

Abstract
Substance use disorder is a chronic condition of compulsive drug seeking and use that is mediated by stable changes in central reward pathways. Repeated use of abused drugs causes persistent alterations in gene expression responsible for the long-term behavioral and structural changes. Recently, it has been suggested that epigenetic mechanisms are responsible in part for these drug-induced changes in gene expression. One of the alluring aspects of epigenetic regulation of gene expression is that epigenetic mechanisms may provide transient and potentially stable conditions that in turn may ultimately participate in the molecular mechanisms required for neuronal changes subserving long-lasting changes in behavior. This review describes epigenetic mechanisms of gene regulation and then discusses the emerging role of epigenetics in drug-induced plasticity and behavior. Understanding these mechanisms that establish and maintain drug-dependent plasticity changes may lead to deeper understanding of substance use disorders as well as novel approaches to treatment.
Keywords: Chromatin, Cocaine, CREB, CBP, Drug-induced plasticity, Histone deacetylase
Introduction
Drug addiction is a disorder of compulsive drug seeking and use despite the associated negative consequences. It is a chronic condition such that even after long periods of abstinence, addicted individuals remain prone to relapse. Drugs of abuse are known to change neuronal structure and function in specific brain regions, resulting in persistent changes at the molecular, cellular, systems, and behavioral levels. For example, cocaine produces long-term brain changes that underlie addiction in part by activating certain genes. These changes are very long lasting; understanding how they are established and maintained is a key open question.
Preclinical research has identified neuroadaptive changes induced by drugs of abuse. These alterations in structural and behavioral plasticity are still evident up to 1 year after drug treatment in rodents [1]. Changes in gene expression are thought to underlie these structural and behavioral changes. Transcription factors such as ΔFosB and cyclic adenosine monophosphate response element–binding (CREB) protein regulate gene expression implicated in drug-induced plasticity [2]. A transcription factor is defined as a regulatory protein that binds to specific sequences in DNA and affects transcription of specific genes. However, transcription is not occurring on naked DNA, but rather in the context of chromatin, providing an additional level of complexity that is not yet fully understood. Chromatin is the protein complex that condenses and organizes genomic DNA (described in more detail subsequently). Thus, gene expression requires the orchestrated effort of not only transcription factors, but also very specific enzymatic protein complexes that modify chromatin structure to regulate gene expression.
Recently it has been shown that transcription factors implicated in regulating gene expression associated with drug-dependent plasticity can recruit chromatin-modifying enzymes that lead to long-lasting changes in gene expression [3]. Why is it critical to understand these chromatin-modifying mechanisms involved in the regulation of gene expression? The answer is that these mechanisms establish and maintain gene expression profiles that dictate cellular function in a persistent manner [4].
As chromatin-modifying mechanisms can change gene expression without changing the DNA sequence of regulatory elements driving gene expression, chromatin modification is an epigenetic mechanism of gene regulation (the prefix epi- meaning on, upon, or above). The preclinical research showing that these epigenetic mechanisms may be involved in drug-dependent changes in plasticity and behavior is critical for our understanding of drug addiction. These findings have profound implications for our understanding of the mechanisms that establish and maintain drug-dependent plasticity changes, and for potential novel avenues for treatment of drug addiction.
The purpose of this review is to examine the contribution of current literature to our understanding of the role of epigenetic mechanisms involved in substance use disorders. We begin by providing critical background in the field of epigenetics. Next, we describe the transcription factors important for mediating gene expression underlying drug-induced plasticity and behavior. Then we discuss the recruitment of chromatin-modifying enzymes by these transcription factors and the resulting effect on behavior. We conclude with a short summary on the implications of these findings and their impact on understanding and treating drug addiction.
Defining Epigenetics
Epigenetics is the study of changes in the regulation of gene expression and gene-product activity that are not dependent on DNA sequence. The term epigenetics was originally coined by C. H. Waddington in 1942 to describe the examination of “causal mechanisms” whereby “the genes of the genotype bring about phenotypic effects” [5]. The term has now taken on several new definitions, especially in the neurosciences. In non-neuroscience fields, the term refers to a stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence [6]. In a recent book on epigenetics by Allis et al. [7•], one can find two different definitions. In one section, epigenetics is defined as “change in phenotype that is heritable but does not involve DNA mutation.” In another section, epigenetics is defined as “changes in gene transcription through modulation of chromatin, which is not brought about by changes in the DNA sequence.” Notably, the term heritable is not part and parcel of the latter definition. As neuroscientists are by definition interested in the function of neurons, which are postmitotic differentiated cells, the definition of epigenetics normally used by neuroscientists has also dropped the heritable component [8–10••, 11].
As the number of publications relating to epigenetics rose from 50 in 1989 to nearly 6000 in 2008, it is obvious that regardless of how researchers define epigenetics, it has taken a central position in research. The National Institutes of Health describes epigenetics as “refer[ring] to both heritable changes in gene activity and expression (in the progeny of cells or of individuals) and also stable, long-term alterations in the transcriptional potential of a cell that are not necessarily heritable” [12]. This is the definition of epigenetics to which we adhere in this review.
Chromatin Modification, Chromatin Remodeling, Histone Variants, and DNA Methylation
The basic repeating unit of chromatin is the nucleosome, which is made up of a histone octamer that interacts with approximately 147 bp of genomic DNA per nucleosome. The histone octamer contains pairs of histone H2A, H2B, H3, and H4. The aminoterminal tails of these core histone proteins are the sites of several post-translational modifications (eg, acetylation, methylation, phosphorylation) carried out by an equally large number of histone-modifying enzymes (eg, acetyltransferases, deacetylases, methyltransferases, demethylases, kinases) [13•]. The manipulation of chromatin via the addition of functional groups to histone tails is referred to as chromatin modification, which serves two main purposes. The first is to provide recruitment signals for nonhistone proteins (eg, transcription factors) involved in transcriptional activation and silencing [13•, 14]. The second is to relax chromatin by disrupting contacts between nucleosomes and interactions between histone tails and genomic DNA (Fig. 1) [13•]. The functional consequence of these modifications is discussed in the next section. Importantly, chromatin modification should not be confused with chromatin remodeling, which refers to adenosine triphosphate-dependent enzymatic complexes (eg, SWI/SNF, ISWI, INO80, NURD) that restructure, mobilize, and remove nucleosomes to regulate access to genomic DNA for transcriptional activation [15, 16]. Chromatin structure also may be manipulated and regulated via histone variant incorporation (eg, H3.3, macroH2A, H2AZ, H2AX) [17]. Thus, epigenetic regulation of transcription may be thought of as the coordinated interplay of mechanisms (eg, chromatin-modifying enzymes, nucleosome remodeling complexes, histone variant incorporation, DNA methylation) that translates incoming signaling events by altering chromatin structure in a specific and precise manner that in turn regulates gene expression profiles for defined cellular functions. An important question then is how these mechanisms function with regard to drug-dependent changes in plasticity and behavior.
Fig. 1.
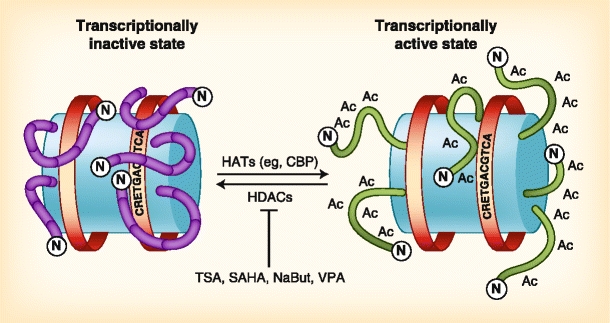
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) have opposing activities. Nucleosomes, shown as blue barrels, are composed of histone octamers that are involved in binding DNA. Histone N-terminal tails, shown in purple (left nucleosome) or in green (right nucleosome), contain residues that directly interact with the genomic DNA. In a transcriptionally silent state (left nucleosome), positively charged lysine residues interact with the negatively charged DNA phosphate backbone, whereas in a transcriptionally active state (right nucleosome), lysine residues are modified by acetyl groups that neutralize a lysine’s positive charge. Transcriptional coactivators such as cyclic adenosine monophosphate response element–binding protein (CBP) are histone acetyltransferases that acetylate lysine residues on histone N-terminal tails. This acetylated state correlates with transcriptional activation. The opposing activity is carried out by HDACs, which remove acetyl groups from lysine residues, correlating with transcriptional silencing. Competitive inhibitors of HDACs (eg, trichostatin A [TSA], suberoylanilide hydroxamic acid [SAHA], sodium butyrate [NaBut], and valproic acid [VPA]) directly interact with and prevent HDACs from deacetylating lysines, thus inducing a hyperacetylated and transcriptionally active state
Emerging Role of Epigenetics in Drug-induced Plasticity and Behavior
To investigate the mechanisms involved in the establishment and maintenance of drug dependence, it is necessary to work with an animal model. Rodents are the most tractable system in that genetic and pharmacologic manipulations allow for the elucidation of pathways underlying drug-induced behavior. Several rodent behavioral paradigms are used to measure different aspects of drug-seeking and drug-taking behaviors. These models are described in Table 1. Most work on epigenetic mechanisms with respect to drug abuse has been done in cocaine research; thus, findings are described primarily for this drug.
Table 1.
Preclinical models of drug addiction
| Locomotor sensitization |
| Psychomotor sensitization is observed across many species, including humans, and models some features of addiction. The acute motor-activating response produced by administration of psychostimulants is augmented with repeated exposure, which has been shown in rodents to be associated with augmented drug reward and increased vulnerability to relapse [46]. Behavioral sensitization can be separated into two components: induction and expression. Induction of sensitization is the progressive increase in locomotor activity during the repeated drug treatment. Expression of sensitization is demonstrated following challenge with a low dose of psychostimulant after a drug-free period. This response can persist up to 1 year after drug treatment. |
| Conditioned place preference |
| Conditioned place preference is used to evaluate the rewarding or aversive motivational effects of drugs [47]. The primary effects of a drug are repeatedly paired with an environmental context that acquires the motivational properties of the drug. Animals receive saline injections in an alternative context. Following multiple pairings, a choice test allows the animal unrestricted access to both contexts in the absence of drug. An increase in time spent in the drug-paired context relative to that before drug conditioning, or compared with the saline-paired context, is evidence that the drug was rewarding. |
| Self-administration |
| Self-administration is a method in preclinical drug abuse research that best models drug consumption in humans [48]. The paradigm is based on principles of operant and classical conditioning in which animals control the delivery of drug infusions by performing an operant response (eg, pressing a lever or actively sniffing a photobeam-triggered hole). Motivation to administer drugs is assessed by progressively increasing the number of responses required to receive a single infusion of drug. Drug self-administration has the most direct connection to addictive behavior in a natural environment. |
| Extinction and reinstatement |
| Extinction training is repeated re-exposure to the drug-paired environment after removal of the unconditioned stimulus (eg, drug) until the response is diminished to preconditioning levels. This is an active learning process by which contextual associations no longer predict drug availability. Reinstatement of drug-seeking behavior in laboratory animals is induced by conditions described to trigger drug craving and relapse in humans, such as acute re-exposure to the drug, drug-associated cues, or stress [48]. Reinstatement behavior is measured by the recovery of operant responding (self-administration) or time spent in drug-paired context (conditioned place preference) after extinction of these responses. |
Transcription Factors Known to Mediate Gene Expression Underlying Drug-induced Plasticity and Behavior
A common mechanism of many abused drugs is the activation of certain transcription factors that induce patterns of gene expression in central reward pathways, which is distinct from that of nonaddictive drugs [18]. Through the regulation of genes, transcription factors can lead to adaptive changes in neuronal structure and function [19]. The best-characterized transcription factors in drug addiction are CREB and ΔFosB [20]. Temporally, CREB is induced rapidly following each drug treatment and then returns to baseline after a few hours. In contrast, ΔFosB accumulates slowly after each drug exposure and is highly stable for months after cessation. In addition to temporal differences in activation, these transcription factors also have been shown to mediate distinct aspects of drug addiction.
In animal models, experimental approaches that artificially increase or decrease CREB function demonstrate that its activation leads to a negative state of decreased reward and increased drug tolerance and dependence (Table 2) [20]. CREB is induced in reward-related brain regions, such as the nucleus accumbens (NAc), not only by drug administration but also in response to drug-paired cues using the cocaine conditioned place preference (CPP) model (Table 1) [21]. To further characterize the contribution of CREB to reward-related behaviors, several approaches have been used. Using herpes simplex virus to deliver wild-type (normal form) CREB or a mutant form of CREB (mCREB), transient overexpression can be induced when the virus is infused into a specific brain region (eg, the NAc). mCREB can bind to DNA but cannot recruit transcriptional machinery because a mutation in the kinase inducible domain prevents phosphorylation of CREB, which normally results in the activation of CREB. Thus, the mutation in mCREB effectively impairs CREB-mediated transcription [22]. Behavioral studies using viral overexpression of the wild-type CREB have found that increased CREB expression in the NAc results in a decrease in cocaine’s rewarding value as measured by CPP [22, 23]. Conversely, expression of mCREB leads to enhanced cocaine reward at very low drug doses [22, 23]. These findings suggest that CREB activity in the NAc decreases cocaine reward, although the role of CREB in reward may be region specific. Opposing reward behaviors are observed when CREB is infused into two distinct subregions of the ventral tegmental area, a dopaminergic nucleus, suggesting that it may have different roles within the reward circuitry [24].
Table 2.
Effects of CREB on reward-related behaviors
| Study | Drug exposure | Model | Effect of CREB | Effect on behavior |
|---|---|---|---|---|
| Carlezon et al. [22] | Cocaine CPP | HSV-CREB | ↑ pCREB NAcSh | ↓ CPP (aversion at low dose) |
| HSV-mCREB | ↓ pCREB NAcSh | ↑ CPP at low dose | ||
| Pliakas et al. [23] | Cocaine CPP | HSV-CREB | ↑ pCREB NAcSh | ↓ CPP (aversion at low dose) |
| HSV-mCREB | ↓ pCREB NAcSh | ↑ CPP at low dose | ||
| Olson et al. [24] | Cocaine CPP | HSV-CREB | ↑ pCREB VTA | ↑ CPP in rostral, ↓ CPP in caudal portion |
| HSV-mCREB | ↓ pCREB VTA | ↓ CPP in rostral, ↑ CPP in caudal portion | ||
| Fasano et al. [26] | Cocaine CPP, sensitization | Str-KCREB | ↓ pCREB DStr | ↑ CPP and locomotor sensitization at low dose |
| Walters and Blendy [25] | Cocaine CPP, sensitization | CREBαΔ | ↓ pCREB | ↑ CPP, ↑ expression of locomotor sensitization |
| Valverde et al. [49] | Cocaine CPP | CREBαΔ | ↓ pCREB | No change in CPP at high dose |
| Choi et al. [26] | Cocaine self-administration | CREB antisense | ↓ pCREB NAc | ↓ cocaine intake |
Analysis of CREB’s effects on behavior has been conducted in discrete brain regions such as the nucleus accumbens shell, ventral tegmental area, and dorsal striatum
CPP conditioned place preference, CREB cyclic adenosine monophosphate response element–binding protein, DStr dorsal striatum, HSV herpes simplex virus, KCREB mutant form of CREB that heterodimerizes with the wild-type form; mCREB mutant form of CREB, NAc nucleus accumbens, NAcSh NAc shell, pCREB phosphorylated (activated) form of CREB, Str striatum, VTA ventral tegmental area
Genetic manipulations also result in impaired CREB activity. CREBαΔ mutant mice lack the α and Δ isoforms of CREB, drastically reducing its activity [25]. Transgenic mice expressing KCREB, a mutant form of CREB that heterodimerizes with the wild-type form, prevent DNA binding primarily in the dorsal striatum and reduce CREB-mediated transcription [26]. These genetic models with decreased CREB activity show enhanced CPP and sensitization to cocaine compared with wild-type controls [25, 26], further supporting CREB mediation of the negative aspects of cocaine. However, a reduction in NAc CREB by antisense infusions reduces cocaine self-administration behavior [27]. Although this may seem paradoxical, the dissociation may be a result of the behavior tasks measuring different stages of addiction. The CPP and sensitization studies tested for changes during the acquisition phase, whereas the self-administration study was conducted in animals that had already acquired and maintained drug response. Overall, these findings suggest that CREB influences early drug-seeking behavior.
ΔFosB, which accumulates slowly after each drug exposure, seems to mediate the positive rewarding and sensitizing effects of abused drugs. Inducible ΔFosB overexpression results in increased cocaine-induced locomotion and CPP [28]. Furthermore, it increases the motivation to self-administer cocaine [29]. ΔFosB is one of the most persistently upregulated proteins after drug exposure, but it is still degraded and returns to predrug levels after 1 month to 2 months [20]. This suggests that the ΔFosB protein itself does not maintain drug dependence; instead, persistent alterations in ΔFosB-regulated genes may be responsible.
Transcription Factors Implicated in Drug-induced Plasticity and Behavior Recruit Chromatin-modifying Enzymes
Drug-associated plasticity is a product of gene activation triggered in part by the previously described transcription factors. However, gene expression also requires the coordinated effort of enzymatic protein complexes that modify chromatin structure. Cocaine exposure causes an overall increase in gene expression in the NAc, and many of these genes are regulated by CREB and ΔFosB [30]. Because there are many possible histone modifications and a staggering combinatorial complexity to the possible patterns of histone modifications present at any given promoter (the specific upstream sequence in DNA that coordinates expression of a gene), new and powerful techniques have been developed to begin to understand this complexity and what it may predict with respect to the expression of a given gene. One such technique, called chromatin immunoprecipitation, involves the use of antibodies that recognize a single specific histone modification. Chromatin containing the specific histone modification of interest can be isolated from the rest of the chromatin by being pulled away (immunoprecipitated) using these antibodies. Then one can use two different approaches to examine which genes have increases or decreases in that histone modification at their promoter. One approach is a direct method in which one already has a target gene of interest, and quantitative polymerase chain reaction can be used to amplify the promoter from the chromatin that has been pulled down by the specific antibody. This direct approach reveals how specific histone modifications change before and after drug exposure and how these changes in histone modifications correlate with altered gene expression [31, 32]. A newer approach involves using the chromatin that has been pulled down by specific antibodies to hybridize it against microarrays containing promoter regions of about 20,000 genes. This approach has provided a more global understanding of cocaine-regulated gene targets. In general, cocaine-activated genes are marked by increased histone acetylation, whereas genes that are repressed following cocaine are marked by histone methylation [33••]. In combination with examining the presence of specific transcription factors at the promoters of genes, it is possible to gain an understanding of how different sets of genes are regulated in different ways [33••].
CREB is the best-characterized transcription factor shown to direct a histone-modifying enzyme to the gene promoter. Phosphorylated CREB at the gene promoter region recruits CREB-binding protein (CBP), a potent histone acetyltransferase and transcriptional coactivator that recruits the basal transcriptional machinery [34]. CBP acetylates histones, allowing chromatin structure to relax and increase expression of target genes. The regulation of gene expression by chromatin modification induced by acute cocaine has been shown for many immediate early genes, such as c-fos and FosB [31, 32]. Infusions of CREB antisense or transgenic CBP mutant mice (that demonstrate decreased CBP function) show decreased CBP occupancy of the FosB promoter and reduced stimulant-induced Fos activation [32, 35]. Furthermore, the drug-induced temporal pattern of acetylation parallels the induction of mRNA expression [31]. Whereas a single dose of cocaine activates c-fos, repeated treatment is necessary to induce bdnf and cdk5 genes. This pattern of gene activation is also seen with acetylation, and chronically upregulated genes show significant acetylation days to weeks after the last cocaine treatment [31].
ΔFosB was recently shown to recruit chromatin-modifying enzymes to cocaine-regulated genes. For example, the promoters of genes chronically upregulated following drug treatment, such as cdk5, are ΔFosB bound and additionally contain histone modifications. ΔFosB facilitates the recruitment of transcriptional activators such as histone acetyltransferases, as well as Brg1-containing SWI/SNF chromatin remodeling proteins [31]. SWI/SNF is an adenosine triphosphate-dependent chromatin remodeling complex that mobilizes the histone octamers. This movement permits binding by protein complexes necessary for initiating transcription. Overall, these findings demonstrate that CREB and ΔFosB can recruit histone-modifying enzymes that may mediate the persistent molecular changes caused by drugs.
Effect of Histone Deacetylase Inhibition on Drug-induced Plasticity and Behavior
Drugs of abuse enhance histone acetylation by increasing histone acetyltransferase or decreasing histone deacetylase (HDAC) activity (Fig. 1). For example, cocaine-self-administering animals have reduced HDAC activity in the frontal cortex and NAc, resulting in increased histone acetylation [36]. As shown in Fig. 1, a histone hyperacetylated state may be achieved by using HDAC inhibitors (eg, sodium butyrate, trichostatin A), which increase global histone acetylation. HDAC inhibition has been shown to potentiate cocaine-induced locomotion and reward [31, 36, 37••, 38]. HDAC inhibition also has been shown to increase drug intake in animals maintained on cocaine self-administration but may reduce intake when given before acquisition [36, 39]. Conversely, overexpression of HDAC4 or HDAC5, but not HDAC9, in the NAc attenuates cocaine CPP, further supporting the hypothesis that chromatin modification is critical to the regulation of gene expression required for changes in drug-seeking behavior [31, 37••]. Importantly, experiments using individual HDACs provide critical insight into which HDACs mediate drug-induced plasticity. Table 3 summarizes the effects of HDAC inhibition in models of drug addiction.
Table 3.
Effects of histone acetylation on reward-related behaviors
| Study | Drug exposure | Model | Effect of histone acetylation | Effect on behavior |
|---|---|---|---|---|
| Levine et al. [32] | Cocaine sensitization | CBP haploinsufficient mice | ↓ (↓HAT) | ↓ cocaine sensitization |
| Kumar et al. [31] | Cocaine CPP | HSV-HDAC4 | ↓ (↑HDAC4 NAc) | ↓ CPP |
| TSA | ↑ (↓HDAC) | ↑ CPP | ||
| Renthal et al. [37••] | Cocaine CPP | SAHA (continuous intra-NAc) | ↑ (↓ HDAC NAc) | ↑ CPP |
| HSV-HDAC5 | ↓ (↑HDAC5 NAc) | ↓ CPP | ||
| HSV-HDAC9 | ↓ (↑HDAC9 NAc) | No change CPP | ||
| Romieu et al. [36] | Cocaine self-administration | TSA | ↑ (↓HDAC) | ↓ cocaine intake, early acquisition |
| Cocaine sensitization | TSA 4 d prior to cocaine | ↑ (↓HDAC) | ↓ cocaine locomotor sensitization | |
| Sun et al. [39] | Cocaine self-administration | Sodium butyrate | ↑ (↓HDAC) | ↑ cocaine intake, maintenance |
| Sanchis-Segura et al. [38] | Cocaine sensitization | Sodium butyrate | ↑ (↓HDAC) | ↑ cocaine-induced locomotion |
| Malvaez et al. [42••] | Extinction of cocaine CPP | Sodium butyrate | ↑ (↓HDAC) | ↑ extinction of CPP |
CBP CREB-binding protein, CPP conditioned place preference, CREB cyclic adenosine monophosphate response element–binding protein, HAT histone acetyltransferase, HDAC histone deacetylase, HSV herpes simplex virus, NAc nucleus accumbens, SAHA suberoylanilide hydroxamic acid, TSA trichostatin A
Sirtuins, a third class of HDACs, can deacetylate histones and nonhistone proteins, such as transcription factors (eg, nuclear factor-κB) [40]. Chronic cocaine exposure increases expression of Sirt1 and Sirt2. This increased expression correlates with increased acetylation at the Sirt1 and Sirt2 promoters, as well as increased sirtuin enzymatic activity [33••]. Pharmacologic manipulation of sirtuins leads to interesting changes in cocaine-induced behavior. A sirtuin activator enhances CPP using a low dose of cocaine, whereas a sirtuin inhibitor attenuates CPP and reduces cocaine self-administration behavior [33••]. A single exposure to cocaine does not alter sirtuin activity; however, upregulation of the sirtuins after chronic cocaine may mediate the stable neuroadaptive changes involved in maintaining addiction. The inhibitors of class I and II HDACs described previously increase behavioral sensitivity to cocaine, but the inhibitors of class III HDACs (sirtuins) reduce sensitivity, suggesting that they may work through mechanisms beyond acetylating histones.
Behavioral and Epigenetic Mechanisms in Extinction of Cocaine-induced Memories
Nonspecific HDAC inhibitors were recently shown to enhance synaptic plasticity and long-term memory [10••]. For example, we recently demonstrated that the HDAC inhibitor sodium butyrate can transform a learning event that does not lead to long-term memory into a learning event that does result in significant long-term memory [41••]. Furthermore, sodium butyrate also generated a form of long-term memory that persisted beyond the point at which normal memory failed [41••]. These demonstrations of modulating memory via HDAC inhibition have considerable therapeutic potential for many cognitive disorders.
For example, an exciting application for the modulation of long-term memory via HDAC inhibition in substance disorders is the pharmacologic enhancement of extinction learning. After establishing a preference using the CPP model, animals can extinguish drug-seeking behaviors after repeated re-exposure to the drug-paired environment in the absence of the drug. Over time, animals no longer demonstrate drug-seeking behavior in response to the environment, although a prime with drug administration reinstates this action. A recent study from our laboratory showed that HDAC inhibition after establishing a CPP significantly facilitated extinction of drug-seeking behavior [42••]. Animals treated with an HDAC inhibitor extinguished cocaine-induced CPP more quickly and to a greater extent than did vehicle-treated animals. Even more remarkable, the extinction of cocaine seeking via HDAC inhibition modulates extinction learning such that reinstatement behavior is significantly attenuated [42••], suggesting that the HDAC inhibition-dependent extinction may be persistent. These findings suggest that in a normal situation, behavioral extinction of drug-seeking behavior, as measured by CPP, is subject to relapse-like behavior (reinstatement). However, behavioral extinction in combination with HDAC inhibition-dependent facilitation of extinction may generate a form of extinction that is much more persistent and thus refractive to relapse-like behavior.
Although further research is needed, this behavioral and epigenetic approach to extinction of drug-seeking behavior may be valuable in treating drug dependence. One caveat is that HDAC inhibition enhances initial learning and memory as well and, as discussed previously, has been shown to potentiate cocaine-induced locomotion and reward [31, 37••, 38]. Thus, HDAC inhibition would have to be used only during extinction consolidation processes.
Clinical Implications
Examining epigenetic mechanisms in human tissue poses a much greater challenge compared with animal models. Substance abusers vary in drug intake and exposure period and often use multiple drugs and have comorbid psychiatric conditions. Despite these challenges, some studies have identified genes altered among cocaine users, many of which overlap with gene expression profiles in animal models [43•]. Recently, alterations in cortical gene expression showed corresponding changes in histone and DNA methylation in these genes in postmortem brains of patients with schizophrenia [44••]. Tsankova et al. [45••] provided an excellent review on epigenetic regulation in psychiatric disorders, including depression and other disorders. With the development of powerful new techniques such as genome-wide microarrays of chromatin modifications, researchers can identify new gene targets underlying the pathophysiology of addiction and compare those of animal models to human postmortem brain data.
Conclusions
We have presented some of the growing body of evidence for epigenetic mechanisms involved in mediating the development and maintenance of substance abuse. Repeated use of abused drugs causes persistent alterations in gene expression responsible for the long-term behavioral and structural changes. Specific transcription factors and chromatin-modifying enzymes regulate drug-induced gene expression during acquisition and maintenance of drug seeking. These changes may underlie the transition from recreational use to compulsive drug-taking behavior, and pharmacologic tools (eg, HDAC inhibitors) in conjunction with behavioral therapy may aid in reversing this process.
Acknowledgments
The authors wish to thank Steve Boikess, Matt Lattal, Melissa Malvaez, and Antonio Wood for their helpful discussions and critical reading of this manuscript. This work was supported by the National Institute of Drug Abuse (R01DA025922 to Dr. Wood) and the National Institute of Mental Health (R01MH081004 to Dr. Wood). The authors wish to apologize to their colleagues whose work was not included in this review due to space limitations.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
- 1.Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):33–46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 2.Nestler EJ. Molecular mechanisms of drug addiction. Neuropharmacology. 2004;47(Suppl 1):24–32. doi: 10.1016/j.neuropharm.2004.06.031. [DOI] [PubMed] [Google Scholar]
- 3.Renthal W, Carle TL, Maze I, et al. Delta FosB mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. J Neurosci. 2008;28:7344–7349. doi: 10.1523/JNEUROSCI.1043-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turner BM. Cellular memory and the histone code. Cell. 2002;111:285–291. doi: 10.1016/S0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- 5.Haig D. The (dual) origin of epigenetics. Cold Spring Harb Symp Quant Biol. 2004;69:67–70. doi: 10.1101/sqb.2004.69.67. [DOI] [PubMed] [Google Scholar]
- 6.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.• Allis CD, Jenuwein T, Reinberg D: Epigenetics. Edited by Caparros ML. Cold Spring Harbor, NY: Cold Spring Harbor Press; 2007. This is an excellent reference for understanding the development of the field of epigenetics and classic examples of epigenetic mechanisms in many disciplines.
- 8.Levenson JM, Sweatt JD. Epigenetic mechanisms of memory formation. Nat Rev Neurosci. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- 9.Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol. 2008;8:57–64. doi: 10.1016/j.coph.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.•• Barrett RM, Wood MA: Beyond transcription factors: the role of chromatin modifying enzymes in regulating transcription required for memory. Learn Mem 2008, 15:460–467. This review highlights the current research in understanding the epigenetic mechanisms involved in long-term memory formation. [DOI] [PMC free article] [PubMed]
- 11.Gräff J, Mansuy IM. Epigenetic codes in cognition and behavior. Behav Brain Res. 2008;192:70–87. doi: 10.1016/j.bbr.2008.01.021. [DOI] [PubMed] [Google Scholar]
- 12.NIH Roadmap for Medical Research: Epigenomics overview. Available at http://nihroadmap.nih.gov/epigenomics/index.asp. Accessed October 2009.
- 13.• Kouzarides T: Chromatin modifications and their function. Cell 2007, 128:693–705. This review highlights the different functions of chromatin modifications in regulating chromatin structure and the recruitment of specialized factors that “read” these modifications to regulate gene expression. [DOI] [PubMed]
- 14.Taverna SD, Li H, Ruthenburg AJ, et al. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saha A, Wittmeyer J, Cairns BR. Chromatin remodelling: the industrial revolution of DNA around histones. Nat Rev Mol Cell Biol. 2006;7:437–447. doi: 10.1038/nrm1945. [DOI] [PubMed] [Google Scholar]
- 16.Saha A, Wittmeyer J, Cairns BR. Mechanisms for nucleosome movement by ATP-dependent chromatin remodeling complexes. Results Probl Cell Differ. 2006;41:127–148. doi: 10.1007/400_005. [DOI] [PubMed] [Google Scholar]
- 17.Ausió J. Histone variants—the structure behind the function. Brief Funct Genomic Proteomic. 2006;5:228–243. doi: 10.1093/bfgp/ell020. [DOI] [PubMed] [Google Scholar]
- 18.Valjent E, Pagès C, Hervé D, et al. Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci. 2004;19:1826–1836. doi: 10.1111/j.1460-9568.2004.03278.x. [DOI] [PubMed] [Google Scholar]
- 19.Kalivas PW, Toda S, Bowers MS, et al. The temporal sequence of changes in gene expression by drugs of abuse. Methods Mol Med. 2003;79:3–11. doi: 10.1385/1-59259-358-5:03. [DOI] [PubMed] [Google Scholar]
- 20.Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- 21.Miller CA, Marshall JF. Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron. 2005;47:873–884. doi: 10.1016/j.neuron.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 22.Carlezon WA, Jr, Thome J, Olson VG, et al. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- 23.Pliakas AM, Carlson RR, Neve RL, et al. Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J Neurosci. 2001;21:7397–7403. doi: 10.1523/JNEUROSCI.21-18-07397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olson VG, Zabetian CP, Bolanos CA, et al. Regulation of drug reward by cAMP response element-binding protein: evidence for two functionally distinct subregions of the ventral tegmental area. J Neurosci. 2005;25:5553–5562. doi: 10.1523/JNEUROSCI.0345-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walters CL, Blendy JA. Different requirements for cAMP response element binding protein in positive and negative reinforcing properties of drugs of abuse. J Neurosci. 2001;21:9438–9444. doi: 10.1523/JNEUROSCI.21-23-09438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fasano S, Pittenger C, Brambilla R. Inhibition of CREB activity in the dorsal portion of the striatum potentiates behavioral responses to drugs of abuse. Front Behav Neurosci. 2009;3:29. doi: 10.3389/neuro.08.029.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi KH, Whisler K, Graham DL, Self DW. Antisense-induced reduction in nucleus accumbens cyclic AMP response element binding protein attenuates cocaine reinforcement. Neuroscience. 2006;137:373–383. doi: 10.1016/j.neuroscience.2005.10.049. [DOI] [PubMed] [Google Scholar]
- 28.Kelz MB, Chen J, Carlezon WA, Jr, et al. Expression of the transcription factor deltaFosB in the brain controls sensitivity to cocaine. Nature. 1999;401:272–276. doi: 10.1038/45790. [DOI] [PubMed] [Google Scholar]
- 29.Colby CR, Whisler K, Steffen C, et al. Striatal cell type-specific overexpression of DeltaFosB enhances incentive for cocaine. J Neurosci. 2003;23:2488–2493. doi: 10.1523/JNEUROSCI.23-06-02488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat Neurosci. 2003;6:1208–1215. doi: 10.1038/nn1143. [DOI] [PubMed] [Google Scholar]
- 31.Kumar A, Choi KH, Renthal W, et al. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 32.Levine AA, Guan Z, Barco A, et al. CREB-binding protein controls response to cocaine by acetylating histones at the fosB promoter in the mouse striatum. Proc Natl Acad Sci U S A. 2005;102:19186–19191. doi: 10.1073/pnas.0509735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.•• Renthal W, Kumar A, Xiao G, et al.: Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron 2009, 62:335–348. This study examines how coordinate gene regulation is affected by cocaine using a genome-wide ChIP-chip approach. This approach revealed a critical role for a type of histone deacetylase called sirtuins in cocaine-mediated behavior. [DOI] [PMC free article] [PubMed]
- 34.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 35.Konradi C, Cole RL, Heckers S, Hyman SE. Amphetamine regulates gene expression in rat striatum via transcription factor CREB. J Neurosci. 1994;14:5623–5634. doi: 10.1523/JNEUROSCI.14-09-05623.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Romieu P, Host L, Gobaille S, et al. Histone deacetylase inhibitors decrease cocaine but not sucrose self-administration in rats. J Neurosci. 2008;28:9342–9348. doi: 10.1523/JNEUROSCI.0379-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.•• Renthal W, Maze I, Krishnan V, et al.: Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 2007, 56:517–529. This elegant study demonstrated a critical role for a specific histone deacetylase (HDAC5) in molecular mechanisms underlying behavioral changes observed in animal models of addiction and depression. [DOI] [PubMed]
- 38.Sanchis-Segura C, Lopez-Atalaya JP, Barco A. Selective boosting of transcriptional and behavioral responses to drugs of abuse by histone deacetylase inhibition. Neuropsychopharmacology. 2009;34:2642–2654. doi: 10.1038/npp.2009.125. [DOI] [PubMed] [Google Scholar]
- 39.Sun J, Wang L, Jiang B, et al. The effects of sodium butyrate, an inhibitor of histone deacetylase, on the cocaine- and sucrose-maintained self-administration in rats. Neurosci Lett. 2008;441:72–76. doi: 10.1016/j.neulet.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 40.Denu JM. The Sir 2 family of protein deacetylases. Curr Opin Chem Biol. 2005;9:431–440. doi: 10.1016/j.cbpa.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 41.•• Stefanko DP, Barrett RM, Ly AR, et al.: Modulation of long-term memory for object recognition via HDAC inhibition. Proc Natl Acad Sci U S A 2009, 106:9447–9452. This study demonstrated that HDAC inhibition can transform a learning event that does not lead to long-term memory into one that does. It also demonstrated that HDAC inhibition can generate a form of long-term memory that persists beyond the point at which normal memory fails. [DOI] [PMC free article] [PubMed]
- 42.•• Malvaez M, Sanchis-Segura C, Vo D, et al.: Modulation of chromatin modification facilitates extinction of cocaine-induced conditioned place preference. Biol Psychiatry 2010, 67:36–43. This study demonstrated that HDAC inhibition during extinction learning can significantly facilitate extinction of drug-seeking behavior. Furthermore, HDAC inhibition generated a form of extinction that was persistent and refractive to drug-primed reinstatement. [DOI] [PMC free article] [PubMed]
- 43.• Lull ME, Freeman WM, Vrana KE, Mash DC: Correlating human and animal studies of cocaine abuse and gene expression. Ann N Y Acad Sci 2008, 1141:58–75. This review integrates human and animal studies of cocaine-induced gene expression, bridging the gap between these fields. [DOI] [PubMed]
- 44.•• Akbarian S: The molecular pathology of schizophrenia—focus on histone and DNA modifications. Brain Res Bull 2009 Sep 1 (Epub ahead of print). This review discusses epigenetic mechanisms identified for schizophrenia, especially with regard to studying these mechanisms from postmortem tissue and interpreting their function and role in the molecular pathology of schizophrenia.
- 45.•• Tsankova N, Renthal W, Kumar A, Nestler EJ: Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 2007, 8:355–367. This review provides a comprehensive—yet easily accessible—view into the epigenetic mechanisms contributing to several psychiatric disorders. [DOI] [PubMed]
- 46.Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-P. [DOI] [PubMed] [Google Scholar]
- 47.Bardo MT, Bevins RA. Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology (Berl) 2000;153:31–43. doi: 10.1007/s002130000569. [DOI] [PubMed] [Google Scholar]
- 48.Shaham Y, Shalev U, Lu L, et al. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (Berl) 2003;168:3–20. doi: 10.1007/s00213-002-1224-x. [DOI] [PubMed] [Google Scholar]
- 49.Valverde O, Mantamadiotis T, Torrecilla M, et al. Modulation of anxiety-like behavior and morphine dependence in CREB-deficient mice. Neuropsychopharmacology. 2004;29:1122–1133. doi: 10.1038/sj.npp.1300416. [DOI] [PubMed] [Google Scholar]