

Abstract
Background & Aims
Colon cancer is one of the best-understood neoplasms from a genetic perspective, yet it remains the second most common cause of cancer-related death. Post-transcriptional regulation mediated by RNA binding proteins or microRNAs coordinately targets multiple genes, holding promise involved in colon cancer initiation and development. Here we studied the role of RNA binding protein QKI in colon cancer.
Methods
We observed the expression pattern of QKI in normal colon and colon cancers through RT-PCR and Western blot. Bisulfite-sequencing and methylation specific PCR were applied for QKI promoter methylation analysis. We used enterocyte differentiation markers and soft agar assay to test the role of QKI in colon differentiation and colon cancer development. 3′ UTR reporter assay and RNA-IP were used to confirm the interaction between QKI and β-catenin or p27.
Results
QKI is significantly downregulated and even absent in some colon cancers, which at least partially due to the promoter hypermethylation. Forced expression of QKI in the colon cancer cells increased the expression of enterocyte differentiation marker IAP and lactase, together with the cell cycle accumulation in G1 phase, enhancement of p27Kip1 protein level and membrane localized β-catenin. Finally, QKI overexpression reduced the proliferation and tumorigenesis ability.
Conclusions
Our study establishes that QKI functions as a principal regulator in the differentiation of colon epithelium and a suppressor of carcinogenesis through coordinately targeting multiple genes associated with cell growth and differentiation, whose deregulation by methylation in colon is involved in cancer onset and progress.
Introduction
Colon cancer is one of the most common cancers worldwide, causing half a million deaths each year.1 The model of colorectal tumorigenesis includes several genetic changes that are required for cancer initiation and progression, such as the APC pathway, β-catenin/Tcf pathway, RAS/RAF pathway and p53 pathway.2 Even though the above genetic changes have been characterized in most of the colon cancer patients, the initial factors corresponding to these changes besides the genetic reasons are not elucidated and it is yet to be established whether other important pathways are involved.
Differentiation deficiency is one of the key characteristics of cancer. Besides the ordinary role in cell cycle regulation, p27 also plays an important role in promoting intestinal cell differentiation3–5 and initiates senescence as a barrier of carcinogenesis.6 Although mutations of p27 are rarely reported, reduced expression of p27 is a common feature of colon cancer.7–9 Another key component of colon epithelial differentiation is Wnt/β-catenin signal. Upon Wnt stimulation, nuclear translocation of membrane-bound β-catenin is induced, which plays a key role in cell-fate determination. Tight somatic regulation of this signal is essential. The transactivation of β-catenin/TCF target genes induced by Wnt pathway mutations constitutes the primary transforming event in colorectal cancer (CRC). Disruption of β-catenin/TCF-4 activity in CRC cells induces G1 arrest and intestinal differentiation program.10 In contrast, the membrane bound β-catenin is an indicator of the differentiation status,11 which in turn facilitates the differentiation.
Nowadays, posttranscriptional regulation are believed to be involved in the regulation of a wide variety of fundamental cellular processes, including cell growth and cell cycle progression, differentiation, and apoptosis. The RNA binding protein QKI belongs to the evolutionarily conserved STAR family and the QKI gene produces a diverse set of proteins by alternative splicing.12 The three well studied isoforms (QKI-5, -6, and -7) appear to have different roles in development. They are constructed with the same 311-amino acid body but have different carboxyl tails consisting of 30, 8, and 14 amino acids, respectively. QKI-5 is prominently expressed in early embryogenesis, whose mutation is believed to be responsible for the lethality at around 9.5 days of gestation13. In contrast, QKI-6 and -7 are mainly expressed in CNS in late development when myelination starts. QKI-6 and -7 are believed to play a fundamental role in myelination through coordinately regulating mRNA targets such as MAG, p27, MBP.5, 14–16 Besides these validated targets, Galarneau A et al has defined the QRE as a bipartite consensus sequence NACUAAY-N(1-20)-UAAY and predicted nearly 1,430 new putative mRNA targets, in which both p27 and β-catenin are included.17 QKI are involved in the regulation of mRNA stability, nuclear retention, RNA transportation, and translational modulation through interaction with QREs locating in the UTR of target mRNAs in the heterodimer or homodimer form.14 The lethal phenotype in QKI knock-out mice18 and its widespread expression in different cell types12, 19 underscore the central importance of this gene in regulation of normal cellular functions. Furthermore, evidence of alterations of QKI expression in neoplastic tissues,20–23 together with the identification of QKI as an important player in embryogenesis 23 and a critical regulator of p27,15 suggests that aberration of this molecule may contribute to uncontrolled cell growth and transformation. Besides, tyrosine phosphorylation of QKI represses its RNA binding activity,24, 25 while excessive tyrosine kinase activity is obviously detected in multiple cancers,26–28 leading to the possibility of aberrant QKI function in cancer cells.
It is thus interesting to test whether this RNA binding protein QKI is present in normal GI tract and its relevance with the colorectal carcinogenesis. In order to answer these questions, we have studied the expression of QKI, its related targets and their interaction by in vivo and in vitro models. Our data demonstrate for the first time that RNA binding protein QKI is normally expressed in the colon epithelium in young individuals while absent or downregulated in colon cancers due to aberrant hypermethylation. Furthermore, by increasing the expression of p27 and membrane bound β-catenin, QKI facilitates the differentiation of colon epithelial cells. Together, the data implicate QKI as one of the key regulators of cell cycle withdrawal and cell differentiation of the gastrointestinal epithelium, whose reduced expression from promoter hypermethylation may contribute to gastrointestinal cancer initiation.
Materials and Methods
Tissue sample collection
Frozen tumor and normal colon mucosa were collected from colectomy specimens from Tangdu Hospital, the University of the Fourth Military Medical University. Tissue was frozen in liquid nitrogen within 30 min of resection. Nonneoplastic mucosa from colon was dissected free of muscle and histologically confirmed to be tumor free by frozen section. For cancer tissue, tumor purity of over 70% was confirmed by frozen section for each case before submission for DNA and protein extraction. Information on the patient’s age and sex, as well as the Duke’s tumor stage is collected. This study was approved by the Ethics Committee of the Fourth Military Medical University.
Immunohistochemistry
Tumors were fixed in 10% neutralized formalin and embedded in paraffin blocks. Sections (4 μm) were prepared for hematoxylin/eosin staining and also for immunohistochemical examination. For immunohistochemistry analysis, endogenous peroxidases were blocked using 0.75% H2O2 in PBS for 30 min, followed by incubation with Serum-Free Protein Block. Incubation with primary antibody anti-QKI (1:300) was performed for 24 h at 4 °C. Immunodetection was performed in a three-step protocol, using streptavidin–horseradish peroxidase complex, with visualization by 3, 3-diaminobenzidine (DAB).
Methylation assay
Total genomic DNA was isolated from multiple cell lines and both tumor and normal colon epithelium samples using the DNA extraction kit according to the manufacturer’s instruction. Genomic DNA (2 μg) was then modified with bisulfite as described before. Bisulfite-modified DNA (2 μL) was then used as template DNA for PCR amplification with the external nest PCR primers corresponding to the region unaffected by the methylation status. Equal aliquots of the amplicon were then amplified with internal nest PCR primers corresponding to methylated promoter region only (Primer MSP internal 1) (Table S1) or the region unaffected by the methylation status (Primer MSP internal 2) (Table S1). The latter two internal resulting PCR products were then electrophoresed on a 1.2% agarose gel. To compare the methylation proportion, the band of the MSP internal 2 was served as a loading control. For tissue samples, the intensity of the methylated and the loading control bands were compared, with the basepairs of the amplicons and the cycles of individual PCR reactions being considered. When the relative ratio of the methylated to the total is more than 50%, methylation is considered to be high, while it is considered to be low when the ratio less than 50%. Samples with no obvious bands were considered as no methylation.
Plasmid construction, virus packaging and infection
For the construction of the β-catenin 3′ UTR and the Δβ-catenin 3′ UTR1 and Δβ-catenin 3′ UTR2 reporter vectors, the entire β-catenin 3′ UTR region or the region without the putative QREs were amplified by RT-PCR from the mRNA of normal gastric tissue using the primers listed in Table S1. The PCR products were digested with indicated restriction enzymes before ligated into our previously modified pGL3-control vector, in which EcoRI, EcoRV, NdeI, PstI are inserted downstream the XbaI site. The pcDNA3.1(+)-QKI-5, 6, 7 were constructed as described before29. Stealth™ siRNAs targeting QKI are synthesized from Invitrogen and was dissolved in DEPC-treated H2O at a concentration of 20 μM as a stock. For the construction of Topflash and Fopflash reporter, the synthetic reversely complementary nucleotides covering 3 repeated TCF binding sites or the mutated TCF binding sites (Table S1) were annealed and cloned into the pGL3-control vector. Construction and infection of the control and QKI overexpression adenovirus was done as previously described15.
Reporter assay
Twenty-four hours after transfection, cells were lysed using passive lysis buffer and analyzed for firefly and Renilla luciferase activities using the dual-luciferase reagent assay kit (Promega) according to the manufacturer’s instructions. Values were expressed as means±SD from at least three independent experiments. Since QKI overexpression might alter the internal control pRL-TK due to alternative splicing,30 all the fold changes in these vectors by QKI were normalized to the changes in pGL3-SV40. Statistical analysis was performed using Student’s t test. A value of p < 0.05 was considered as significant difference.
Measurement of IAP activity
The cells were washed once with phosphate-buffered saline before trypsinization. And then the cells were centrifuged at low speed and the supernatant was discarded. The pellet was lysed with buffer same as the Western blot at 4°C. The activity of IAP was measured according to the instruction. Protein concentration was estimated by the Pierce BCA Kit according to the manual instruction. Fold induction of the enzyme activities are calculated.
RNA-IP
RNA-IP was done as described before.15 The RNA in the immunoprecipitated complex and the RNA in the previously saved input fraction were extracted. Specific primers were applied for detection of the target mRNAs (Table S1).
Soft-agar assay
Efficiency of plating in soft agarose was determined by plating the cells in DMEM containing 0.33% agarose (Indubiose A37; HAA, IBF, France) plus 10% FBS, over a layer of 0.5% agarose in the same culture medium. Colonies were counted two weeks after plating. The cultures were examined with the inverted microscope.
Statistics
All the experiments are done at least in triplicates and the data are expressed as means ± SD. Student t test is applied for statistics analysis. P <0.05 is considered as significantly different.
Results
Expression pattern of QKI in normal colon and colon cancers
To establish the role of QKI in the differentiation of colon epithelium, we first analyzed the expression of QKI in normal colon and colon cancers. Expression levels of QKI were compared among normal gastrointenstinal epithelial cell lines (GES-1 and IEC-6) and cancerous cell lines (HCT116, HT29, SW480, SW620 and Colo205 cells). High expression of QKI in normal GES-1 and IEC-6, median level in HT29 cells, and low level in HCT116, SW480 and SW620 were observed both at RNA and protein levels, while null expression was found in the poorly differentiated Colo205 cells (Fig 1A, B). Furthermore, immunohistochemistry analysis in tissue samples revealed that QKI expression was detectable in about 60% of the adjacent normal colon samples (6/10), while only in 30% of colon cancer samples (3/10) and with a relatively lower level. Representative data are shown in Fig 1C. Similar results were observed through western blot analysis (Fig 1D). It is important to note that there is a doublet for QKI in the western blot assay, with the molecular weight corresponding to QKI5 (upper band) and QKI6 (lower band) respectively. Besides, RT-PCR with specific primers revealed that both QKI5 and QKI6 are expressed in colon cells (Fig S1). Strikingly, in the QKI positive cancer tissue samples, QKI6 were greatly suppressed or nearly absent (Fig 1D). All of these implicate that expression of QKI, especially QKI6 are aberrantly reduced in the colon cancer.
Fig 1. Expression pattern of QKI in normal and neoplastic colonic epithelium.


A, RNA levels of QKI in multiple colon cancer cell lines, the normal small intestinal epithelial IEC-6 cell line, and normal gastric epithelium GES-1 were quantified by comparative RT-PCR. Adherent cells were cultured to reach confluence and harvested for examination. β-actin served as an internal control to ensure equal loading.
B, Protein levels of QKI in the above cell lines were detected by Western blot and GAPDH served as an internal control to ensure equal loading.
C, Immunostaining for QKI in normal and cancerous colon epithelium in 10 human colon cancer specimens and paired adjacent normal tissue collected at surgical resection. QKI expression was much lower in cancerous tissues than that in adjacent normal tissue. Representative data is provided.
D, Protein levels of QKI in the above patients were detected by Western blot assay and β-actin served as a loading control.
Methylation of QKI promoter contributes to the deregulation of QKI in cancer cells
To deliberate the underlying mechanisms responsible for deregulation of QKI in cancer cells, we analyzed the promoter region of QKI. QKI promoter region is rich in CpG islands, especially in the 500bp upstream the putative transcription start site, leading us to ask whether CpG methylation accounts for the low and even null expression of QKI in colon cancers. Human intestinal epithelial cell lines listed above, colon cancers and corresponding adjacent normal controls of the 10 patients analyzed above were collected for methylation analysis.
Consistent with the varied expression of QKI in cell lines, methylation status of QKI promoter varied in these cell lines, which is reversely related with the expression level (Fig 2A). Although methylation was found in the adjacent normal epithelium in about half of the patients, low or no methylation was found in normal colon epithelium while abundant methylation was found in the corresponding cancer samples in the remaining (Fig 2B). Reanalysis of the information of the 10 patients (Table 1) indicated that those patients with no or low methylation in the QKI promoter region in the normal colon were relatively younger than those patients with abundant methylation. Taken together, we can see that aging and carcinogenesis should be the risk factors responsible for methylation of QKI promoters.
Fig 2. Methylation status of QKI promoter region in normal and cancerous colon tissues.

A, Methylation status of QKI promoter region in multiple human cell lines. Methylated promoter and the total level (served as a loading control) of each cell line were amplified with specific primers.
B, Methylation status of QKI promoter region in normal and cancerous colon tissues.
Table 1.
Clinical information of the patients in Fig 2B.
| Patient ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Gender | M | M | F | F | M | M | M | F | M | F |
| Age (yr) | 72 | 79 | 46 | 65 | 61 | 42 | 45 | 67 | 37 | 53 |
| Methylation (Normal/Tumor) | H/H | H/H | N/N | N/H | N/H | L/H | L/H | H/H | N/N | N/N |
Methylation status of each sample was expressed as the ratio of the methylated products to the total products. L, low frequency of methylation; H, high frequency of methylation; N, no obvious methylation. M, male; F, Female.
Strikingly, 5-aza-dC treatment of the hypermethylated colo205 cells for 7 days rescued the expression of QKI (Fig S2A), highly suggesting that hypermethylation of QKI lead to the reduced expression of QKI.
Differentiation promoting role of QKI
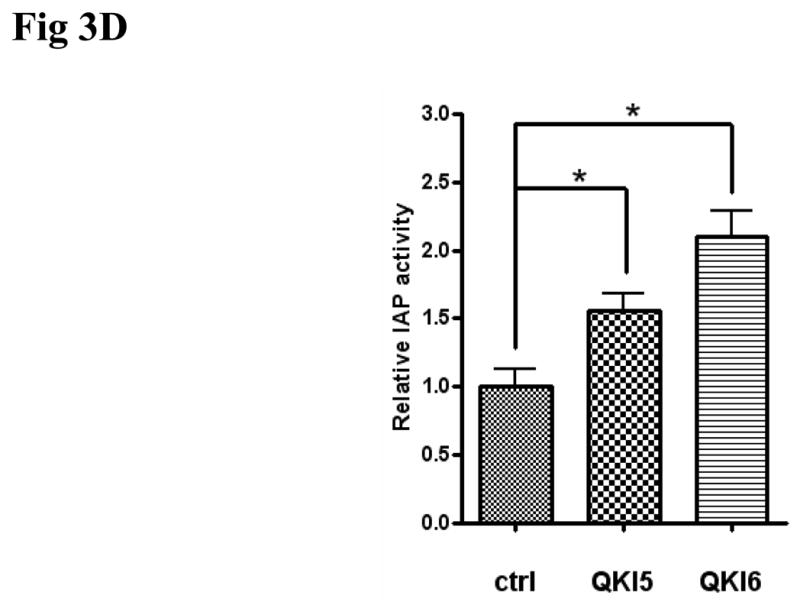
With the rescue of QKI expression under 5-aza-dC treatment, intestinal differentiation markers, such as lactase and IAP were also significantly upregulated (Fig S2A, B), suggesting a differentiating role of QKI in colon epithelium. In addition, post-confluence growth for two days induced early differentiation of HT29 cells, as observed by the increased expression of p27 (Fig 3A). Coincidentally, increased the expression of both QKI5 and QKI6 were observed (Fig 3A). To this end, HT29 cells were transfected with QKI expressing vector. Overexpression of either QKI5 or QKI6 in HT29 cells (Fig 3B) facilitates the differentiation of colon cancer cell as indicated by the IAP activity and the expression of Lactase and E-cadherin (Fig 3C and 3D). Accordingly, knockdown of QKI through RNAi reduced the expression of lactase and IAP (Fig S3A, B and C).
Fig 3. Differentiation promoting role of QKI.

A, QKI expression under post-confluence growth induced differentiation. Cells were seeded at 80% confluence at day 0 or continued to grow for additional 1 day or 2 days. QKI expression was significantly upregulated in colon cancer cell HT29 after 2 day culture, which is correlated with the increased expression of p27. α-tubulin serves as a control to ensure equal loading.
B, Analysis of QKI overexpression. Cells were infected with the indicated adenovirus and QKI expression was analyzed by Western blot.
C, QKI overexpression efficiently induced the expression of Lactase and E-cadherin. Compared with the control, infection of Ad-QKI5 or Ad-QKI6 in HT29 cells for 4 days significantly upregulated the expression of enterocyte marker Lactase and E-cadherin. QKI overexpression does not change the β-catenin mRNA level. Here is the representative data of three different experiments.
D, Effects of QKI on the expression of IAP. HT29 cells were infected with Ad-QKI5 or Ad-QKI6 or the control for 3 days before harvest for IAP detection. Data are expressed as means±SD (n=3). * P<0.05.
Increased p27 expression attributed to the function of QKI
As an RNA binding protein, QKI acts through modulating the expression of its targets at post-transcriptional level. Among the thousands of putative QKI targets, p27 and β-catenin are most fascinating and highly possible to be involved in our model. In line with the confluence induced early differentiation of HT29 cells, the stability of the p27 mRNA increased significantly at high density (Fig 4A, B), which should contribute at least partially to the increased protein level (Fig 3A). Previous finding suggested that QKI6 increased the expression of p27 through enhancing the RNA stability,15 we observed that overexpression of QKI6 increased p27 expression in our model too(data not shown). What’s more, QKI5 expression also increased p27 expression only if HT29 cells were induced to differentiation by confluence (Fig S4A). Since QKI6 was expressed at high density (Fig 3A) and QKI5/6 can form heterodimers,14 it is reasonable to deduce that enhancement of p27 by QKI5 might dependent on QKI6 or other cell context. Finally, RNA-IP assay confirmed the direct interaction between p27 and QKI5 (Fig S4B). In addition, knock-down of QKI reduced high density induced p27 mRNA stability partially (Fig 4A, B). Our data revealed that QKI5 and 6 are involved in the increased mRNA stability of p27 under confluence.
Fig 4. QKI enhances the mRNA stability of p27 at high confluence condition.

A, p27 mRNA stability in HT29 cells at low density, high density and high density with QKI knockdown. Cells cultured in the above condition, were further treated with Actinomycin D at time 0, RNA was extracted at indicated time to examine the RNA stability. Increased mRNA stability was detected in high density culture, which is partially blocked by QKI RNAi. Data presented here is a representative of 3 independent experiments.
B, Quantification of the data from Fig 5A. Intensity of the bands were analyzed by ImageJ and expressed as mean±SD. * P<0.05.
QKI expression repressed the cytoplasmic and nuclear β-catenin
Besides p27 upregulation, we observed the relationship between QKI and β-catenin. Results from the subcellular fraction revealed that overexpression of QKI reduced the cytosol and nucleus fraction of β-catenin (Fig 5A). Consistently, overexpression of QKI reduced the luciferase activity of Topflash under such confluent condition, with no significant change to Fopflash reporter (Fig 5B). Whereas the mRNA level of β-catenin was not affected by forced expression of QKI (Fig 3C), implicating the regulation might be at the layer of translational efficiency. In this regard, serial reporter vectors containing both of the two potential QREs, only the proximal QRE, and none of the QREs were constructed (Fig 5C). Over expression of QKI5 or QKI6 decreased the activity of β-catenin 3′ UTR and Δβ-catenin 3′UTR1, while Δβ-catenin 3′UTR2 with no QREs did not responded to QKI, suggesting the proximal QRE was the true responsive QRE (Fig 5C). In order to test the possibility of direct interaction between β-catenin and QKI, RNA-IP assay was employed. Forced expression of flag-tagged QKI5 (Fig 5D) or flag-tagged QKI6 (Fig S6) co-precipitates with β-catenin mRNA, suggesting a direct interaction between them.
Fig 5. QKI alters the subcellular distribution of β-catenin.


A, QKI overexpression decreased the cytoplasmic and nuclear β-catenin, while increased the membrane bound β-catenin. α-tubulin and Lamin B served as internal controls.
B, QKI expression reduced the Topflash activity. Topflash or Fopflash reporter plasmid and 50 ng internal control vector pRL-TK were cotransfected with the indicated plasmids and 36h later cells were lysed and luciferase activity were examined as the protocol. Fold induction by QKI (Topflash/Fopflash) were calculated and expressed as means±SD (n=3).* P<0.05.
C, QKI regulates β-catenin expression through 3′UTR. 200ng β-catenin 3′UTR, Δβ-catenin 3′UTR1, or the Δβ-catenin 3′UTR2 reporter and 50 ng internal control vector pRL-TK were cotransfected with the indicated plasmids for 24h before luciferase activity examination. β-catenin 3′UTR and the Δβ-catenin 3′UTR1 responded similarly to QKI overexpression, while Δβ-catenin 3′UTR2 did not respond to QKI overexpression. Fold induction were calculated and expressed as means±SD (n=3). * P<0.05.
D, Direct interaction between β-catenin and QKI. pcDNA3.1-3× flag-QKI5 transfected HT29 cells were immunoprecipitated with anti-flag antibody or the negative control IgG. The presence of β-catenin mRNA in the immunoprecipitation was detected by RT-PCR and visualized by ethidium bromide staining.
In contrast to the reduction of cytosol and nucleus β-catenin by QKI, both QKI5 and QKI6 increased the membrane bound β-catenin (Fig 5A). To test whether this is an indirect effect of differentiation, subcellular distribution of β-catenin was tested in HT29 cells cultured both in low density and confluence induced differentiation. β-Catenin localized mainly in the cytosol and nucleus when cells were scattered, in contrast, membrane localization of β-catenin was overt when cells began confluent (Fig S5), suggesting that differentiation and E-cadherin mediated cell contact increased the membrane bound β-catenin. In light of the increased E-cadherin by QKI (Fig 3C), increased membrane bound β-catenin might be a mirror of differentiation and increased E-cadherin.
Tumor suppressing role of QKI in colon cancer cells
In view of the above data, we hypothesize that QKI might affect the carcinogenesis through coordinately regulating the differentiation and proliferation of intestinal epithelial cells. Overexpression of QKI, especially QKI6 reduced the cell proliferating ability both in HT29 cells and in HCT116 cells as indicated by MTT assay (Fig 6A). Anchorage-independent growth is one of the key characteristics of transformed cells.31 To this end, HCT116 cells infected with Ad-EGFP, Ad-QKI5, Ad-QKI6 were cultured in semisolid medium. Colonies of Ad-QKI6 infected cells are much smaller and fewer, compared with the control (Fig 6B, 6C). These data implicate a tumor suppressing role of QKI in colon epithelium.
Fig 6. Tumor suppressive role of QKI.


A, Growth curves of HT29 and HCT116 cells infected with indicated adenovirus (n=3). QKI, especially QKI6 significantly blocked the growth of the tumor cells.
B, Colony formation of HCT116 cells infected with indicated adenovirus in semisolid medium (n=3). Cells on soft agar plates were grown for 2 weeks before colonies were visualized microscopically. A representative view of them is shown.
C, Quantification of colony formation data from B. Colonies were counted in a blinded fashion. Those with a diameter of >50μm are defined as large and those <50μm as small colonies. * p<0.05.
Discussion
Our study investigates the mechanism by which QKI regulates colon epithelial cell differentiation and the role of its aberrant expression in colon cancer evolution. The data demonstrate for the first time that QKI expression, especially QKI6, is widely distributed in normal colon epithelium while absent or very lowly expressed in colon cancer cells due to high methylation of the promoter region. QKI expression in differentiating HT29 cells resulted in sustained accumulation of p27kip1, decreased cytoplasmic and nuclear β-catenin and increased membrane bound β-catenin, facilitating the colon epithelial differentiation.
In line with the ability of HDAC inhibitors to induce intestinal cell maturation in colon cancer cell lines, aberrant epigenetic modification is a common characteristic of colon cancer.32 Here we identified the methylation of QKI promoter region accounts for the low or no expression of QKI in colon cancer patients, which is unique from previously reported LOH in gastric cancer and glioma,20, 21 pointing out the general role of RNA binding protein QKI in multiple cancer formation. Different from the usual tumor suppressor, which is aberrantly methylated in cancer samples while unmethylated in the normal counterpart. QKI methylation is nearly equal in normal and cancerous cells in the aged patients. In other words, besides carcinogenesis, QKI methylation also tends to occur in old subjects, which is reminiscent of the susceptibility of colon cancer in aged subjects. Parallel with the hypermethylation of QKI in aged subjects, increased or aberrant methylation has been observed in stem cells undergoing too many cycles33 and aging subjects.34 In combination, we can conclude that age-dependent methylation of QKI promoter is one of but not the only one of the causes of QKI promoter methylation and its subsequent repression leads to colon epithelium differentiation defect, which sensitizes the subjects for cancer onset and evolution. In this view, QKI expression and its promoter methylation should be of potential value to predict colorectal carcinogenesis. Additional studies at a much larger scale are needed to draw a full conclusion.
Although previously it has been defined that QKI, mainly QKI6 and QKI7, can stabilize the mRNA of p27 and subsequently enhanced the expression of p27 in oligodendrocytes, it is still largely unknown whether this regulation conserved in cancer cells. Here we found that in cancer cell HT29, in addition to QKI6, QKI5 stabilize the p27 mRNA only when the cells undergoing contact inhibition. It may be due to the accumulation of p27 mRNA under the contact inhibition or due to the posttranslational modification of QKI by the altered signal cascades under the contact inhibition condition. Our study here furthers the understanding of the interaction between QKI and p27 and its role in the colon epithelial differentiation.
Besides p27, here we also identified β-catenin as a novel target of QKI, which is also involved in the colon epithelial differentiation. It is well established that activation of Wnt signal plays a significant role in colon carcinogenesis through increasing the nuclear and cytoplasmic form of β-catenin. Our data provided the first evidence that QKI was able to decrease the cytoplasmic and nuclear form of β-catenin, acting as a tumor suppressor. Regarding to the underlying mechanism, we showed a highly possible interaction between QKI and a proximal QRE located in the β-catenin 3′UTR through RNA-IP and reporter assay, further confirmation of this interaction through EMSA is still needed, which is undergoing now. QKI expression decreased the β-catenin 3′UTR reporter activity, which is not via decreasing the RNA stability. Nonetheless, the detailed mechanism for increased membrane bound β-catenin and dereased cytoplasmic and nuclear β-catenin by QKI overexpression is still lacking from the present study. It is widely accepted that RNA localization is important for the establishment and maintenance of polarity in multiple cell types.35, 36 Localized mRNAs are usually transported along microtubules or actin filaments and become anchored at their destination to some specifc subcellular structures. Upon reaching the destination, translational repression apparatus are released from the mRNA for local translation.37 Previously, it is reported there is different and asymmetric localization of β-catenin mRNA in the migratory and contact inhibition cells.38 Here we have addressed that β-catenin was bound by QKI, combining the previous finding that QKI acts through translational repression39 and QKI itself shuttles between the cytosol and nucleus; we favor a role of QKI in coordinately regulating the transportation and repressing the translation of β-catenin before it reaches the destination. Confirmation of this hypothesis by further necessary experiment should provide additional strategies to control aberrant wnt signal.
It is important to note that QKI5 and QKI6 function similarly in the regulation of p27 and β-catenin and promoting cell differentiation, while significant different roles of QKI5 and QKI6 in the regulation of tumor growth was observed by the MTT assay and soft agar assay. Previously, both coordinate and antagonistic functions between QKI5 and QKI6 have been reported.29, 40 On one hand, QKI5 and QKI6 can form heterodimers and they shared similar target binding activity and specificity. On the other hand, different effects on the same targets might be produced by QKI5 and QKI6 due to their different subcellular localizations.41 It is thus reasonable to deduce that both coordinate and different functions of QKI5 and 6 should occur in intestine development. Detailed elucidation of the dynamics of QKI isoforms during the differentiation from intestinal stem cells to the different differentiated stages will shed light on this STAR molecule in intestine development.
In summary, we have identified the RNA binding protein QKI as a critical regulator of colon epithelial differentiation, whose aberrant reduction due to methylation might facilitate the colon carcinogenesis.
Supplementary Material
Acknowledgments
This work was supported by Foundation for excellent Ph.D students of FMMU (200706), National Science Foundation of China (NSF: 30470387; NSF: 30570396; NSF: C030200303) and National Key Basic Research and Development Program 2009CB521706.
Abbreviations
- CNS
central nervous system
- HDAC
histone deacetylase
- IAP
intestinal alkaline phosphatase
- MAG
Myelin associated glycoprotein
- MBP
myelin basic protein
- QKI
quaking
- QRE
QKI response element
- SB
sodium butyrate
- STAR
signal transduction and activator of RNA
- UTR
untranslated region
Footnotes
Author contribution: Guodong Yang, acquisition of data and drafting of the manuscript;
Haiyan Fu, acquisition of data;
Jie Zhang, acquisition of data;
Xiaozhao Lu, acquisition of data;
Fang Yu, acquisition of data;
Liang Jin, acquisition of data;
Liyuan Bai, acquisition of data;
Bo Huang, acquisition of data;
Lan Shen, acquisition of data;
Yue Feng, critical revision of the manuscript for important intellectual content, technical and material support;
Libo Yao, critical revision of the manuscript for important intellectual content and study supervision;
Zifan Lu, study concept and design, obtained funding, critical revision of the manuscript for important intellectual content and study supervision.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gaudette LA, Altmayer CA, Wysocki M, Gao RN. Cancer incidence and mortality across Canada. Health Rep. 1998;10:51–66. [PubMed] [Google Scholar]
- 2.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 3.Yamamoto H, Soh JW, Shirin H, Xing WQ, Lim JT, Yao Y, Slosberg E, Tomita N, Schieren I, Weinstein IB. Comparative effects of overexpression of p27Kip1 and p21Cip1/Waf1 on growth and differentiation in human colon carcinoma cells. Oncogene. 1999;18:103–15. doi: 10.1038/sj.onc.1202269. [DOI] [PubMed] [Google Scholar]
- 4.Quaroni A, Tian JQ, Seth P, Ap Rhys C. p27Kip1 is an inducer of intestinal epithelial cell differentiation. AJP - Cell Physiology. 2000;279:C1045–1057. doi: 10.1152/ajpcell.2000.279.4.C1045. [DOI] [PubMed] [Google Scholar]
- 5.Deschenes C, Vezina A, Beaulieu J-F, Rivard N. Role of p27Kip1 in human intestinal cell differentiation. Gastroenterology. 2001;120:423–438. doi: 10.1053/gast.2001.21199. [DOI] [PubMed] [Google Scholar]
- 6.Majumder PK, Grisanzio C, O’Connell F, Barry M, Brito JM, Xu Q, Guney I, Berger R, Herman P, Bikoff R, Fedele G, Baek WK, Wang S, Ellwood-Yen K, Wu H, Sawyers CL, Signoretti S, Hahn WC, Loda M, Sellers WR. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell. 2008;14:146–55. doi: 10.1016/j.ccr.2008.06.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tao L, Kramer PM, Wang W, Yang S, Lubet RA, Steele VE, Pereira MA. Altered expression of c-myc, p16 and p27 in rat colon tumors and its reversal by short-term treatment with chemopreventive agents. Carcinogenesis. 2002;23:1447–1454. doi: 10.1093/carcin/23.9.1447. [DOI] [PubMed] [Google Scholar]
- 8.Palmqvist R, SR, Oberg A, Landberg G. Prognostic significance of p27(Kip1) expression in colorectal cancer: a clinico-pathological characterization. The Journal of Pathology. 1999;188:18–23. doi: 10.1002/(SICI)1096-9896(199905)188:1<18::AID-PATH311>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 9.Alessandro Sgambato CR, Faraglia Beatrice, Merico Marta, Ardito Raffaele, Schinzari Giovanni, Romano Gianpiero, Cittadini Achille RM. Reduced expression and altered subcellular localization of the cyclin-dependent kinase inhibitor p27(Kip1) in human colon cancer. Molecular Carcinogenesis. 1999;26:172–179. doi: 10.1002/(sici)1098-2744(199911)26:3<172::aid-mc6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 10.van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, Tjon-Pon-Fong M, Moerer P, van den Born M, Soete G, Pals S, Eilers M, Medema R, Clevers H. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–50. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 11.Ramesh S, Nash J, McCulloch PG. Reduction in membranous expression of beta-catenin and increased cytoplasmic E-cadherin expression predict poor survival in gastric cancer. Br J Cancer. 1999;81:1392–7. doi: 10.1038/sj.bjc.6693437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kondo T, Furuta T, Mitsunaga K, Ebersole TA, Shichiri M, Wu J, Artzt K, Yamamura K, Abe K. Genomic organization and expression analysis of the mouse qkI locus. Mamm Genome. 1999;10:662–9. doi: 10.1007/s003359901068. [DOI] [PubMed] [Google Scholar]
- 13.Cox RD, Hugill A, Shedlovsky A, Noveroske JK, Best S, Justice MJ, Lehrach H, Dove WF. Contrasting effects of ENU induced embryonic lethal mutations of the quaking gene. Genomics. 1999;57:333–41. doi: 10.1006/geno.1999.5804. [DOI] [PubMed] [Google Scholar]
- 14.Larocque DRS. QUAKING KH domain proteins as regulators of glial cell fate and myelination. RNA Biol. 2005;2:37–40. doi: 10.4161/rna.2.2.1603. [DOI] [PubMed] [Google Scholar]
- 15.Larocque D, Galarneau A, Liu HN, Scott M, Almazan G, Richard S. Protection of p27(Kip1) mRNA by quaking RNA binding proteins promotes oligodendrocyte differentiation. Nat Neurosci. 2005;8:27–33. doi: 10.1038/nn1359. [DOI] [PubMed] [Google Scholar]
- 16.Larocque D, Pilotte J, Chen T, Cloutier F, Massie B, Pedraza L, Couture R, Lasko P, Almazan G, Richard S. Nuclear retention of MBP mRNAs in the quaking viable mice. Neuron. 2002;36:815–29. doi: 10.1016/s0896-6273(02)01055-3. [DOI] [PubMed] [Google Scholar]
- 17.Galarneau A, Richard S. Target RNA motif and target mRNAs of the Quaking STAR protein. Nat Struct Mol Biol. 2005;12:691–8. doi: 10.1038/nsmb963. [DOI] [PubMed] [Google Scholar]
- 18.Li Z, Takakura N, Oike Y, Imanaka T, Araki K, Suda T, Kaname T, Kondo T, Abe K, Yamamura K. Defective smooth muscle development in qkI-deficient mice. Dev Growth Differ. 2003;45:449–62. doi: 10.1111/j.1440-169x.2003.00712.x. [DOI] [PubMed] [Google Scholar]
- 19.Janice K, Noveroske LL, Gaussin Vinciane, Northrop Jennifer L, Nakamura Hisashi, Hirschi Karen K, Justice Monica J. Quaking is essential for blood vessel development. Genesis. 2002;32:218–230. doi: 10.1002/gene.10060. [DOI] [PubMed] [Google Scholar]
- 20.Li ZZ, Kondo T, Murata T, Ebersole TA, Nishi T, Tada K, Ushio Y, Yamamura K, Abe K. Expression of Hqk encoding a KH RNA binding protein is altered in human glioma. Jpn J Cancer Res. 2002;93:167–77. doi: 10.1111/j.1349-7006.2002.tb01255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ichimura K, Mungall AJ, Fiegler H, Pearson DM, Dunham I, Carter NP, Collins VP. Small regions of overlapping deletions on 6q26 in human astrocytic tumours identified using chromosome 6 tile path array-CGH. Oncogene. 2006;25:1261–71. doi: 10.1038/sj.onc.1209156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mulholland PJ, Fiegler H, Mazzanti C, Gorman P, Sasieni P, Adams J, Jones TA, Babbage JW, Vatcheva R, Ichimura K, East P, Poullikas C, Collins VP, Carter NP, Tomlinson IP, Sheer D. Genomic profiling identifies discrete deletions associated with translocations in glioblastoma multiforme. Cell Cycle. 2006;5:783–91. doi: 10.4161/cc.5.7.2631. [DOI] [PubMed] [Google Scholar]
- 23.Ebersole TA, Chen Q, Justice MJ, Artzt K. The quaking gene product necessary in embryogenesis and myelination combines features of RNA binding and signal transduction proteins. Nat Genet. 1996;12:260–5. doi: 10.1038/ng0396-260. [DOI] [PubMed] [Google Scholar]
- 24.Lu Z, Ku L, Chen Y, Feng Y. Developmental abnormalities of myelin basic protein expression in fyn knock-out brain reveal a role of Fyn in posttranscriptional regulation. J Biol Chem. 2005;280:389–95. doi: 10.1074/jbc.M405973200. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Lu Z, Ku L, Chen Y, Wang H, Feng Y. Tyrosine phosphorylation of QKI mediates developmental signals to regulate mRNA metabolism. EMBO J. 2003;22:1801–10. doi: 10.1093/emboj/cdg171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wagh PK, Peace BE, Waltz SE. Met-related receptor tyrosine kinase Ron in tumor growth and metastasis. Adv Cancer Res. 2008;100:1–33. doi: 10.1016/S0065-230X(08)00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolpin BM, Mayer RJ. Systemic treatment of colorectal cancer. Gastroenterology. 2008;134:1296–310. doi: 10.1053/j.gastro.2008.02.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong SF. Cetuximab: an epidermal growth factor receptor monoclonal antibody for the treatment of colorectal cancer. Clin Ther. 2005;27:684–94. doi: 10.1016/j.clinthera.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Chen Y, Tian D, Ku L, Osterhout DJ, Feng Y. The selective RNA-binding protein quaking I (QKI) is necessary and sufficient for promoting oligodendroglia differentiation. J Biol Chem. 2007;282:23553–60. doi: 10.1074/jbc.M702045200. [DOI] [PubMed] [Google Scholar]
- 30.Wu JI, Reed RB, Grabowski PJ, Artzt K. Function of quaking in myelination: regulation of alternative splicing. Proc Natl Acad Sci U S A. 2002;99:4233–8. doi: 10.1073/pnas.072090399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freedman VH, Shin SI. Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell. 1974;3:355–9. doi: 10.1016/0092-8674(74)90050-6. [DOI] [PubMed] [Google Scholar]
- 32.Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135:1079–99. doi: 10.1053/j.gastro.2008.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, Gnirke A, Jaenisch R, Lander ES. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim JY, Siegmund KD, Tavare S, Shibata D. Age-related human small intestine methylation: evidence for stem cell niches. BMC Med. 2005;3:10. doi: 10.1186/1741-7015-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jansen RP. mRNA localization: message on the move. Nat Rev Mol Cell Biol. 2001;2:247–56. doi: 10.1038/35067016. [DOI] [PubMed] [Google Scholar]
- 36.St Johnston D. The intracellular localization of messenger RNAs. Cell. 1995;81:161–70. doi: 10.1016/0092-8674(95)90324-0. [DOI] [PubMed] [Google Scholar]
- 37.Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, Greenberg ME. A brain-specific microRNA regulates dendritic spine development. Nature. 2006;439:283–9. doi: 10.1038/nature04367. [DOI] [PubMed] [Google Scholar]
- 38.Mili S, Moissoglu K, Macara IG. Genome-wide screen reveals APC-associated RNAs enriched in cell protrusions. Nature. 2008;453:115–9. doi: 10.1038/nature06888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schumacher B, Hanazawa M, Lee MH, Nayak S, Volkmann K, Hofmann ER, Hengartner M, Schedl T, Gartner A. Translational repression of C. elegans p53 by GLD-1 regulates DNA damage-induced apoptosis. Cell. 2005;120:357–68. doi: 10.1016/j.cell.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 40.Pilotte J, Larocque D, Richard S. Nuclear translocation controlled by alternatively spliced isoforms inactivates the QUAKING apoptotic inducer. Genes Dev. 2001;15:845–58. doi: 10.1101/gad.860301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu J, Zhou L, Tonissen K, Tee R, Artzt K. The Quaking I-5 Protein (QKI-5) Has a Novel Nuclear Localization Signal and Shuttles between the Nucleus and the Cytoplasm. Journal of Biological Chemistry. 1999;274:29202–29210. doi: 10.1074/jbc.274.41.29202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.