the cKO brain (D). The corpus callosum (CC) was greatly reduced in the cKO brain (F, H) compared to the control (E, G). G and H show higher magnifications of the white box in E and F, respectively. Similar results were obtained from three different pairs of animals from different litters. Scale bars: A–F=500μm; G,H=50μm
Figure 4.
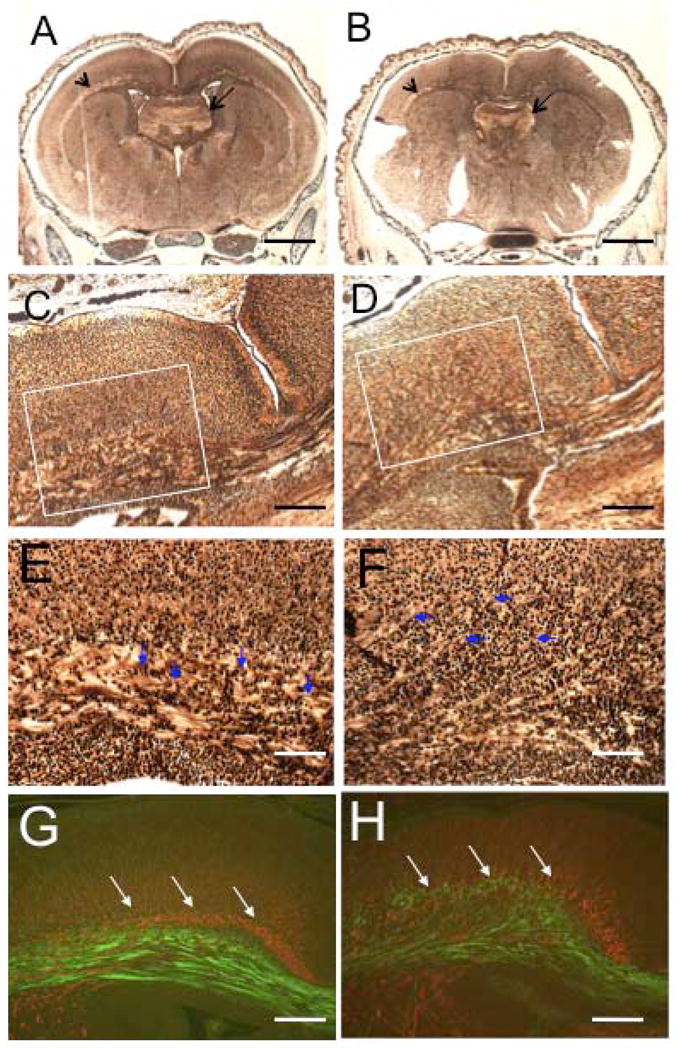
Thalamocortical fibers and the hippocampal commissure are abnormal in the cKO brain. (A–F) Silver-stained sections of control (A, C, E) and cKO (B, D, F) brains. The hippocampal commissure (arrows in A, B) and thalamocortical fibers (arrowheads in A, B) were significantly reduced in the cKO brains (B, D) compared to the control brains (A, C). E and F show higher magnifications of the white box in C and D, respectively. Note the much-looser structure of the cKO fibers as well as the individual axons extending tangentially into the cortical plate (E, F, arrowheads). Immunofluorescent staining of control (G) and cKO (H) sections with α-internexin and TAG-1 antibodies revealed a pronounced disorganization of the thalamocortical fibers (red) as well as corticofugal fibers (green) in the mutant brain. These results were seen with three pairs of animals from different litters. Scale bars:A, B=500μm; C, D, G, H=50μm; E, F=25μm
An abnormal laminar pattern was also evident in the hippocampus. The apparent depth of the pyramidal neuronal layer was greatly decreased in cKO brains compared with control brains (Fig. 5A,B). The cKO hippocampus also displayed striking heterotopia consisting of an additional pyramidal layer positioned on the outside of the primary pyramidal layer (Fig. 5A,B). These heterotopia were especially pronounced in the CA1–CA2 regions. This feature is consistent with a defect in migration of newborn pyramidal neurons from the ventricular zone, which normally occurs tangentially and also laterally in the CA1-to-CA3 direction. The pyramidal cell layer in more caudal regions of the cKO hippocampus displayed characteristic undulations absent in the control brains (Fig. 5C,D arrows). The dentate gyrus was also less compact in the cKO brain, further demonstrating defects in cell positioning within laminar structures in the brain.
Figure 5.
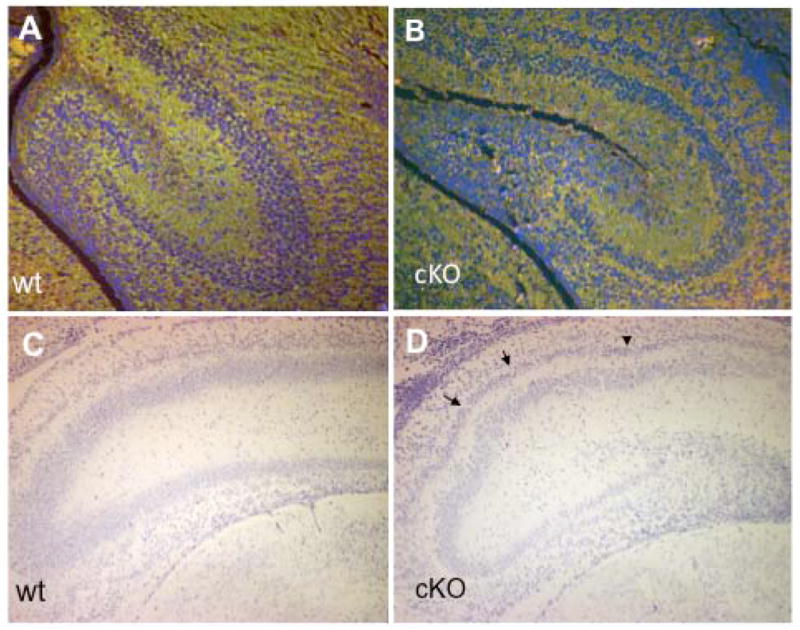
Pyramidal cell heterotopia in the cKO hippocampus. Control (A, C) or mutant (B, D) P0 coronal brain sections were triple-stained with CU119 antibody (green), monoclonal tubulin antibody (green), and Hoechst dye (blue; A,B), or stained with Nissl dye (C,D). Compared to the control hippocampus, the pyramidal cell layer in the mutant hippocampus was less compact and narrower (A, B). The mutant hippocampus also contained a second pyramidal layer (B, D). At a more caudal section level, the pyramidal layers in the cKO hippocampus showed undulations (D, arrowheads) that were absent in the control brain (C). Similar results were obtained from three pairs of animals from different litters. Scale bars: A–D=50μm
MACF1 protein expression in the cKO brain
To delineate the patterns of MACF1 isoform expression in normal and cKO brains, we studied coronal P0 brain sections by immunocytochemistry. In the cortices of control mice, CU119 antibody stained multiple populations of neuronal cell bodies and processes (Fig. 6A–C). In the cKO cortex, the intensity of the cell body staining was significantly diminished and the process staining was almost entirely eliminated (Fig. 6D–F). The anti-ABD antibody (Antolik et al., 2007) showed a similar staining pattern as CU119, with intense process staining and some cell body staining in the cortex of control mice, while no significant staining was detected in cKO cortical sections (Supplemental Fig. 3). These data confirm the immunoblotting data (Fig. 1H–I) and demonstrate that the MACF1a isoform is eliminated in the cKO cortex, but MACF1c is still present. These studies further confirmed the structural disorganization of the cKO cortex, with a disturbed cellular distribution and marked thinning of the marginal zone.
Figure 6.

MACF1 expression in the mouse cortex. Coronal sections of control (A–C) or Macf1 cKO (D–F) P0 brains were stained with CU119 polyclonal antibody (A, D), monoclonal tubulin antibody (B, E), and Hoechst (C, F). C, F, superimposed double images, with MACF1 staining in green and tubulin staining in red. Similar results were obtained from three pairs of animals from different litters. Scale bars: A–F=25μm
Immunostaining with CU119 antibodies revealed MACF1 reactivity throughout the hippocampi of control animals, with uniform staining detected in most cell bodies and processes of the pyramidal layer, as well as in the dentate gyrus. In the cKO hippocampi, CU119 staining was diminished but not eliminated (Supplemental Fig. 4). The anti-ABD antibody showed more intense staining in the processes of the pyramidal layer, as well as some cell body staining and this staining was almost entirely eliminated in the hippocampi of cKO mice (Supplemental Fig. 4).
Mixed layering of projection neurons in the cKO cortex
To determine the nature of the putative migration defect in the cKO cortex, we examined the expression of specific layer markers. The transcription factor T-brain 1 (Tbr1) is expressed in layer 6 neurons (Hevner et al., 2001). Another transcription factor, Ctip2 (COUP-TF interacting protein 2) is specifically upregulated in layer 5 neurons (Arlotta et al., 2005). Double immunostaining of control cortices revealed distinct adjacent layers of labeled cells, with virtually all Ctip2-postitive layer 5 neurons located peripherally to the Tbr1-positive layer 6 neurons (Fig. 7A). In contrast, the Ctip2 and Tbr1-positive layers partially overlapped in cKO cortices, albeit the relative positioning of the two layers was preserved (Fig. 7B). These data suggest that cortical layer identities are partially mixed in cKO animals, possibly because of a migration defect. Staining with Cux1 which labels the upper layers of the cortex (Nieto et al., 2004) also showed that the layers were mostly formed in the control mice, but that many Cux1 positive cells were still present in the intermediate zone (Fig. 7C, D). These studies also confirm that the cells in the intermediate zone shown in the hematoxylin/eosin staining (Fig. 2) are composed of late-born neurons that are delayed in migration. In the control hippocampi, Tbr1-positive cells were located in a broad band corresponding to the pyramidal cell layer, with a thin band of Ctip2-positive cells located on the periphery. In contrast, Tbr1 localized to the double pyramidal layer in the cKO hippocampi, whereas Ctip2 staining was found in only a few isolated cells scattered throughout both pyramidal layers (Fig. 7E,F).
Figure 7.
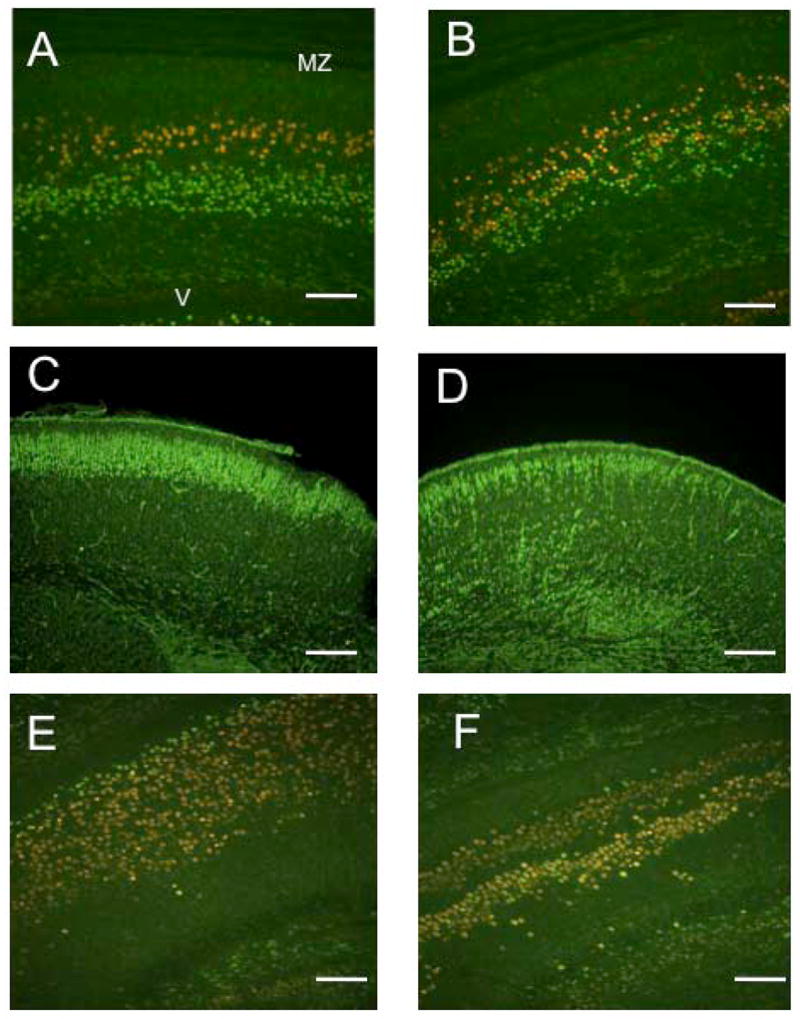
Cortical layers are mixed in the cKO brain. Control (A) or mutant (B) coronal sections were double-stained with Ctip2 (red) and Tbr1 (green) antibodies. In the control cortices, Ctip2 and Tbr1-positive neurons formed distinct layers. In the mutant cortices, the two layers were partially mixed. Staining with Cux-1, which labels cortical layers 2–4 in the control cortex (C), shows that in the mutant cortices (D), many Cux-1 positive cells have not completed migration. Similar to the cortex, staining with Ctip2 (red) and Tbr1 (green) antibodies of control (E) and mutant (F) hippocampal sections shows a mixing of the two layer markers and also confirms the heterotopia observed before. Similar results were obtained for three pairs of animals from different litters. Scale bars: A, B, E, F=50μm; C, D=100μm
To ascertain whether the preplate was split correctly in cKO cortices, we stained E17.5 cKO brain sections with reelin and CS56 antibodies. Reelin is a signaling molecule that is expressed by Cajal-Retzius cells in the marginal zone and guides the migration and final location of cortical projection neurons (D’Arcangelo et al., 1997). CS56 monoclonal antibody recognizes chondroitin sulfate proteoglycans (CSPGs) that are expressed in subplate neurons (Bicknese et al., 1994). CS56 revealed a deep layer of cells in both control and cKO cortices, although the mutant cortex also displayed significant staining in the ventricular zone (Fig. 8A, B, brackets). Reelin-positive cells were detected in the marginal zones of both control and cKO cortices (Fig. 8C, D, arrows). There was little reelin or CS56 staining in the cortical plates of either control or cKO cortices. These data indicate that the preplate is split correctly in the cKO cortex.
Figure 8.
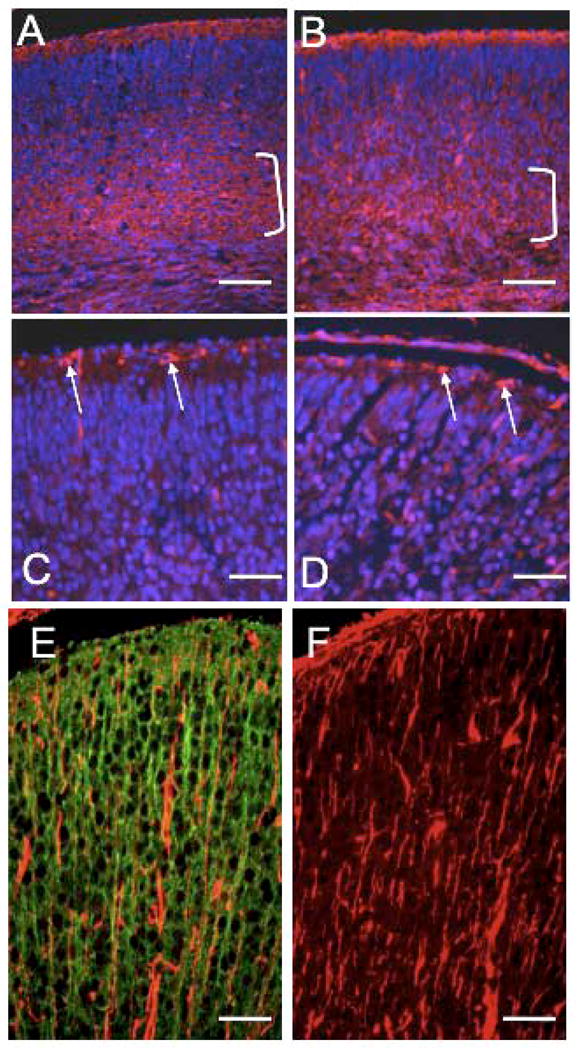
Cortical plate splitting and the radial glia are unaffected in Macf1 cKO brains. Coronal sections of control (A, C, E) or mutant (B, D, F) E17.5 brains were stained with CS-56 (A, B), reelin (C, D), or vimentin (E, F) antibodies (red), and co-stained with Hoechst nuclear dye (blue; A–D only) or CU119 antibody (green, E only). Brackets in A and B indicate subplate staining. Arrows in C and D indicate reelin-positive Cajal-Retzius cells in the marginal zone. E and F show vimentin-positive processes of radial glial cells, which did not colocalize with CU119-positive neuronal processes (E). Similar results were obtained for three pairs of animals from different litters. Scale bars: A–F=50μm
One possibility that could explain the apparent defects in neuronal migration in the brains of cKO animals is a primary deficit in radial glia, which gives rise to, and subsequently directs the migration of cortical projection neurons and hippocampal pyramidal cells (Malatesta et al., 2008). We visualized the radial glia in control and cKO brains by staining E17.5 coronal sections with polyclonal antibodies against the intermediate filament protein vimentin and brain lipid binding protein (BLBP) (Feng et al., 1994; Yang et al., 1993). There were no significant differences in vimentin or BLBP staining between cKO and control cortices (Fig. 8E, F and data not shown). Double labeling of control cortical sections with vimentin and CU119 MACF1 antibody showed distinct staining patterns, suggesting that radial glia do not express MACF1. The knockout of MACF1 should therefore have no direct effect on the radial glial cells (Fig. 8E). Taken together, these data suggest that the cell migration defect in the cKO brain is not due to defects in radial glia.
Defects in neuronal migration in the cKO cortex
To obtain further insights into the nature of the cortical abnormalities in cKO mutants, we performed neuronal birth-dating experiments. BrdU was injected into pregnant females at E12 or E14 and the brains of the embryos were examined at E18. When BrdU was injected at E12, no significant differences were found in the location of most of the labeled nuclei between control and mutant cortices in the cortical plates (Fig. 9A, B and E), although there were some cells left in the IZ and VZ in the cKO animals. The majority of positive cells were localized in the deeper layers, as expected from the inside-out pattern of the normal course of radial neuronal migration. In contrast, when BrdU was injected at E14, the distributions of the labeled cells in the two types of cortices were different. In control cortices most of the positive cells were located in the superficial layers (Fig. 9C and E), whereas in the mutant cortices the labeled nuclei were distributed more evenly throughout the cortical plate (Fig. 9D and E). There were also more labeled nuclei still present in the intermediate zone of the cKO cortices as compared to controls. These data indicate that cortical neurons in cKO mice: (1) follow a normal inside-out pattern of migration; (2) exhibit abnormal positioning in traversing the cortical plate, at least when born relatively late.
Figure 9.

Cortical neuronal migration is delayed in Macf1 nestin-cKO brains. Pregnant Macf1F/F females were injected with BrdU at E12 (A, B) or E14 (C, D). The embryos were dissected and their brains fixed at E18. Coronal sections of control (A, C) or mutant (B, D) littermates were stained with BrdU monoclonal antibody and secondary HRP conjugates, and counterstained with eosin. Early-born neurons (E12) migrate into the deeper layers of the cortical plate in both control and mutant cortices (dark dots in A, B). In contrast, late-born neurons (E14) reach the upper levels only in the control cortex (C), whereas in the mutant cortex they are scattered throughout the cortical plate (D). (E) Histograms of the distributions of labeled nuclei in 6 equal bins covering the cortical plate. Differences for the corresponding bins with P-values <0.05 (calculated by Student’s t-test) are marked with asterisks. Three different pairs of animals from different litters were analyzed. Scale bars: A–D=50μm.
DISCUSSION
MACF1 and other plakins are multifunctional cytolinker proteins that have been implicated in cell migration, cell signaling, tissue integrity and maintenance, and axonal extension (Ruhrberg and Watt, 1997; Sonnenberg and Liem, 2007). Macf1-null embryos do not progress past E10, making it difficult to study tissue-specific MACF1 functions in developing mice. Here, we report the generation of a Cre/loxP-mediated conditional Macf1 knockout mouse and describe the phenotype resulting from MACF1a inactivation in the nervous system.
Because the Macf1 gene contains multiple translation initiation codons separated by extended stretches of DNA (Karakesisoglou et al., 2000; Leung et al., 1999), targeting all of them by a single construct was not readily feasible. Instead, we chose to target the ABD region, which is present in both MACF1 isoforms that were known at the time this study was conceived. The phenotype of Macf1R/R embryos (i.e. embryos carrying two fully recombined alleles in all tissues) appears to be as severe as that of the previously described Macf1 knockout embryos (Chen et al., 2006; Kodama et al., 2003). However, our subsequent analysis of MACF1 protein expression in the brains of the cKO mice revealed the existence of a novel isoform lacking the ABD domain. This isoform, which we have termed MACF1c, is not affected by the conditional knockout, suggesting that it has a separate initiation codon downstream of the entire ABD segment. Therefore, our Macf1 cKO mice fortuitously represent a nervous-system-specific knockout of MACF1a, which appears crucial for normal brain development. It is interesting to note that the MACF1 homologue, BPAG1 also has an initiation codon downstream of the ABD segment, although the resulting transcript, BPAG1e is expressed only in the epithelia (Sonnenberg and Liem, 2007).
Macf1 cKO animals die within 24–36 hrs after birth, apparently from respiratory distress. The innervation and structure of the diaphragm appeared normal in Macf1 cKO mice, suggesting that the lethality was due to a central, rather than peripheral, neurologic deficiency. We discovered multiple developmental defects in cKO brains, including a disorganization of the cerebral cortex, heterotopia of the hippocampal pyramidal layer, aplasia of the corpus callosum and anterior and hippocampal commissures, altered shapes of the lateral ventricles, and hypotrophy and disorganization of thalamocortical fibers.
The cortices of Macf1 cKO mice exhibit a perturbed layer structure suggesting a defect in cortical migration. Cortical neurons with different layer identities failed to separate fully into physically distinct layers in the cKO mice as determined by staining for markers specific for layers 5 and 6. BrdU labeling at different gestation times suggests that the defect is due to an inability of later born neurons to pass the earlier born ones, leading to a layer positioning defect. While most early-born cortical neurons reached their destinations in the deeper layers, some late-born cells failed to traverse into the outer layers of the cortex and are located throughout the cortical plate, as well as the intermediate zone. Since the early-born neurons have a shorter distance to migrate compared to the late-born neurons, the simplest explanation is that, due to a reduced migration rate, some late-born neurons do not have enough time to reach their destinations by the time of birth. This interpretation is consistent with the layer marker staining data, although we cannot rule out that migration guidance defects are (also) at play. Our observation that some thalamocortical axons do not extend correctly in the mutant cortex provides circumstantial evidence that axonal and, conceivably, cell migration targeting is affected in the absence of MACF1a. It is possible that the MACF1a cKO phenotype described here results from a combination of a cell-autonomous migration/extension defect and a non-cell-autonomous guidance defect. The various degrees of aplasia in the development of several major axonal tracts (anterior and hippocampal commissures, corpus callosum, and thalamocortical fibers) in the absence of MACF1a may indicate a defect in axonal guidance. An alternative explanation is that the numbers of neurons involved in the formation of these tracts are reduced. Similar considerations apply to the cell migration defects found in the mutant hippocampus.
The phenotype of the nervous-system-specific MACF1 cKO mouse is very different from the dt mouse, which is a knock-out of the related plakin BPAG1. BPAG1 is expressed in the nervous system and its nervous system isoform, BPAG1a, has the same domains as MACF1a (Leung et al., 2001). Since both proteins are expressed in the same neurons during development, it is therefore unlikely that they are functionally redundant. BPAG1 knock-out causes sensory neuron degeneration with neurofilamentous aggregation and microtubule disorganization. The absence of a protein that has the ability to bind to and bundle MTs could account for these abnormalities. In Drosophila, the MACF1 homologue, shortstop or shot is necessary for axon elongation, and the ABD and MTBD of shot are required in the same molecule for axon extension. Mutant shot axons navigate along the sensory axon substrate, but then terminate prematurely. In the MACF1 cKO mice, axon outgrowth can still occur, as in the α-internexin-positive and silver-stained thalamocortical fibers, but their disorganization leads to the possibility that axon guidance may be affected by the absence of MACF1. A recent paper also describes a role for MACF1 in axonal extension, where knock-down of MACF1 resulted in 25–35% decrease in axon length (Sanchez-Soriano et al., 2009).
The microtubule-associated proteins MAP1b, MAP2, and tau have been shown to regulate synergistically multiple aspects of brain development (Gonzalez-Billault et al., 2005; Takei et al., 2000; Teng et al., 2001). Map2;map1b and map1b;tau double knockout mice, as well as some map1b-null animals, exhibit phenotypes that have similarities to the Macf1 cKO mouse. Thus, map1b;tau mutant mice display a cortical migration defect, an amorphous pyramidal cell layer in the hippocampus together with some heterotopia, altered lateral ventricles, hypotrophic anterior commissures, and a dysgenesis of the corpus callosum. They display a diminished viability, but no clear-cut perinatal lethality. Cultured primary neurons from these animals have shorter processes and growth cones with splayed MT arrays, primarily in axons (Takei et al., 2000). Map1b;map2 double knockout mice also show cortical and hippocampal migration defects, altered lateral ventricles, hypotrophic anterior commissure, and a Probst bundle instead of a normal corpus callosum. Like the Macf1 cKO mice, they die shortly after birth (P0). Primary cultures from these mice also show shorter processes (primarily dendrites) and splayed growth cones (Teng et al., 2001). Map1b-null mice have a background-specific phenotype, with some strains appearing normal and others showing defects that are similar to, but are less pronounced than the two double knockouts (Gonzalez-Billault et al., 2005; Takei et al., 2000). These phenotypes have been interpreted as resulting from changes in MT dynamic/stabilization properties, consistent with the known functions of the MAPs involved, although they may also have an effect on axonal transport. Cortical migration and differentiation defects have also been described as a result of deficiency in the multi-subunit histone acetyltransferase Elongator complex, which was thought to affect the acetylation of α-tubulin (Creppe et al., 2009). The authors suggest that this change in acetylation could affect binding of motor proteins that regulate transport in axons and dendrites. Given that MACF1 also acts as a scaffolding protein that interacts with MTs, it will be interesting to determine if the levels of acetylated tubulin are also altered in the MACF1 cKO mice.
The MAP knockouts also share many of the features of the Macf1 cKO hippocampus. Map1b;tau and map1b;map2 double knockouts, and to a lesser extent map1b-null mice, exhibit heterotopia, undulations, partial breaks (splitting), and a decreased density of the pyramidal cell layer. Pyramidal cell heterotopia has also been found in the hippocampi of dcx (doublecortin) mutant mice (Corbo et al., 2002). In Macf1 cKO hippocampi, we observed pronounced undulations and a greatly reduced cell density in the pyramidal layer as well as striking single-band heterotopia contouring the outside of the main layer, with thickness diminishing in the CA1–CA3 direction. In contrast, the heterotopia in the dcx mice are located mostly in the CA3 area, on both sides of the apparent main layer. The hippocampal heterotopia in the other MAP mutants vary in extent, number, and location, but seldom extend over more than one CA area. In Macf1 cKO hippocampi, we did not see any layer splitting, appearing as linear breaks, that has been reported in some of these MAP mutants. Severe undulations, or “kinks”, found in the main pyramidal layer of map1b;tau mice (Takei et al., 2000) were also absent in Macf1 cKO hippocampi. In addition, the heterotopia in Macf1cKO hippocampi appeared to be significantly more pronounced than in the MAP mutants. In summary, while the overall morphology of the hippocampal defects in Macf1 cKO mice shares many features with those described in other MAP knockout mice, the details are unique in the extent of the heterotopia and continuity/regularity of the two resulting layers.
Although Macf1 cKO mice share several features with MAP mutants, an important finding of the present study is that the MACF1c isoform, which is apparently identical to MACF1a except for the absence of an ABD, cannot compensate for the lack of MACF1a in the mouse brain. The MACF1c isoform is expected to contain the microtubule binding domain and should therefore still act as a MAP. This finding provides the first direct in vivo evidence that the ability to interact with the actin cytoskeleton is indispensable for MACF1 function in the nervous system. In migrating fibroblasts, the actin cytoskeleton plays crucial roles in the formation of leading-edge membrane protrusions, such as filopodia and lamellipodia. The actin reorganization underlying these formations is regulated by Rho family GTPases such as Cdc42, Rac1, and RhoA (Hall, 2005). Similarly, migrating cortical neurons extend the leading process towards the pial surface and retract their cell bodies in a stepwise fashion (Nadarajah et al., 2001). Multiple actin regulators have been shown to affect radial neuronal migration in the cortex, including VASP, Rac1, Tiam1, Cdc42, RhoA, and cofilin (Chen et al., 2007; Goh et al., 2002; Kawauchi et al., 2003, 2006; Kholmanskikh et al., 2006; Konno et al., 2005; Nguyen et al., 2006). The actin-crosslinking protein filamin A is mutated in human patients with periventricular heterotopia, a cortical migration disorder (Fox et al., 1998; Nagano et al., 2004). Given that the microtubule and actin filament systems operate in unison in migrating cells, it appears likely that bridging factors are involved in their integrated functioning in axonal extension and neuronal migration. This study identifies MACF1 as one of the coordinating regulators.
One pivotal role of MACF1a in the developing nervous system may be to link the MTs and actin filaments in the growth cone and in the axon shaft. At the signaling level, multiple factors are known to co-regulate MTs and actin in these processes, e.g., Cdk5 and JNK (Kawauchi and Hoshino, 2008). MACF1a, however, is likely to mediate a physical interaction between the two types of filaments. Besides the presence of both types of binding domains, the evidence for this is two-fold: (1) in Macf1-null primary fibroblasts, extending MTs fail to coalign with actin filaments at the plasma membrane (Kodama et al., 2003); (2) MACF1a enhances MT-actin colocalization in transfected cells (Leung et al., 1999). A recent study utilizing a similar strategy to inactivate MACF1 in skin epidermis showed that MACF1 plays an essential role in regulating the dynamics of actin-microtubule-focal adhesion interactions during keratinocyte migration, possibly through both cytoskeleton-binding domains and an intrinsic actin-activated ATPase activity (Wu et al., 2008). However, the possible involvement of MACF1c or other MACF1 isoforms not amenable to inactivation through the removal of the ABD was not considered in this report.
The precise mechanisms of MACF1a functions in neuronal migration and growth cone extension will require further analysis. The conditional Macf1 targeting model reported here can be used to study the roles of MACF1a in specific brain regions and cell populations, and will likely reveal MACF1 involvement in additional processes.
MATERIALS AND METHODS
Generation of Macf1 conditional knockout mice
To construct a conditional Macf1 targeting vector (Fig. 1A), a 1.2-kb fragment encompassing exons 6 and 7 of the mouse Macf1 gene (GenBank accession #AF150755) was amplified from C57BL/6J genomic DNA with primers GTCGACCAGCCAACCTTGGACCTCTCTTCC (forward) and GTCGACGCCAGATAGAATTTTCAGAACACG (reverse) and ligated into the SalI site of pEasyFlox vector (Schenten et al., 2002). A ~5.0-kb large arm sequence was amplified with forward primer CTCGAGCTCATGAGAGACCACCACCTTACAG and reverse primer CTCGAGGCAGCCCTATTGATGACAACTCCAG and subcloned into the vector at the XhoI site. A ~1.9-kb short arm sequence was amplified with primers TTCTATCTGGCAGGATCCCTTGTTCTGACC (forward) TTGCGGCCGCCCCAGGGAATGTCAGACCACC (reverse), digested with BamHI and NotI, and subcloned into the targeting construct. CSL3 ES cell electroporation, selection, and expansion were performed at the Columbia University Transgenic Animal Core Facility as described before (Chen et al., 2006). ES cell clones were screened by PCR and positive colonies verified by Southern blotting. Two positive clones were microinjected into C57BL/6J blastocysts. The neo cassette was excised in vivo by crossing F1 female mice heterozygous for the targeted allele with E2a-Cre males (Lakso et al., 1996). Nestin-Cre mice (B6.Cg-Tg(Nes-cre)1Kln/J) were obtained from Jackson Laboratories (Tronche et al., 1999). To achieve Macf1 gene inactivation in the nervous system, Macf1F/F females were crossed with Macf1+/RCre males, resulting in a quarter of the progeny being Macf1F/RCre.
Southern blotting, genomic PCR, RT-PCR
Southern blotting was performed as described before (Ching et al., 1999). Briefly, genomic DNA from tail biopsies was extracted by proteinase K treatment followed by phenol-chloroform extractions, digested with either EcoRI (to detect the vector-targeted allele) or XbaI (to detect the floxed allele), separated on 0.8% agarose gels, and transferred onto Zeta-Probe GT membranes (BioRad, Hercules, CA). The targeted allele was detected in EcoRI-digested preparations as a ~5–kb band and the wild-type allele as a ~12–kb band. The floxed allele in XbaI-digested genomic preparations was detected as a ~5.5-kb band and the wild-type allele as a ~9-kb band. The probe template for both types of blots was amplified with primers GCTAGAATTGCTGCTGCAGATTGC (forward) and GGAAGGAAGTAGCATAGTGTTTGTG (reverse) (Fig 1A).
PCR genotyping of ES cells and targeted F1 animals was performed with forward primer GGCTGCTAAAGCGCATGCTCCAGAC and reverse primer GCAATCTGCAGCAGCAATTCTAGC (primer pair 1 in Fig. 1A). To genotype animals carrying floxed and recombined alleles, or to monitor F->R allele conversion in the brains of conditional knockout animals, genomic PCR was performed with forward primer CATCAGAAGAGATCAACCAACC and reverse primer AGGCAGCAGGCTATCCTAGAGC (primer pair 2). This reaction yields expected products of ~1.8 kb (WT), ~1.9 kb (F), and ~0.6 kb (R). Cre genotypes were determined with forward primer CACCAAAATTTGCCTGCATTACC and reverse primer CGCCATCTTCCAGCAGGCGCACC. All mice used in experimental matings and their offspring, including embryos, were genotyped by allele-specific qPCR (Transnetyx, Inc.). To examine MACF1 expression by RT-PCR, total RNA from mouse tissues was isolated with TRIzol reagent (Invitrogen), reverse-transcribed with SuperScript II polymerase (Invitrogen), and the resulting first-strand DNA was amplified with forward primer TTCCACAGGCTGCAGAATGTGCAG, located in exon 4, and reverse primer ATGCTGCCTGATCCAGGGAATGAG, located in exon 9 (primer pair 3). The expected WT/F product of this reaction is ~0.6 kb; the expected R product is ~0.3 kb.
Histology
Mouse brains were fixed with 4% paraformaldehyde and embedded in paraffin. Five 5-μm coronal sections were collected every 100 μm throughout the brain. One of the 5 sections of each level was stained with Bielschowsky’s silver stain as follows. Sections were deparaffinized and rehydrated and placed in pre-warmed 10% silver nitrate for 15 minutes. The slides were washed 3 times with distilled water and placed into precipitate-free silver solution for 30 minutes at 40°C. The slides were transferred into developer solution for 1 minute. After incubating the slides in 1% ammonium hydroxide for 1 min and in 5% sodium thiosulfate for 5 minutes, the sections were dehydrated, cleared, and mounted onto resinous medium. Another one of the 5 sections of each level was stained with 0.1% Cresyl violet solution for Nissl body staining after regular deparaffinization and rehydration. After staining, the slides were dehydrated and cleared before being mounted onto permanent mounting medium. Paraffin sections were also stained by routine Hematoxilin & Eosin staining procedures.
Immunohistochemistry
E17.5 and P0 brains were fixed in 4% paraformaldehyde, embedded in paraffin, and 5-μm sections were cut. After antigen retrieval and rehydration, sections were blocked with 2% normal goat serum for 30 min and incubated with primary antibodies overnight at 4°C. The sections were washed 3 times with PBS and incubated with AlexaFluor-488/594 secondary antibodies (Invitrogen) for 1 hr at room temperature. After washing, the sections were sealed in Aquamount medium (Lerner Laboratories) with coverslips. Some sections were also incubated with Hoechst DNA dye solution (Invitrogen).
Western blotting
Mouse brains were homogenized in ice-cold PBS containing 1% SDS and cOmplete™ protease inhibitors (Roche) and centrifuged at 16,000 rpm in an Eppendorf tabletop microcentrifuge. The supernatant was collected, supplemented with SDS denaturing buffer, and boiled. Brain lysate proteins were resolved by SDS-PAGE on bipartite minigels (top gel, 6%; bottom gel, 15%) for 1.5 hrs at 30 mA, and transferred onto Immobilon-P membrane (Millipore). The membrane was blocked with 5% fat-free milk in PBS/0.05% Tween-20 and incubated with primary antibodies in blocking solution overnight at 4°C. After washing with PBS/Tween-20, the membrane was incubated with AlexaFluor-680-conjugated goat-anti-mouse (Invitrogen) and IRDye-800-conjugated goat-anti-rabbit (Rockland Immunochemicals) antibodies for 1 hr, washed with PBS/Tween-20 and PBS, and the immunoreactive proteins were visualized on the Odyssey infrared scanner (LICOR Biosciences).
Antibodies
The following commercial antibodies were used: Ctip2, rat polyclonal (Abcam), Cux1 rabbit polyclonal (Cruz Biotechnology), TBr1, rabbit polyclonal (Chemicon), reelin monoclonal (Chemicon), chondroitin sulfate proteoglycan (CSPG) mouse monoclonal CS-56 (Sigma), BrdU mouse monoclonal BU-33 (Sigma), neuron specific tubulin, mouse monoclonal TuJ1 (Covance), neurofilament heavy chain, mouse monoclonal N52 (Sigma), brain lipid binding protein (BLBP) polyclonal (Abcam) and Vimentin, mouse monoclonal vim13.2 (Sigma). α-Internexin antibodies have been previously described (Kaplan et al., 1990) and the rabbit anti-TAG1 was generously provided by Dr. Thomas Jessell (Columbia University). Polyclonal antibodies against the MACF1 plakin domain (CU119) and ABD have been described (Antolik et al., 2007; Lin et al., 2005). The latter antibody was a generous gift from Dr. Robert Bloch (Univ. of Maryland).
Neuron tracing
E12 or E14 pregnant females were injected with 50 μg/g BrdU (Sigma) intraperitoneally as described previously (Teng et al., 2001). The females were sacrificed at E18, the embryos dissected, and the brains were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned as above. The sections were deparaffinized in xylene and serial alcohols, and the endogenous peroxidase activity was quenched by incubation in 0.3% H2O2 in methanol for 3 min. The sections were washed in PBS and blocked with 2% normal goat serum for 1 hr. The sections were then incubated with a monoclonal BrdU antibody (Sigma) overnight at 4°C. After washing, the sections were incubated with EnVision™ secondary antibodies conjugated to horseradish peroxidase (Dako) for 1 hr at room temperature. The bound conjugate was visualized by incubation with DAB+ chromogen (Dako). The sections were counterstained with eosin, dehydrated in serial alcohols and xylene, and mounted in Permount medium.
For quantitative measurements of the distributions of BrdU-labeled nuclei, comparable sections were chosen at the level of the anterior commissure and divided into 6 equal horizontal bins approximately corresponding to the different cortical layers. Labeled nuclei in each bin were counted and the data were plotted as percentages of the total number of labeled nuclei. Sections from 2 mice were counted for each genotype and time point.
Diaphragm staining
Diaphragms were dissected from euthanized newborn pups (P0) and fixed for 1.5 hrs in 2% paraformaldehyde on ice. The diaphragms were washed with PBS and incubated in PBS containing 100 mM glycine for 15 min to quench any residual fixative. The diaphragms were then incubated in PBS containing 1% Triton X100 and 10% normal goat serum for 1 hr, followed by an overnight incubation with NFH monoclonal antibody (Sigma) and α-bungarotoxin-AlexaFluor488 (Invitrogen) in blocking solution at 4°C. After washing, the diaphragms were incubated with an AlexaFluor594-conjugated goat-anti-mouse antibody (Invitrogen) for 1 hr, washed in PBS, mounted onto slides in Aquamount medium (Lerner Laboratories), and flattened with weights placed on top of the coverslips overnight.
Supplementary Material
Innervation of the diaphragm is unaffected in Macf1 nestin-cKO mice. Control (A, C, E) or mutant (B, D, F) diaphragms were dissected from P0 pups and stained with neurofilament heavy chain antibodies (A, B; red in E, F) and α-bungarotoxin (C, D; green in E, F) to reveal phrenic nerves and muscle endplates, respectively. Arrows point to lower and upper branches of the phrenic nerves (A, B). E and F, higher magnifications of the superimposed images in A and C, and B and D, respectively. Scale bars, 200 μm.
Cell distributions across the cortex, but not their total numbers, are different in cKO brains. Bins were selected in the cortical sections presented in Fig. 2(A, control; B, cKO), and the numbers of stained cells were counted and plotted. The total number of cells in the cKO cortex was similar to that in the control cortex but their distribution was “flat”, without the peaks in the cortical plate and subventricular zone present in the control cortex. Scale bars, 50μm
MACF1 ABD is eliminated in the cKO cortex. Coronal sections of control (A) or mutant (B) brains were stained with MACF1-ABD polyclonal antibody. Scale bars, 25μm
MACF1 staining in the hippocampus. Coronal sections of control (A, C) and Macf1 cKO (B, D) hippocampi were stained with CU119 and MACF1-ABD polyclonal antibody. Scale bars, 50μm
Acknowledgments
This work was supported by NIH grants NS40784 and NS47711 to RKHL. The authors are grateful to Dr. Robert Burgess (Jackson Laboratory) for technical advice, Dr. Robert Bloch (University of Maryland) for the generous gift of the MACF-ABD antibodies, Dr. Tom Jessell (Columbia University) for the TAG-1 antibody and especially Dr. Phyllis Faust (Columbia University) for helpful discussions and critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antolik C, Catino DH, O’Neill AM, Resneck WG, Ursitti JA, Bloch RJ. The actin binding domain of ACF7 binds directly to the tetratricopeptide repeat domains of rapsyn. Neuroscience. 2007;145:56–65. doi: 10.1016/j.neuroscience.2006.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Berg DK, Kelly RB, Sargent PB, Williamson P, Hall ZW. Binding of a-bungarotoxin to acetylcholine receptors in mammalian muscle (snake venom-denervated muscle-neonatal muscle-rat diaphragm-SDS-polyacrylamide gel electrophoresis) Proc Natl Acad Sci U S A. 1972;69:147–151. doi: 10.1073/pnas.69.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicknese AR, Sheppard AM, O’Leary DD, Pearlman AL. Thalamocortical axons extend along a chondroitin sulfate proteoglycan-enriched pathway coincident with the neocortical subplate and distinct from the efferent path. J Neurosci. 1994;14:3500–3510. doi: 10.1523/JNEUROSCI.14-06-03500.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchi B, Kelly LM, Viemari JC, Lafon I, Burnet H, Bevengut M, Tillmanns S, Daniel L, Graf T, Hilaire G, Sieweke MH. MafB deficiency causes defective respiratory rhythmogenesis and fatal central apnea at birth. Nat Neurosci. 2003;6:1091–1100. doi: 10.1038/nn1129. [DOI] [PubMed] [Google Scholar]
- Bosher JM, Hahn BS, Legouis R, Sookhareea S, Weimer RM, Gansmuller A, Chisholm AD, Rose AM, Bessereau JL, Labouesse M. The Caenorhabditis elegans vab-10 spectraplakin isoforms protect the epidermis against internal and external forces. J Cell Biol. 2003;161:757–768. doi: 10.1083/jcb.200302151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Bernier G, Mathieu M, Rossant J, Kothary R. The mouse dystonia musculorum gene is a neural isoform of bullous pemphigoid antigen 1. Nat Genet. 1995;10:301–306. doi: 10.1038/ng0795-301. [DOI] [PubMed] [Google Scholar]
- Chen HJ, Lin CM, Lin CS, Perez-Olle R, Leung CL, Liem RK. The role of microtubule actin cross-linking factor 1 (MACF1) in the Wnt signaling pathway. Genes Dev. 2006;20:1933–1945. doi: 10.1101/gad.1411206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liao G, Waclaw RR, Burns KA, Linquist D, Campbell K, Zheng Y, Kuan CY. Rac1 controls the formation of midline commissures and the competency of tangential migration in ventral telencephalic neurons. J Neurosci. 2007;27:3884–3893. doi: 10.1523/JNEUROSCI.3509-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching GY, Chien CL, Flores R, Liem RK. Overexpression of alpha-internexin causes abnormal neurofilamentous accumulations and motor coordination deficits in transgenic mice. J Neurosci. 1999;19:2974–2986. doi: 10.1523/JNEUROSCI.19-08-02974.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbo JC, Deuel TA, Long JM, LaPorte P, Tsai E, Wynshaw-Boris A, Walsh CA. Doublecortin is required in mice for lamination of the hippocampus but not the neocortex. J Neurosci. 2002;22:7548–7557. doi: 10.1523/JNEUROSCI.22-17-07548.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creppe C, Malinouskaya L, Volvert ML, Gillard M, Close P, Malaise O, Laguesse S, Cornez I, Rahmouni S, Ormenese S, Belachew S, Malgrange B, Chapelle JP, Siebenlist U, Moonen G, Chariot A, Nguyen L. Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cell. 2009;136:551–564. doi: 10.1016/j.cell.2008.11.043. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Curran T. Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. J Neurosci. 1997;17:23–31. doi: 10.1523/JNEUROSCI.17-01-00023.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denaxa M, Chan CH, Schachner M, Parnavelas JG, Karagogeos D. The adhesion molecule TAG-1 mediates the migration of cortical interneurons from the ganglionic eminence along the corticofugal fiber system. Development. 2001;128:4635–4644. doi: 10.1242/dev.128.22.4635. [DOI] [PubMed] [Google Scholar]
- Drabek K, van Ham M, Stepanova T, Draegestein K, van Horssen R, Sayas CL, Akhmanova A, Ten Hagen T, Smits R, Fodde R, Grosveld F, Galjart N. Role of CLASP2 in microtubule stabilization and the regulation of persistent motility. Curr Biol. 2006;16:2259–2264. doi: 10.1016/j.cub.2006.09.065. [DOI] [PubMed] [Google Scholar]
- Feng L, Hatten ME, Heintz N. Brain lipid-binding protein (BLBP): a novel signaling system in the developing mammalian CNS. Neuron. 1994;12:895–908. doi: 10.1016/0896-6273(94)90341-7. [DOI] [PubMed] [Google Scholar]
- Fliegner KH, Kaplan MP, Wood TL, Pintar JE, Liem RK. Expression of the gene for the neuronal intermediate filament protein alpha-internexin coincides with the onset of neuronal differentiation in the developing rat nervous system. J Comp Neurol. 1994;342:161–173. doi: 10.1002/cne.903420202. [DOI] [PubMed] [Google Scholar]
- Fox JW, Lamperti ED, Eksioglu YZ, Hong SE, Feng Y, Graham DA, Scheffer IE, Dobyns WB, Hirsch BA, Radtke RA, Berkovic SF, Huttenlocher PR, Walsh CA. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315–1325. doi: 10.1016/s0896-6273(00)80651-0. [DOI] [PubMed] [Google Scholar]
- Goh KL, Cai L, Cepko CL, Gertler FB. Ena/VASP proteins regulate cortical neuronal positioning. Curr Biol. 2002;12:565–569. doi: 10.1016/s0960-9822(02)00725-x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Billault C, Del Rio JA, Urena JM, Jimenez-Mateos EM, Barallobre MJ, Pascual M, Pujadas L, Simo S, Torre AL, Gavin R, Wandosell F, Soriano E, Avila J. A role of MAP1B in Reelin-dependent neuronal migration. Cereb Cortex. 2005;15:1134–1145. doi: 10.1093/cercor/bhh213. [DOI] [PubMed] [Google Scholar]
- Goryunov D, Adebola A, Jefferson JJ, Leung CL, Messer A, Liem RK. Molecular characterization of the genetic lesion in Dystonia musculorum (dt-Alb) mice. Brain Res. 2007;1140:179–187. doi: 10.1016/j.brainres.2006.04.068. [DOI] [PubMed] [Google Scholar]
- Gressens P. Pathogenesis of migration disorders. Curr Opin Neurol. 2006;19:135–140. doi: 10.1097/01.wco.0000218228.73678.e1. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33:891–895. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- Hatten ME. Central nervous system neuronal migration. Annu Rev Neurosci. 1999;22:511–539. doi: 10.1146/annurev.neuro.22.1.511. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, Rubenstein JL. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/s0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- Jefferson JJ, Leung CL, Liem RK. Dissecting the sequence specific functions of alternative N-terminal isoforms of mouse bullous pemphigoid antigen 1. Exp Cell Res. 2006;312:2712–2725. doi: 10.1016/j.yexcr.2006.04.025. [DOI] [PubMed] [Google Scholar]
- Kaplan MP, Chin SS, Fliegner KH, Liem RK. Alpha-internexin, a novel neuronal intermediate filament protein, precedes the low molecular weight neurofilament protein (NF-L) in the developing rat brain. J Neurosci. 1990;10:2735–2748. doi: 10.1523/JNEUROSCI.10-08-02735.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakesisoglou I, Yang Y, Fuchs E. An epidermal plakin that integrates actin and microtubule networks at cellular junctions. J Cell Biol. 2000;149:195–208. doi: 10.1083/jcb.149.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. Embo J. 2003;22:4190–4201. doi: 10.1093/emboj/cdg413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. Cdk5 phosphorylates and stabilizes p27kip1 contributing to actin organization and cortical neuronal migration. Nat Cell Biol. 2006;8:17–26. doi: 10.1038/ncb1338. [DOI] [PubMed] [Google Scholar]
- Kawauchi T, Hoshino M. Molecular pathways regulating cytoskeletal organization and morphological changes in migrating neurons. Dev Neurosci. 2008;30:36–46. doi: 10.1159/000109850. [DOI] [PubMed] [Google Scholar]
- Kerjan G, Gleeson JG. Genetic mechanisms underlying abnormal neuronal migration in classical lissencephaly. Trends Genet. 2007;23:623–630. doi: 10.1016/j.tig.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Kholmanskikh SS, Koeller HB, Wynshaw-Boris A, Gomez T, Letourneau PC, Ross ME. Calcium-dependent interaction of Lis1 with IQGAP1 and Cdc42 promotes neuronal motility. Nat Neurosci. 2006;9:50–57. doi: 10.1038/nn1619. [DOI] [PubMed] [Google Scholar]
- Kodama A, Karakesisoglou I, Wong E, Vaezi A, Fuchs E. ACF7: an essential integrator of microtubule dynamics. Cell. 2003;115:343–354. doi: 10.1016/s0092-8674(03)00813-4. [DOI] [PubMed] [Google Scholar]
- Konno D, Yoshimura S, Hori K, Maruoka H, Sobue K. Involvement of the phosphatidylinositol 3-kinase/rac1 and cdc42 pathways in radial migration of cortical neurons. J Biol Chem. 2005;280:5082–5088. doi: 10.1074/jbc.M408251200. [DOI] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung CL, Sun D, Zheng M, Knowles DR, Liem RK. Microtubule actin cross-linking factor (MACF): a hybrid of dystonin and dystrophin that can interact with the actin and microtubule cytoskeletons. J Cell Biol. 1999;147:1275–1286. doi: 10.1083/jcb.147.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung CL, Zheng M, Prater SM, Liem RK. The BPAG1 locus: Alternative splicing produces multiple isoforms with distinct cytoskeletal linker domains, including predominant isoforms in neurons and muscles. J Cell Biol. 2001;154:691–697. doi: 10.1083/jcb.200012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CM, Chen HJ, Leung CL, Parry DA, Liem RK. Microtubule actin crosslinking factor 1b: a novel plakin that localizes to the Golgi complex. J Cell Sci. 2005;118:3727–3738. doi: 10.1242/jcs.02510. [DOI] [PubMed] [Google Scholar]
- Malatesta P, Appolloni I, Calzolari F. Radial glia and neural stem cells. Cell Tissue Res. 2008;331:165–178. doi: 10.1007/s00441-007-0481-8. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–150. doi: 10.1038/83967. [DOI] [PubMed] [Google Scholar]
- Nagano T, Morikubo S, Sato M. Filamin A and FILIP (Filamin A-Interacting Protein) regulate cell polarity and motility in neocortical subventricular and intermediate zones during radial migration. J Neurosci. 2004;24:9648–9657. doi: 10.1523/JNEUROSCI.2363-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L, Besson A, Heng JI, Schuurmans C, Teboul L, Parras C, Philpott A, Roberts JM, Guillemot F. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev. 2006;20:1511–1524. doi: 10.1101/gad.377106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto M, Monuki ES, Tang H, Imitola J, Haubst N, Khoury SJ, Cunningham J, Gotz M, Walsh CA. Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II–IV of the cerebral cortex. J Comp Neurol. 2004;479:168–180. doi: 10.1002/cne.20322. [DOI] [PubMed] [Google Scholar]
- Rakic P. Principles of neural cell migration. Experientia. 1990;46:882–891. doi: 10.1007/BF01939380. [DOI] [PubMed] [Google Scholar]
- Ruhrberg C, Watt FM. The plakin family: versatile organizers of cytoskeletal architecture. Curr Opin Genet Dev. 1997;7:392–397. doi: 10.1016/s0959-437x(97)80154-2. [DOI] [PubMed] [Google Scholar]
- Sanchez-Soriano N, Travis M, Dajas-Bailador F, Goncalves-Pimentel C, Whitmarsh AJ, Prokop A. Mouse ACF7 and drosophila short stop modulate filopodia formation and microtubule organisation during neuronal growth. J Cell Sci. 2009;122:2534–2542. doi: 10.1242/jcs.046268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenten D, Gerlach VL, Guo C, Velasco-Miguel S, Hladik CL, White CL, Friedberg EC, Rajewsky K, Esposito G. DNA polymerase kappa deficiency does not affect somatic hypermutation in mice. Eur J Immunol. 2002;32:3152–3160. doi: 10.1002/1521-4141(200211)32:11<3152::AID-IMMU3152>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Sonnenberg A, Liem RK. Plakins in development and disease. Exp Cell Res. 2007;313:2189–2203. doi: 10.1016/j.yexcr.2007.03.039. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Prokop A, Yamamoto M, Sugimura K, Uemura T, Betschinger J, Knoblich JA, Volk T. Shortstop recruits EB1/APC1 and promotes microtubule assembly at the muscle-tendon junction. Curr Biol. 2003;13:1086–1095. doi: 10.1016/s0960-9822(03)00416-0. [DOI] [PubMed] [Google Scholar]
- Sun D, Leung CL, Liem RK. Characterization of the microtubule binding domain of microtubule actin crosslinking factor (MACF): identification of a novel group of microtubule associated proteins. J Cell Sci. 2001;114:161–172. doi: 10.1242/jcs.114.1.161. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zhang J, Kraeft SK, Auclair D, Chang MS, Liu Y, Sutherland R, Salgia R, Griffin JD, Ferland LH, Chen LB. Molecular cloning and characterization of human trabeculin-alpha, a giant protein defining a new family of actin-binding proteins. J Biol Chem. 1999;274:33522–33530. doi: 10.1074/jbc.274.47.33522. [DOI] [PubMed] [Google Scholar]
- Takei Y, Teng J, Harada A, Hirokawa N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol. 2000;150:989–1000. doi: 10.1083/jcb.150.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng J, Takei Y, Harada A, Nakata T, Chen J, Hirokawa N. Synergistic effects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth, and microtubule organization. J Cell Biol. 2001;155:65–76. doi: 10.1083/jcb.200106025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Wu X, Kodama A, Fuchs E. ACF7 regulates cytoskeletal-focal adhesion dynamics and migration and has ATPase activity. Cell. 2008;135:137–148. doi: 10.1016/j.cell.2008.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HY, Lieska N, Shao D, Kriho V, Pappas GD. Immunotyping of radial glia and their glial derivatives during development of the rat spinal cord. J Neurocytol. 1993;22:558–571. doi: 10.1007/BF01189043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Innervation of the diaphragm is unaffected in Macf1 nestin-cKO mice. Control (A, C, E) or mutant (B, D, F) diaphragms were dissected from P0 pups and stained with neurofilament heavy chain antibodies (A, B; red in E, F) and α-bungarotoxin (C, D; green in E, F) to reveal phrenic nerves and muscle endplates, respectively. Arrows point to lower and upper branches of the phrenic nerves (A, B). E and F, higher magnifications of the superimposed images in A and C, and B and D, respectively. Scale bars, 200 μm.
Cell distributions across the cortex, but not their total numbers, are different in cKO brains. Bins were selected in the cortical sections presented in Fig. 2(A, control; B, cKO), and the numbers of stained cells were counted and plotted. The total number of cells in the cKO cortex was similar to that in the control cortex but their distribution was “flat”, without the peaks in the cortical plate and subventricular zone present in the control cortex. Scale bars, 50μm
MACF1 ABD is eliminated in the cKO cortex. Coronal sections of control (A) or mutant (B) brains were stained with MACF1-ABD polyclonal antibody. Scale bars, 25μm
MACF1 staining in the hippocampus. Coronal sections of control (A, C) and Macf1 cKO (B, D) hippocampi were stained with CU119 and MACF1-ABD polyclonal antibody. Scale bars, 50μm