

Abstract
Oculodentodigital dysplasia (ODDD) is a dominant negatively inherited disorder with variable but characteristic anomalies of the fingers and toes, eyes, face and teeth, which are caused by mutations in the connexin 43 (Cx43) gene. All mutations analyzed so far have a negative influence on the conductance through gap junctional channels and hemichannels, as well as trafficking of Cx43 protein in transfected cells. In this study, we inserted the human Cx43G138R point mutation into the mouse Cx43 gene and generated mice conditionally expressing this mutation. All ODDD phenotypic manifestations observed in humans, including syndactyly and enamel hypoplasia as well as craniofacial, bone and heart anomalies, were also observed with significant penetrance in Cx43G138R mice. When this mutation was specifically expressed in cardiomyocytes, characteristic alterations in the electrocardiogram and spontaneous arrhythmias were recorded. In vitro studies with Cx43G138R-expressing cells revealed loss of the Cx43 P2 phosphorylation state, which was also absent in the mutated hearts. This loss has previously been associated with gap junctional dysfunction and increased cellular ATP release. The Cx43G138R mutated mice show significantly increased arrhythmogeneity ex vivo in Langendorff experiments with explanted hearts and in vivo in particular under hypoxic conditions. Our results suggest that the increased activity of ATP-releasing channels in Cx43G138R mutated cardiomyocytes may further reduce the already decreased gap junctional communication and thus aggravate arrhythmogenesis in the mouse mutant.
INTRODUCTION
Oculodentodigital dysplasia (ODDD) is an autosomal dominantly inherited disorder characterized by anomalies of face, eyes, limbs and teeth with a high manifestation and variable expression (1). The most common symptoms are syndactyly, enamel hypoplasia, microdontia, microcornea and craniofacial, skeletal and skin alterations (2-6). Furthermore, some neurological and cardiac symptoms were described, such as spastic paraparesis, ataxia, progressive leukodystrophy (7) and arrhythmias, which were discussed as the reason for the sudden cardiac death in an affected family (8). ODDD is caused by mutations in the GJA1 gene, coding for connexin43 (Cx43) protein (8), which is expressed in many cell types and has essential functions during embryonic development. Cx43 is a member of the connexin family that comprises 21 different proteins in humans and 20 in mice (9). Connexin proteins show characteristic domains including the cytoplasmic N-terminus, four transmembrane and two extracellular regions, a cytoplasmic loop and the cytoplasmic C-terminal region. Connexins oligomerize into hexameric connexons or hemichannels that can dock to each other in contacting plasma membranes forming gap junction channels (GJCs). These channels facilitate electric and metabolic communication between cells of the cardiovascular, the central nervous system and other tissues. GJCs allow the diffusional exchange of metabolites such as sugars, amino acids, nucleotides and ions (10) or second messengers such as cAMP, Ca2+, NAD+ or IP3 between contacting cells (11).
Recent data suggest that unpaired hemichannels can transiently open under cellular stress, i.e. hypoxia (12). These hemichannels have been proposed to be also permeable to second messengers such as ATP or NAD+, indicating their potential involvement in cellular signaling (11-13). However, the molecular identity of hemichannels and their physiological role are still controversially discussed (14). For example, besides connexin hemichannels, also pannexins and the purinergic P2X7 receptor have been suggested to be involved in the cellular release of ATP (14).
Several of the known Cx43 mutations causing ODDD have already been analyzed after expression in different cell lines. These mutations impaired Cx43-mediated gap junctional conductance [I130T, K134E, G138R; G21R; F52dup, R202H, L90V, Y17S and A40V (15-17)], disturbed [Y17S, G21R, A40V, F52dup, L90V and I130T (18)] or enhanced the activity of ATP release [I31M, G138R and G143S (19)]. Three of these Cx43 mutant proteins [F52dup, R202H and H194P (18,19)] were not found in the plasma membrane, suggesting defective oligomerization or trafficking. Others were only located in the plasma membrane when co-expressed with wild-type Cx43 (Cx43WT). The phenotypic variability of ODDD between different mutations (interfamiliar variability) was discussed as an altered selectivity of heterologous GJCs containing wild-type Cx43 and the mutated form (Cx43R202H) (17) or as a difference in dominant potency between the mutations (G21R and G138R) (20). However, the molecular mechanisms underlying for the intrafamiliar variability are still unclear.
A mouse model for ODDD, the Gja1Jrt/+ mouse, has been already described (21). These mice, originated from a screen after N-ethyl-N-nitroso urea mutagenesis, express the Cx43G60S mutation and phenocopy many clinical ODDD symptoms. However, the Cx43G60S mutation has so far not been found in patients and is constitutively expressed in the transgenic mice. Thus, phenotypic alterations due to expression of the Cx43 mutation in different cell types cannot be dissected.
In this study, we generated a targeted, conditional mouse model for ODDD expressing the Cx43G138R mutation. Previously, it has been shown (15) that the Cx43G138R protein was expressed in contacting membranes and found in communication-deficient plaques, thus excluding a trafficking defect as being responsible for the observed lack of gap junctional communication. After ubiquitous expression, the Cx43+/G138R mice showed all phenotypes known for ODDD in human patients with a corresponding (intrafamiliar) variability. Additionally, we investigated the influence of the mutated Cx43 protein on the function of GJCs and ATP release in stably transfected HeLa cells, mouse embryonic stem cells and embryonic ventricular cardiomyocytes. We demonstrated that GJCs and ATP release from Cx43G138R-expressing cells exhibited different properties, compared with Cx43WT cells. In accordance with a previous report (16), we found a lack of intercellular coupling via Cx43G138R homotypic GJCs, whereas Cx43G138R formed functional heterotypic channels with Cx43WT. Furthermore, we demonstrate enhanced activity of ATP-releasing channels in Cx43G138R cells which in addition to the strongly reduced gap junction intercellular communication could contribute to the severe ventricular tachycardias observed in mutant hearts.
RESULTS
Generation of Cx43G138R-expressing mice
In order to investigate the influence of Cx43G138R mutation in different tissues, we generated mice in which the cell type-specific activation of this mutated gene could be accomplished by Cre-recombinase-mediated deletion of the loxP-flanked Cx43WT region (Fig. 1A). The Cre-recombinase activity led to the expression of the Cx43G138R-internal ribosomal entry site (IRES)-eGFP cassette (Fig. 1C) under the control of the Cx43 regulatory elements. For the selection of vector containing HM1 embryonic stem (ES) cells, the neomycin resistance gene flanked by frt sites was cloned directly after the polyA signal of Cx43WT. One of 251 ES cell clones had undergone correct homologous recombination and was used for blastocyst injections. The integration into the Cx43 locus was demonstrated in the different genotypes of the Cx43floxG138R mouse line before and after Cre-mediated recombination (Fig. 1D). The Cx43+/floxG138R mice were mated with PGK-, alphaMyHC-or Nestin-Cre-expressing mice for ubiquitous, cardiac myocyte or early neuron-specific activation of the mutated gene. Furthermore, the cell type-specific transcription of the Cx43G138R-IRES-eGFP cassette, as a long 4.4 kb mRNA, was found in heart but not in brain after the alphaMyHC-Cre activity (Fig. 1E). The mice were genotyped by polymerase chain reaction (PCR), as shown in Figure 1F.
Figure 1.
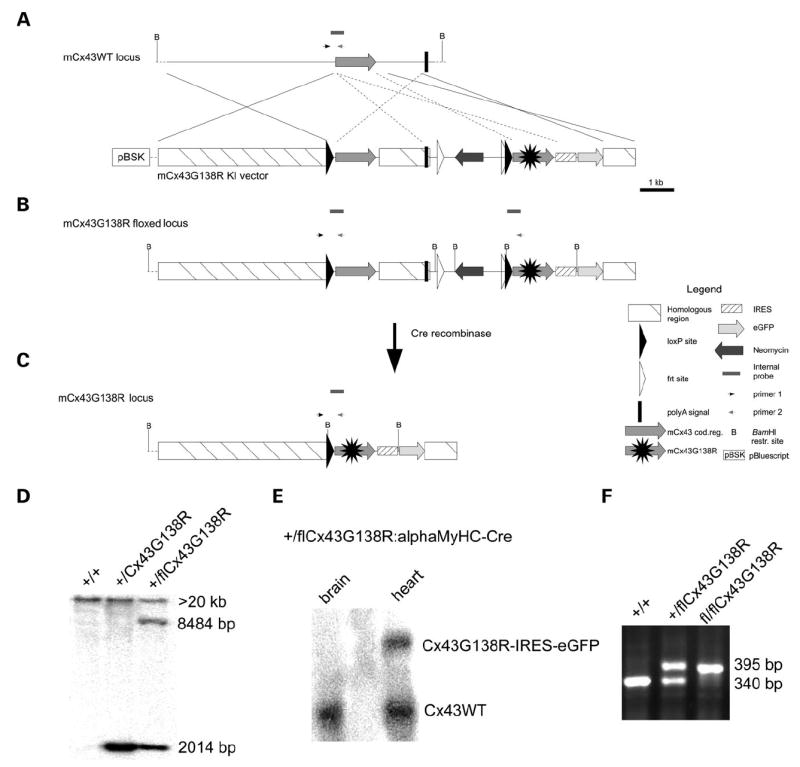
Generation of the conditionally Cx43G138R mutated mice by homologous recombination. (A) Homologous recombination of the targeting vector into the genomic locus of Cx43. (B) The resulting recombinant chromosome corresponds to the floxed state. (C) After Cre-mediated recombination, sequences between both loxP sites were excised resulting in the Cx43G138R gene expression. (D) Southern blot analysis indicating homologous recombination in mice. The 8484 bp band is missing in the middle lane, demonstrating the deletion of loxP-flanked regions on the DNA. (E) Northern blot analysis indicates the conditional transcription of the mutated Cx43 construct in the heart, but not in the brain after mating with alphaMyHC-Cre mice. (F) PCR genotyping using the primers as shown in A–C.
Morphologic abnormalities in Cx43G138R heterozygous mice equal those in ODDD patients
The ubiquitous and heterozygous expression of the Cx43G138R mutation resulted in mice exhibiting different ODDD characteristics with variable penetrance and appearance. The observed phenotypic manifestations were syndactyly in 70% (Fig. 2A–E), enamel hypoplasia in 80% (F and G), craniofacial abnormalities in 70% (H) and sparse hair in 30% (I) of all mutants. Syndactyly affected only the soft tissue (type III syndactyly) (Fig. 2E) and was observed in the second, third and fourth digits on all limbs. The reduction of dental enamel was obvious as translucent teeth and their faster abrasion with age. Furthermore, the craniofacial anomalies described in ODDD patients, i.e. the prominent and depressed nasal bridge and microcephaly (8), could largely be observed in the Cx43+/G138R mice. The morphologic skull analysis of 3–9-week-old mutants (Fig. 2H) revealed an obvious compactness of the facial region and differences in the angle of the nasal bone, the zygomatic arch, the eye pits and the size of the mandible, similar to as reported in ODDD patients (8). Sparse hair, an infrequent symptom of ODDD, was rarely observed and became more pronounced in adult mice.
Figure 2.
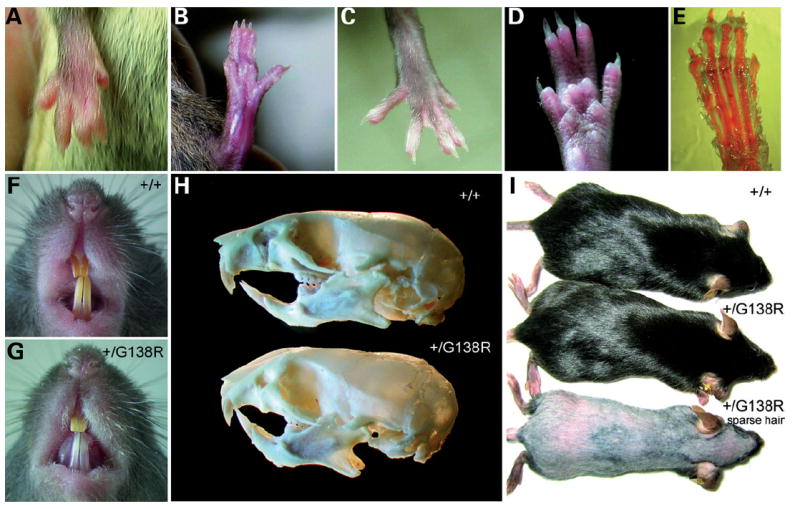
Morphological anomalies of the Cx43+/G138R ubiquitously expressing mice. (A–D) Syndactyly of all types. (E) Alcian blue and alizarin red staining for bones and cartilage indicates that bones are not affected by syndactyly. (F and G) Comparison of teeth reveals enamel hypoplasia in mutants (G). (H) Skull bones in mutant mice show significant changes in the craniofacial regions (bottom). (I) Sparse hair could be observed in a small number of mutants but never in wild-type littermates.
Bone morphology of Cx43 +/G138R mice is similar to the one observed in ODDD patients
Hypoplasias of the phalages, broad tubular bones and hyperostosis of the skull have also been reported in patients with ODDD (8). In Cx43+/G138R mice, a significant osteopenia was observed. Seven males per genotype group were used for analysis. The results show significantly lower trabecular bone volume in ODDD relative to wild-type littermates, indicating a reduction in trabecular bone mass. Accordingly, trabecular spacing was increased and the osteoblast number was not significantly decreased in the mutant mice (Fig. 3), even though trabecular thickness was not different. In all investigated mutants, a high variability was observed.
Figure 3.
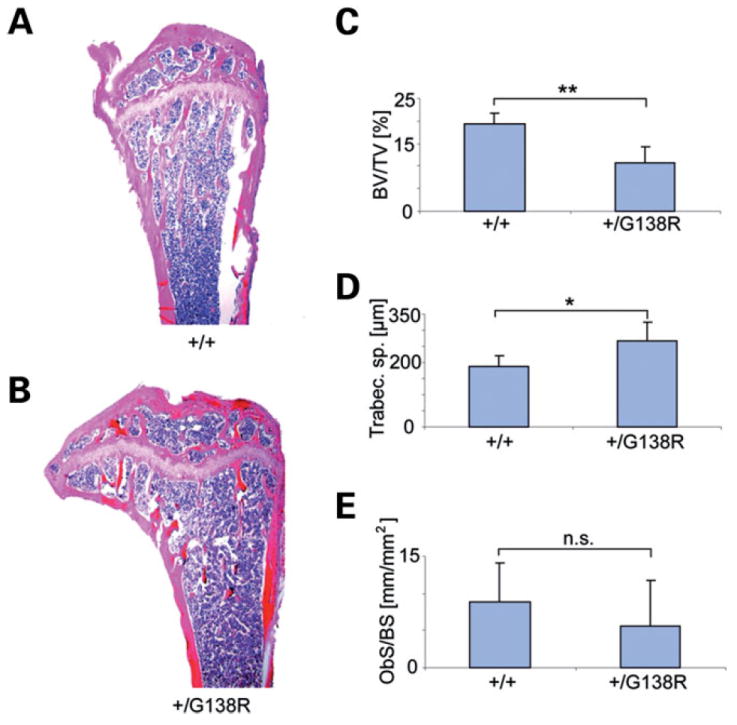
Skeletal phenotype of the ODDD-mutated mice. Comparison of wild-type (A) and Cx43+/G138R (B) tibial epiphysis and metaphysis shows more rarefied trabecular structure in the ODDD mutant relative to a wild-type littermate. Histomorphometric analysis reveals lower bone volume/total volume (**P < 0.001) (C), an increase in trabecular space (*P = 0.01) (D) and a non-significant (n.s.) decrease in osteoblast number (E) in mutants.
The Cx43G138R mutation frequently results in premature death because of cardiac expression
The matings of Cx43+/floxG138R male mice with PGK-Cre female mice (n = 32) yielded a reduction in the Mendelian ratio from 50 to 23% of mutants in a litter (Fig. 4A). The mutants were Cx43+/G138R:PGK-Cre and Cx43+/G138R because of the activity of active Cre protein on the oocyte level (22). The observed embryonic lethality of 54% in mutants could be narrowed to a period between ED 14.5 and ED 16.5 (data not shown). In order to ascertain the tissue responsible for the lethality, the Cx43+/floxG138R female mice were mated with alphaMyHC-Cre or Nestin-Cre male mice to specifically activate the mutation in heart or early neurons. Conditional activation of the Cx43G138R mutation in the heart (29 matings) revealed a reduction in the expected number of mutants in a litter (mutants were Cx43+/flox:alphaMyHC-Cre; expected frequency of mutants: 25%, born: 14% and reduction: 44%). Thus, the lethality of mice with cardiac-specific mutation was similar to that observed with the ubiquitous mutants (44–54%; P = 0.1) and occurred at the same embryonic stage.
Figure 4.
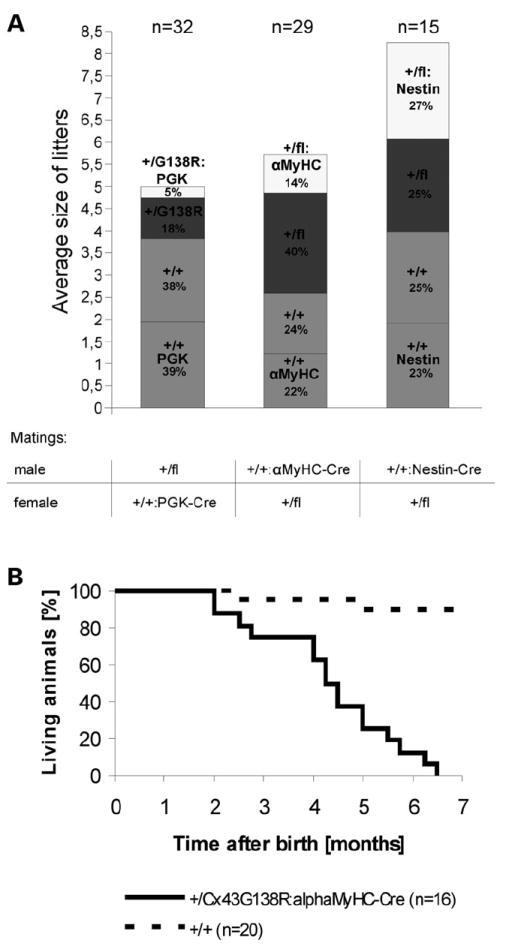
Genotype statistics for progeny from heterozygous Cx43floxG138R mice after mating with PGK-, alphaMyHC-or Nestin-Cre mice and premature death in adult cardiac-specific mutants. (A) The ubiquitous expression of the mutated gene using PGK-Cre mice results in the reduction of the number of littermates from the expected 50–23% of mutants in a litter. The conditional activation of the Cx43G138R mutation in the heart (alpha-MyHC-Cre mating) led to a smaller reduction in the expected number of mutants per litter from the expected 25–14% born. The activation of the mutation in early neurons does not influence the survival of the mutants (Nestin-Cre mating); (n = number of litters). (B) The Kaplan–Meier survival curve of Cx43+/floxG138R: alphaMyHC-Cre mice (n = 16) in comparison with wild-type mice (n = 20) shows premature death of cardiac-specific mutants between 2 and 6.5 months after birth.
In contrast, the activation of the point-mutated gene in early neurons, using the Nestin-Cre mice (n = 15 matings), did not influence the survival of the heterozygous mutants (mutants were Cx43+/flox:Nestin-Cre). The surviving cardiac-specific mutants died during the first 6.5 months after birth (Fig. 4B). Thus, these data imply that the mutant mice die because of cardiac alterations.
Morphologic development of the heart
As Cx43-deficient mice died at birth due to a failure in pulmonary gas exchange as a result of an altered right ventricular outflow tract (23), it was important to check the heart morphology in our adult mutants (Supplementary Material, Fig. S10A–D). Nine Cx43+/floxG138R:alphaMyHC-Cre hearts, two Cx43+/+:alphaMyHC-Cre hearts and three Cx43+/+ hearts were investigated morphologically. Two of the nine Cx43+/floxG138R:alphaMyHC-Cre hearts were smaller than that of the control group. Microscopically, no structural defects were detected. Both atrial septation and ventricular septation were normal. The pulmonary outlet was open, in contrast to subpulmonary obstruction with abnormal pouch formation in Cx43-deficient mice. Coronary arteries originated from the aorta normally, and their arrangement appeared the same as in wild-type mice.
Cardiac function in cardiac-specific Cx43G138R mutants: spontaneous arrhythmias in vivo and ex vivo disturbed impulse propagation in surface electrocardiograms and no alteration in echocardiography indicates an electrical problem in G138R mutated mice
In two ODDD families, severe cardiac abnormalities have been described with two cases resulting in sudden cardiac death probably due to arrhythmia (8). In our studies, all Cx43+/floxG138R:alphaMyHC-Cre mice died during the first 6.5 months postnatally (n = 16, Fig. 4B). The functional cardiac ex vivo analyses in the Langendorff setup revealed spontaneous and sustained ventricular tachycardias (VT) or other types of cardiac arrhythmias starting directly after excorporation and continuing during perfusion in all mutants investigated (n = 8), but in none of the wild-type animals (n = 6; Fisher’s test: P = 0.0003) (Fig. 5B–I). This prevented analysis of sinus rhythm and application of stimulation protocols to Cx43+/floxG138R: alphaMyHC-Cre hearts. Therefore, no systematic evaluation of conductive properties could be carried out, and the mechanism(s) responsible for the induction of these arrhythmias could not be further explored. Epicardial activation maps revealed a heterogeneous distribution of activity in the ventricles during these episodes. The surface and intercardiac ECG studies showed significant changes in P, PQ, QRS and QTc intervals and R and RS amplitudes. The most prominent change was the broadening of the QRS complex and decrease in the R wave, indicating disturbed impulse propagation in the ventricle accompanied by low voltage ECG (Fig. 5A and Table 1).
Figure 5.
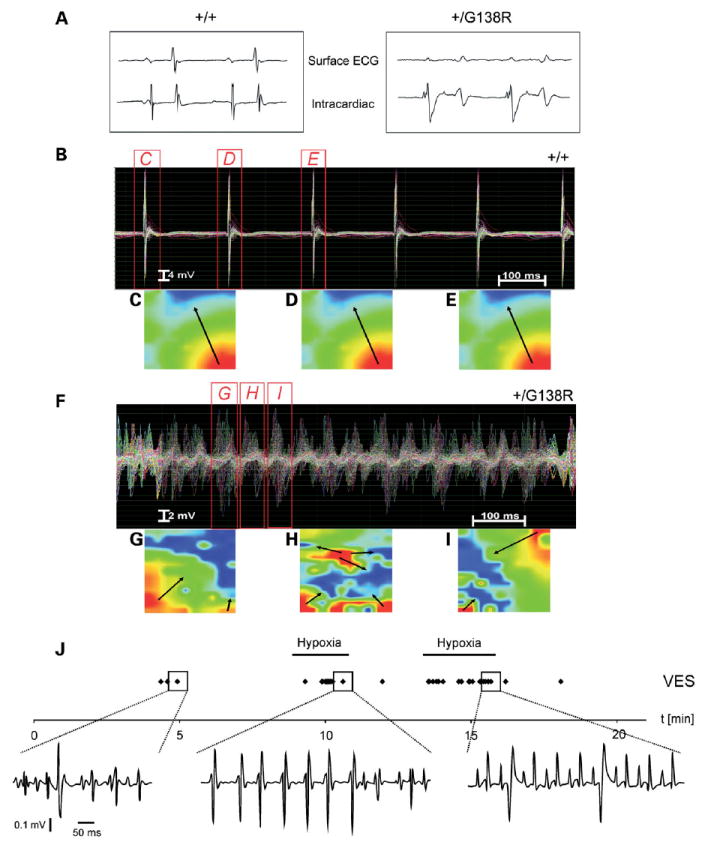
Disturbed impulse propagation and spontaneous cardiac arrhythmias developed in mutated hearts. (A) The disturbed impulse propagation in the ventricle is clearly seen in the striking broadening of the QRS complex in the surface ECG of the mutated hearts. (B) Normal impulse propagation and activation pattern in wild-type (n = 6) but ventricular tachycardias (VT) of all mutated (n = 8) hearts in Langendorff measurements. The cumulative Langendorff recordings of 128 epicardial electrograms clearly show repetitively directed and homologous conduction without signs of conduction disturbances or shift of conduction speed and direction in wild-type hearts during sinus rhythm (B–E) but a chaotic appearance of field potentials in the mutated ventricles during a polymorphic, high-frequency VT (F–I). Such polymorphic, incessant VTs were spontaneously present in all mutant hearts directly after the initiation of Langendorff extracorporal perfusion. EAMs of the left ventricular epicardium at three different time points during sinus beats in wild-type or polymorphic VTs in mutant hearts are shown in (C—E) or (G—I), respectively. The false color-coded reconstruction with red representing earliest and blue representing latest epicardial activation shows a heterogenic distribution of field potentials with inconsistent direction and speed of ventricular conduction in the Cx43G138R mutated mouse heart (arrows) during VT. This indicates different conduction pathways during different VT beats as typically present in polymorphic VT. (J) Cardiac arrhythmias recorded in vivo in cardiac-specific mutants. The frequency of the arrhythmias could be increased by hypoxia (hypoxia bars). Adjustment of O2 to normoxia largely abolished the arrhythmias. Arrhythmic events (VES) are displayed as individual points (VES during normoxia and under hypoxia). Even VT (1 VT ≥4 consecutive VES) could be recorded as a rare arrhythmic event during hypoxia (middle panel).
Table 1.
Results of surface ECG analyses, including low voltage ECG in Cx43G138R mice
| +/+ | +/G138R | P-value | |
|---|---|---|---|
| P (ms) | 14.8 ± 1.3 | 23.0 ± 3.4 | 0.013 |
| PQ (ms) | 42.8 ± 6.3 | 53.0 ± 2.0 | 0.043 |
| QRS (ms) | 14.0 ± 0.8 | 23.8 ± 4.4 | 0.02 |
| QTc (ms) | 30.1 ± 1.6 | 36.7 ± 3.4 | 0.023 |
| R (mV) | 0.55 ± 0.07 | 0.12 ± 0.02 | 0.001 |
| RS (mV) | 1.17 ± 0.25 | 0.28 ± 0.06 | 0.005 |
R, R-wave amplitude measured from the isoelectrical line to the maximum of R-wave; RS, maximal amplitude from the maximum of the R-wave to the most negative spike of the S-wave.
Using ECG recordings in vivo under normal normoxic conditions, we observed spontaneous arrhythmic events such as ventricular extra systole (VES) in the Cx43+/floxG138R:alphaMyHC-Cre mice (Fig. 5J). The frequency and severity of arrhythmic events were strongly increased by hypoxic stimulation (Fig. 5J and Supplementary Material, Fig. S12). In fact, VES developed with 5.1-fold higher probability during application of hypoxia in mutants, whereas no VES could be seen in wild-type littermates. Even VTs (four or more consecutive VES events) could rarely be observed in mutated mice (Fig. 5J, middle panel); none of the mice tested died during the hypoxic stimulations.
Anesthetized mice were also functionally analyzed using echocardiography. This analysis yielded no significant changes to wild-type mice in regard to the heart frequency, left ventricular muscle mass, left ventricular end-diastolic volume and left ventricular ejection fraction (Supplementary Material, Fig. S11).
The Cx43G138R protein is not phosphorylated but expressed in ODDD mutated hearts
Previously, it has been described that the phosphorylation of Cx43 regulates gap junctional and even the activity of ATP-releasing channels (12). We therefore tested whether the phosphorylation of Cx43G138R-containing channels is affected in vitro and in vivo. Immunoprecipitations of Cx43 from 32P-labeled HeLa transfectants (Fig. 6A) and immunoblot analyses of heart lysates (Fig. 6B) clearly showed a partial lack of phosphorylation of the Cx43G138R isoform.
Figure 6.
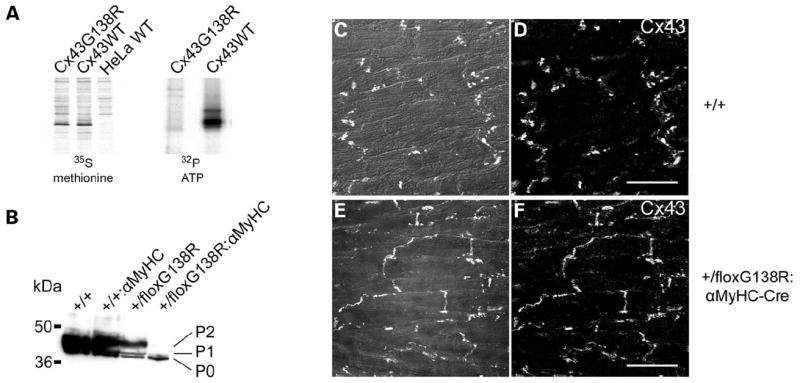
Cx43G138R is not phosphorylated at the P2 state in vitro as well as in vivo and is localized in contacting membranes of cardiomyocytes in the heart. Immunoprecipitation of Cx43 from 32P-labeled HeLa cells and immunoblot analyses of tissue lysates are shown. (A) Equal numbers of Cx43-expressing cells (parallel culture dish tested by 35S methionine labeling, left picture) were labeled with 32P ATP (right picture). Subsequent Cx43 immunoprecipitation and exposure reveal a reduced phosphorylation of Cx43G138R protein by 80%. (B) Immunoblot analyses show lack of P2 phosphorylation of Cx43G138R protein in hearts. (C–F) Immunofluorescence analyses indicate localization of Cx43 in controls (C and D) and in heterozygous Cx43G138R mice (E and F), suggesting an unhindered trafficking of the mutated proteins in vivo. Some dyslocalization of Cx43 protein from intercalated discs to lateral contact membranes is apparent [phase contrast and Cx43 staining merged in (C) and (E); Cx43 staining in (D) and (F); scale bar: 20 μm].
Equal amounts of Cx43WT-or Cx43G138R-expressing cells (see 35S methionine labeling) were labeled with 32P ATP, followed by immunoprecipitation of Cx43. The results clearly indicate that up to 80% of the Cx43G138R protein is not phosphorylated, in comparison with Cx43WT. Immunoblot analyses also showed in Cx43+/floxG138R:alphaMyHC-Cre hearts that the P2 phosphorylation isoform of Cx43 is absent. Immunofluorescence analyses of connexin isoforms in the ventricle revealed a slightly altered localization of these proteins in mutated hearts. Cx43-specific signals were detected in intercalated discs and in lateral contact membranes of ventricular cardiomyocytes (Fig. 6C–F), indicating an unhindered trafficking but a slightly changed localization of the Cx43G138R channels. Additional analyses to determine the expression of Cx40 in the atria and Cx45 in the ventricle, where Cx43 and Cx43G138R are co-expressed, revealed no changes in mutated relative to wild-type hearts (data not shown). This suggests no trans-dominant effects of the mutant protein onto Cx40 and Cx45 in the heart.
The Cx43G138R ODDD mutation leads to a loss of Cx43-mediated conduction, which can be maintained in heterozygous animals by Cx43WT homotypic and Cx43G138R/Cx43WT heterotypic channels
In order to determine the function of the Cx43G138R mutated protein, microinjections of tracers and electrophysiological studies were performed using mouse embryonic cardiomyocytes, ES cells and stably transfected HeLa cells (Fig. 7A–H). The microinjections into cardiomyocytes resulted in a reduction of neurobiotin transfer by 94%, thus indicating that only 6% of connexin-mediated conduction is maintained in Cx43+/G138R cardiomyocytes (Fig. 7I). Furthermore, the neurobiotin transfer in Cx43+/G138R mouse ES cells, which heterozygously expressed the Cx43 mutation, was reduced by 79% (Fig. 7I). Because of possible co-expression of additional connexins in cardiomyocytes (Cx45) or ES cells (Cx45 and Cx31), microinjection analyses using cells expressing only Cx43 isoforms were performed. Thus, HeLa cells stably expressing Cx43G138R alone or in the presence of Cx43WT proteins were investigated. No neurobiotin transfer could be detected in cells expressing only Cx43G138R (n = 15), whereas in cells co-expressing Cx43WT and Cx43G138R, neurobiotin transfer was slightly higher, suggesting a strong reduction or even a loss of Cx43-mediated conductance in Cx43+/G138R mice. Electrophysiological studies using dual whole-cell voltage clamp (24) revealed that all 14 examined HeLa Cx43G138R cell pairs exhibiting eGFP fluorescence (non-fused construct) demonstrated complete absence of coupling. In order to determine whether Cx43-mediated coupling in Cx43+/G138R mice is maintained through Cx43G138R/Cx43WT heterotypic channels besides wild-type homotypic channels, we examined cell–cell coupling in co-cultures of HeLa cells expressing Cx43G138R and Cx43-eGFP (fusion construct). We found well distinguishable and, in many cases, multiple junctional plaques in the contact regions between cells expressing Cx43G138R and cells expressing Cx43-eGFP (Fig. 7J). Figure 7K shows the averaged dependence of junctional conductance (Gj) on transjunctional voltage (Vj) that was obtained by measuring junctional current (Ij) dynamics over time in response to long (150 s) Vj ramps that change from 0 to +120 mV and from 0 to −120 mV [for more details see Fig. 3 in (25)]. In all nine cell pairs examined, Gj–Vj dependence was asymmetric with higher Vj gating sensitivity at positive voltage on the Cx43-eGFP side. GJC gating at the single-channel level (Fig. 7L) shows a tendency to open during a negative voltage step and to close during a positive voltage step, applied to the HeLaCx43-eGFP cell. This gating asymmetry at the single-channel level is in accordance with macroscopic Gj–Vj dependence, as shown in Figure 7K. Figure 7M shows the Ij record of a single channel in a heterotypic junction during application of hyperpolarizing Vj step of 87 mV with a HeLaCx43G138R cell. In the example shown, the single-channel conductance at the open state is ~103 pS, and channels mainly gate between the open state and a substrate or the residual state with a conductance of ~25 pS. We ascribe this kind of gating to the fast gating (26). Besides the fact that most gating transitions are between open and residual states of ~78 pS in magnitude, we also observed, but more rarely, transitions between the open state and the fully closed state (indicated by arrows), which can be ascribed to the slow gating (26). Earlier, it was reported that Cx43-eGFP channels exhibit the slow but not the fast gating mechanism. Therefore, the data shown in Figure 7M should be ascribed to the gating of Cx43G138R hemichannels, which presumably retain both fast and slow gating mechanisms such as wild-type Cx43. Gating to the substate can also be seen in Figure 7M at the negative Vj step. Both macroscopic and microscopic gating properties shown in Figure 7K and M suggest that Cx43G138R hemichannels exhibit higher Vj gating sensitivity than Cx43-eGFP hemichannels and presumably gate at negativity on the cytoplasmic side of Cx43G138R.
Figure 7.
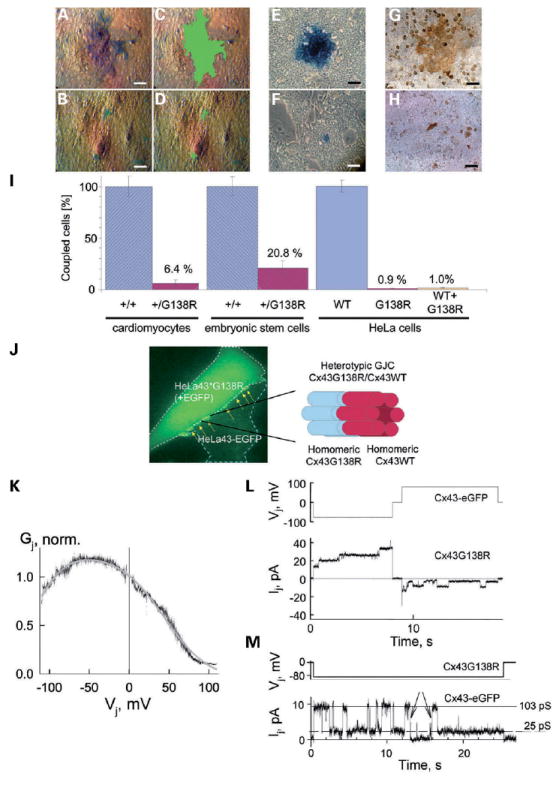
Cx43-mediated coupling is disturbed in ODDD-mutated mice and can be maintained by Cx43/Cx43 homotypic and Cx43G138R-Cx43WT heterotypic channels. Intercellular transfer of microinjected neurobiotin in wild-type Cx43-expressing cells [primary mouse cardiomyocytes (A and C), ES cells (E) and HeLa cells (G)] in comparison with Cx43G138R-expressing cells [primary mouse cardiomyocytes (B and D), ES cells (F) and HeLa cells (H)]. In (C) and (D), the coupling area is outlined in pseudo-color. The diagrams shown in (I) compare neurobiotin transfer in wild-type and mutated cardiomyocytes and stem cells as well as in HeLa cells expressing Cx43WT or Cx43G138R alone or co-expressing Cx43WT and Cx43G138R. A residual coupling in approximately 2.5 cells in contact (~6.4%) was observed. In stem cells dye, transfer was substantially higher (six cells; 20.8%). HeLa cells expressing Cx43G138R alone or in parallel with Cx43WT showed a very low level of dye transfer. Cx43–eGFP/Cx43G138R heterotypic junctions, shown in (J), form junctional plaques (see arrows) in the junctional region between HeLa Cx43G138R cell (left/top) exhibiting homogenous eGFP in cytoplasm and HeLaCx43-eGFP cell (right/bottom) exhibiting punctate staining of eGFP prominently in contacting membranes. (K) Summarized junctional conductance dependence on transjunctional voltage (Gj–Vj) measured in nine cell pairs forming heterotypic Cx43–eGFP/Cx43G138R junctions. These exhibit an asymmetric Vj gating with higher sensitivity at positive voltages on the HeLaCx43-eGFP side. (L) Unitary opening and closing events at the single-channel level of Cx43–eGFP/Cx43G138R heterotypic GJ channels seen as stepwise junctional current (Ij) transitions in response to consecutive negative and positive voltage steps applied to the HeLaCx43-eGFP cell. Channels show a tendency to open during a negative voltage step and to close during a positive voltage step that is in accordance with macroscopic Gj–Vj dependence shown in (K). (M) Single-channel record demonstrating that both fast and slow gating mechanisms operate in Cx43G138R/Cx43 heterotypic GJC. During application of hyperpolarizing Vj step to HeLa Cx43G138R cell, the channel operates mainly between the open state with a conductance of ~103 pSand the residual state with a conductance of ~25 pS. Arrows point totransitions between the open state and the fully closed state, which can be ascribed to the operation of slow gating mechanism.
HeLa cells are known to express intrinsically Cx45, but at very low level (25). If one assumes that the cell–cell coupling shown occurs through Cx43-eGFP/Cx45 heterotypic junctions then (i) the macroscopic Gj/Vj dependence should be more asymmetric with higher voltage sensitivity at positive Vj (27), (ii) single-channel conductance of the open state should be ~55 pS (27) instead of ~100 pS (Fig. 7M), (iii) among 14 examined HeLaCx43G138R cell pairs, at least one or few of them should show coupling with some Cx45 channels instead of full absence of coupling and (iv) junctional plaques should be much smaller than those shown in Figure 7J (25). Therefore, we can strongly argue that observed coupling is not due to the formation of Cx43/Cx45 instead of Cx43/Cx43G138R channels. We suggest that at Vj = 0 mV, only a fraction of Cx43G138R hemichannels are open. At Vj = 0 mV, open probability of the Cx43WT hemichannel is close to 1 and therefore the open probability of Cx43G138R/Cx43WT GJ channels will be limited by the open probability of Cx43G138R hemichannels (pCx43G138R). If, for example, pCx43G138R equals 0.1, then only one of 100 Cx43G138R homotypic channels will be open, which may explain the low functional efficiency of Cx43G138R homotypic junctions.
Cells expressing Cx43G138R show enhanced release of ATP
As recently described, some ODDD mutations can lead to the reduced release of ATP through hemichannels (18). To investigate whether the Cx43G138R mutation alters cellular ATP release, we measured the extracellular ATP concentration and propidium iodide uptake of cultured cells expressing Cx43G138R upon stimulation in calcium-free solution or under hypoxic conditions (Fig. 8). The heterozygously recombined ES cells expressing the ODDD-mutated Cx43 besides Cx43WT released 2.0-fold more ATP than wild-type ES cells cultured in calcium-free medium (Fig. 8A). Cardiomyocytes derived from embryonic Cx43+/floxG138R:alphaMyHC-Cre or wild-type hearts on ED 12.5 or ED 16.5 were tested in the hypoxic incubator. The activity of ATP-releasing channels in oxygen-dependent cardiomyocytes on ED 16.5, but not in the oxygen-independent ED 12.5 cells, could be stimulated by hypoxia (Fig. 8B). A 1.8-fold higher ATP concentration in media from mutant cells was measured. The increased activity of ATP-releasing channels was also confirmed by propidium iodide uptake in embryonic cardiomyocytes stimulated in calcium-free medium (Fig. 8C). The mutated cells exhibited 2.5-fold higher ATP release than wild-type cells.
Figure 8.
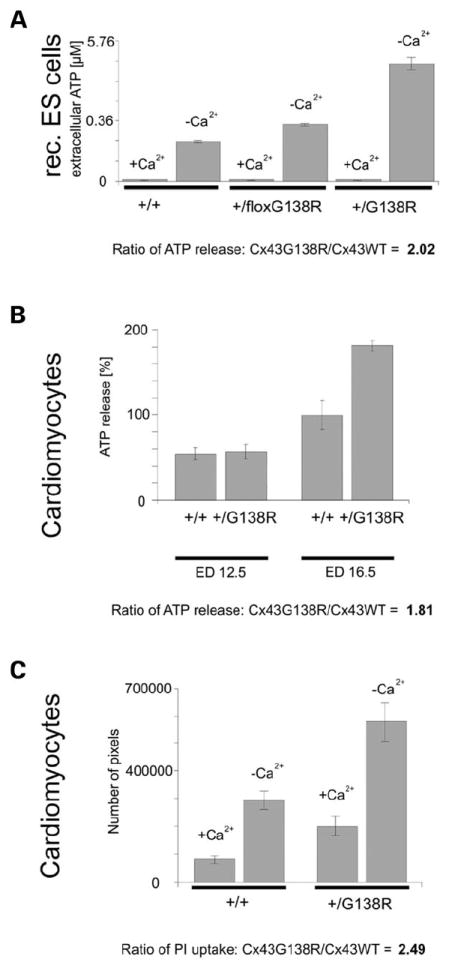
Cx43G138R leads to an increased activity of ATP-releasing channels. Measurements of ATP concentrations in media of cultured cells expressing Cx43G138R (A and B), and tracer uptake studies (C) showed a doubled activity of ATP-releasing channels. (A) Recombined, heterozygous ES cells expressing the mutation showed an increased activity of the ATP-releasing channels. (B) In oxygen-dependent cardiomyocytes (ED 16.5), the activity of ATP-releasing channels could be observed after 1.5 h of stimulation in a hypoxic incubator, but was not detected in oxygen-independent cells derived from younger embryos (ED 12.5). (C) Studies of tracer uptake of Cx43+/floxG138R:alphaMyHC-Cre cardiomyocytes also indicate the doubled activity of these channels before and after stimulation, as already shown in the ATP-release experiments.
The activity of ATP-releasing channels influences the beating frequency of embryonic Cx43+/floxG138R: alphaMyHC-Cre cardiomyocytes
To investigate the potential functional relevance of ATP-releasing channels, we next assessed the beating rates in mutant and wild-type cardiomyocytes. We found that embryonic cardiomyocytes isolated from cardiac-specific mutants exhibited a 1.6-fold higher beating frequency (242 bpm) than wild-type cardiomyocytes (148 bpm; P < 0.001) (Fig. 9). This finding in combination with the increased ATP release and dye uptake (discussed earlier) indicated an ATP release channel-dependent positive chronotropic effect. This was further investigated by using peptides and drugs that have been reported to inhibit the activity of ATP-releasing hemichannels. Flufenaminic acid (FFA) (50 μm) or the mimetic peptide Gap26 (300 μm) blocked the ATP-releasing channels, whereas the activity of GJCs was un-affected (data not shown). Both these pharmacological agents reduced the beating frequency of mutated cardiomyocytes to the level of wild-type cardiomyocytes (FFA: 155 bpm and Gap26: 143 bpm), supporting a functional impact of ATP-releasing channels.
Figure 9.
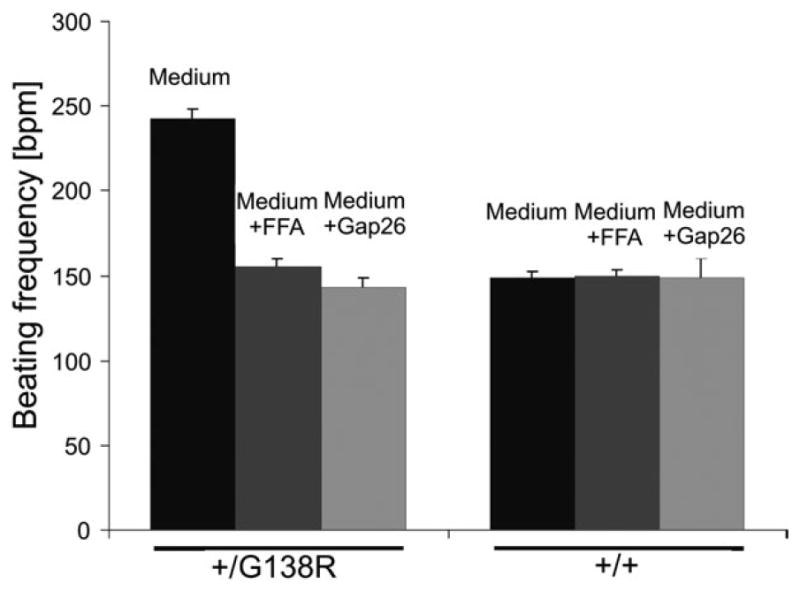
Activity of ATP-releasing channels influences the beating frequency of mutated cardiomyocytes. Mutated cardiomyocytes beat faster than the wild-type ones (P < 0.001). Application of FFA or the mimetic peptide Gap26 at concentrations blocking the ATP-releasing channel activities decreased the beating frequency of mutant cells to the wild-type level (P < 0.001).
DISCUSSION
Here, we present a new conditional mouse model for ODDD, carrying the human Cx43G138R point mutation. The main findings of our study are the occurrence of all common phenotypes of the human-inherited disease ODDD and the heart-related mortality of Cx43G138R heterozygously mutated mice. The mortality of the mice is likely due to the strongly decreased gap junctional function, supporting the proarrhythmic action of the G138R-mutated Cx43 protein. This appears to be associated with a partial lack of phosphorylation. The gap junction blocker FFA at a concentration inhibiting ATP-releasing hemichannels or the mimetic peptide Gap26 as specific blocker of these channels reduced the increased beating frequency of mutated cardiomyocytes in culture to the same level as observed in wild-type cells. This suggests that the spontaneous ventricular arrhythmias observed in Lan-gendorff and in vivo recordings of mutated adult hearts are mainly due to the strongly decreased gap junctional coupling and could be aggravated by the increased release of cellular ATP or other small molecules.
The G138R mutation is localized in the cytoplasmic loop of the membrane-embedded Cx43 protein, i.e. the receptor domain for the C-terminal tail in the ball-and-chain model describing the opening and closing of a GJC (28). This mutation was characterized in transfected cells as a dominant-negative one inhibiting the gap junctional function of wild-type Cx43 (16). Thus, this mutation leads to a lack of GJC activity, although the Cx43 protein is still expressed in the cell membrane. These properties clearly distinguish the Cx43G138R mutation from Cx43 null mutant mice that do not express the Cx43 protein and the Cx43G60S mutation, where the mutated protein is largely excluded from the plasma membrane (21).
In our study, we confirmed the dominance of the G138R mutation using embryonic cardiomyocytes from heterozygous mice and found a strong (~94%) reduction in cell-to-cell permeability of tracer molecules. Furthermore, the loss of the P2 phosphorylation band that was observed in the mutated mouse hearts under in vitro and in vivo conditions has been suggested to be associated with the loss of Cx43 channel function (29,30). Thus, the glycine residue at position 138 of Cx43 seems to contribute to the occurrence of the P2 phosphorylation state and the function of GJCs. A possible explanation for this could be the influence of glycine 138 on the structure of the cytoplasmic loop, which contains two distinct alpha helical regions promoting the binding to the channel closing C-terminal tail (31).
After ubiquitous activation of the mutated Cx43G138R gene in the transgenic mice, all ODDD phenotypic manifestations described in patients including syndactyly, craniofacial, bone and cardiac abnormalities were found. These abnormalities demonstrate the importance of Cx43 during embryonic development and confirm the identity of this ODDD mouse model. Because of high embryonic lethality of the ubiquitously expressed mutant and the necessity to compare mice of the same gender and age for the bone morphological studies, we only tested male mice. Here, significant osteopenia with reduced bone volume and increased trabecular spacing was observed. This is most likely due to a decreased number of osteoblasts in the ODDD mutant mice, although the difference in osteoblast number was not statistically significant, probably due to the high variability of the ODDD phenotype. Notably, expression of Cx43G138R, as well as other ODDD mutants in osteoblast-enriched calvaria cells, does not seem to alter the ability of these cells to differentiate and produce mineralized matrix in vitro (32). Therefore, the dominant-negative effect of the mutant proteins may not be sufficient to completely inhibit wild-type Cx43 in committed osteoblasts, but it may be sufficient to interfere with earlier commitment of stromal cell precursors. A reduced number of osteoblasts in the ODDD transgenic mice would be consistent with this notion.
The conditional approach allowed us to investigate the influence of the G138R mutation in each cell type, where the Cre-recombinase can be specifically activated. Thus, we were able to determine the heart as the causative tissue for the embryonic lethality observed in the mutant, further underlining the importance of Cx43 for normal heart function. In humans, only few ODDD patients have been reported to suffer from a cardiac phenotype. However, the Cx43G60S and, even to a stronger degree, the Cx43G138R mutant mice exhibit cardiac dysfunctions, which have not been reported for ODDD patients. Currently, we cannot exclude that the mouse heart is more sensitive to Cx43 ODDD like mutations than the human heart. On the basis of our report as well as the study by Flenniken et al. (21), it appears possible that some of the ODDD patients may exhibit a cardiac phenotype during hypoxic stress that has not yet been detected. It is also possible that up-regulation of other connexins such as Cx45 in human heart failure (33) can partially compensate for the loss of Cx43 coupling. However, we could not find such an up-regulation of Cx45 in the Cx43G138R mouse mutant.
Because of the embryonic lethality due to cardiac expression of the Cx43G138R mutation, we carried out a detailed investigation of the cardiac phenotype and its molecular basis. To exclude the influence of any other organs such as the central nervous system, we performed all cardiac analyses with mice specifically expressing the Cx43G138R mutation in cardiomyocytes (Cx43+/floxG138R:alphaMyHC-Cre). In addition to the heart-related embryonic lethality, we found premature lethality in adult mutants during 2–6.5 months after birth, as determined by the Kaplan–Meier survival curve (Fig. 4B). The observed in vivo arrhythmias (Fig. 5J), which are particularly pronounced during hypoxic periods (Supplementary Material, Fig. S12), could be responsible or at least associated with the high lethality of ODDD-mutated mice.
The cardiac function in adult mice was determined by surface and intracardiac ECG, Langendorff and in vivo ECG measurements. Suppressed impulse propagation in the ventricle recognized by the broadening of the QRS complex and the lowering of its amplitude are similar to the situation in the Cx43-deficient heart (34). The Cx43-mediated coupling in the heterozygous mouse mutant described in this article is maintained by Cx43G138R-Cx43WT heterotypic and Cx43WT-Cx43WT homotypic channels.
The most striking findings in the Cx43G138R transgenic mice were the development of severe ventricular tachycardias, the missing influence of the mutation on heart morphology, as seen in the Cx43-deficient mice, and the increased activity of ATP-releasing channels. In fact, the predisposition to arrhythmia in Cx43G138R mice can be explained by the uncoupling of ventricular cardiomyocytes, resulting in the loss of directed electrical conduction. This has been discussed for adult Cx43-deficient hearts (34,35), exhibiting ventricular arrhythmias that occur less frequently than in Cx43+/floxG138R: alphaMyHC-Cre mice. In this context, van Rijen et al. (35) showed that only a low percentage of cardiac Cx43-depleted animals developed spontaneous arrhythmias, whereas all of the Cx43G138R mice exhibited spontaneous VTs implicating an enhanced arrhythmogeneity of the G138R mutation versus the Cx43-deficient heart.
The previously published Cx43 mutant by Flenniken et al. (21) showed no relevant QRS-prolongation, low-voltage ECG or long-lasting ventricular arrhythmias, but significantly prolonged P-waves and AV nodal conduction blocks. Analogous conduction defects of the atria and AV node, i.e. P-prolongation and first-degree AV nodal block, occurred in Cx43+/floxG138R:alphaMyHC-Cre mice. In comparison with other mouse strains with alterations of Cx43 expression and function, the Cx43G138R mutant seems to have the strongest impact on cardiac arrhythmogeneity, resulting in prominent alterations of the electrical conduction in atria, AV node and ventricular myocardium accompanied by severely enhanced arrhythmogeneity in vivo and in the isolated heart. These findings point toward more severe alterations of intercellular coupling, conduction properties and other Cx43 functions when compared with all other Cx43 mutants described to date. A possible molecular basis for the striking arrhythmogeneity of G138R mutated hearts may be enhanced activity of the ATP-releasing channels, which form a leak across the plasma membrane. This could lead to a reduction in the resting potential, as already proposed for mCx30.2 (36). This may contribute to enhanced automaticity and initiation of extrasystolic activity. This notion is supported by aggravation of cardiac arrhythmia during short hypoxic phases. Here, the opening times of ATP-releasing channels may be extended, leading to increased uncoupling of cardiomyocytes and thus to ventricular arrhythmia.
The increased activity of ATP-releasing channels was observed in transgenic ES cells and embryonic cardiomyocytes expressing the same amount of both Cx43WT and Cx43G138R proteins. We found under hypoxic conditions an 80% increased ATP release in oxygen-dependent cardiomyocytes (ED 16.5), but not in oxygen-independent ones (ED 12.5).
The physiological relevance of this gain-of-function mutation regarding the ATP-releasing channel activity was studied in cultured cardiomyocytes. Here, we could lower the increased beating frequency of mutated cells to the wild-type level using Gap26, a mimetic peptide with an amino acid sequence corresponding to the region of the first extracellular loop of Cx43 as a blocker of ATP-releasing channels. Furthermore, this experiment clearly shows that the blockade of ATP-releasing channels in wild-type cells does not influence the beating frequency of wild-type ventricular cardiomyocytes. We hypothesize that the increased activity of ATP-releasing channels and the associated increased release of ions could depolarize the membrane potential of the mutated cells. Under this aspect, the ATP-releasing channels in Cx43G138R mutated cells show some analogy to the mutated cardiac potassium channel KCNQ1 (37). This gain-of-function mutation in the KCNQ1 gene, associated with the long-QT syndrome in a Chinese family, was shown to be the molecular basis for the autosomal-dominant atrial fibrillation of the corresponding patients. However, this hypothesis needs to be corroborated by further experiments.
Our results suggest that increased activity of ATP-releasing channels in patients with ODDD mutations can aggravate the predisposition to arrhythmias, which is due to the strong reduction of gap junctional communication (cf. 35). The Cx43G138R mouse model of ODDD will be further used to analyze the effects of increased activity of ATP-releasing channels on bone formation, syndactyly and astrocytic functions.
MATERIALS AND METHODS
All mice used in this study were kept under standard housing conditions with a 12 h/12 h dark–light cycle and with food and water ad libitum. All experiments were carried out in accordance with local and state regulations for research with animals.
Cloning of the conditional Cx43G138R expression vector
The Cx43G138R mutation from a human patient (8) was inserted by PCR mutagenesis in the corresponding mouse gene, cloned into the pBluescript vector and sequenced in both directions by AGOWA (Berlin, Germany). The mutated gene was cloned into a vector containing the IRES and the eGFP cassettes by ClaI/XhoI digestions. The IRES sequence was cloned from pIRES (Clontech Laboratories, CA, USA) by EcoRI/XbaI into pBluescript, digested with SalI and the eGFP-coding DNA from pMJ-Green (38) by BamHI/NotI digestion into the pBluescript-IRES vector.
The final construct consisted of two homologous regions: a 4811 bp intron 5′ upstream fragment and a 922 bp ClaI/EcoRI 3′ downstream fragment from Cx43 genomic DNA. The neomycin selection cassette and its PGK promoter were flanked by frt sites and cloned downstream of the polyA signal of Cx43 by SalI digestion. Both the Cx43 coding region and the selection cassette were flanked by loxP sites, followed by the Cx43G138R-IRES-eGFP cassette (XbaI/EcoRI).
The final conditional vector was partially sequenced by AGOWA after a restriction analysis. The functionality of the loxP and the frt sites was verified by transformation into Cre or Flp recombinase expressing Escherichia coli bacteria (39). The function of the Cx43G138R-IRES-eGFP construct was tested in HeLa cells (described subsequently).
Screening of ES cell clones for homologous recombination
The targeting vector DNA (150 μg) was linearized by NotI digestion and transfected by electroporation (800 V and 3 μF) into murine ES cells (HM1) (40). Selection of vector containing cells was performed using 350 μg/ml G418 (Invitrogen, Karlsruhe, Germany). In order to test for homologous recombination, genomic DNA from the ES cell clones was processed for PCR analysis using a 5′ upstream primer, binding to the eGFP sequence in the targeting vector (eGFP_rev: CAT GGA CGA GCT GTA CAA GTA AAG CG), and an external 3′ downstream primer specific for the 3′ region of Cx43 (Cx43_3′HR: CAC TTG ATA GTC CAC TCT AAG CAA CC). To exclude clones in which recombination occurred partially, a second PCR analysis was performed with cells positive in the first one. Here, we used primers annealing upstream of the 5′ loxP site (Cx43/31for: GCA CTT GGT AGG TAG AGC CTG TCA GGT C) and downstream behind it (Cx43/31rev: CTC CAC GGG TCT GTA CCC ACT GAC CTC). Clones found to be positive in both PCR analyses were tested by Southern blot hybridization. For this purpose, DNA from the PCR-positive clones was digested with HincII or BamHI (both for the hybridization with the internal probe) or HindIII (external probe). After electrophoretic separation on agarose gels, blotting onto Hybond N+ membranes (Amersham Biosciences, Buck, UK) and ultraviolet crosslinking for fixation, hybridization was performed under stringent conditions using the Quick Hyb solution (Stratagene, La Jolla, CA, USA) at 68°C for 1.5 h. A 550 bp HincII fragment from the coding region of Cx43 was used as the internal probe and a 550 bp AvaI fragment of the 3′ homology region served as the external probe.
Generation and genotyping of the Cx43floxG138R mice
The homologously recombined ES cell clone was injected into C57BL/6 blastocysts, as described previously (41). Blastocyst injections of ES cells resulted in mice, showing a high-degree fur-color chimerism which afterwards mated with C57BL/6 mice. Germline transmission of the recombinant allele was checked in agouti offspring by PCR analyses of isolated tail DNA. The heterozygous Cx43+/floxG138R mice were backcrossed with C57BL/6 mice to increase the C57BL/6 genetic background and mated to PGK-Cre (22), alphaMyHC-Cre (42) or Nestin-Cre (43) expressing mice for deletion of the floxed Cx43WT coding DNA and activation of the Cx43G138R allele ubiquitously in cardiomyocytes or in early neurons, respectively. To check for recombination after exposure to Cre-recombinase activity, BamHI digestion and hybridization with the internal probe were performed as described for screening of ES cell clones. Genotyping was performed by the PCR analysis using primers flanking the 5′ upstream loxP site (primer1_loxP: GCA CTT GGT AGG TAG AGC CTG TCA GGT C and primer2_Cx43: GCT TCC CCA AGG CGC TCC AGT CAC CC). All experiments were carried out with littermates of >87.5% C57BL/6 genetic background.
Northern blot hybridization
Hearts and brains from transgenic and control mice were collected in liquid nitrogen. Total RNA was extracted using TRIZOL (Invitrogen, Carlsbad, USA), following the instructions of the manufacturer. Twenty micrograms was electrophoretically separated, blotted and hybridized as described (44). The same 550 bp HincII fragment of the Cx43 coding region that was used for Southern blot hybridizations was also used for Northern blot hybridization.
35S methionine and 32P ATP labeling of proteins
HeLa cells cultured on 35 mm dishes were incubated in 500 μl deficiency medium (Dulbecco’s modified Eagle’s medium without L-glutamine and L-methionine or phosphate, c.c.pro, Neustadt, Germany) for 1 h at 37°C and afterwards with 50 μCi 32P ATP or 10 μCi 35S methionine per 105 cells in same medium for 4 h at 37°C. The labeled cells were washed three times with phosphate-buffered saline (PBS) and harvested in cold RIPA buffer (38).
Immunoblot analysis and immunoprecipitation
Hearts or HeLa cells were solubilized in RIPA buffer and 1× Complete (Roche, Mannheim, Germany) and sonicated three times for 10 s during incubation on ice. For immunoprecipitation, rabbit Cx43 serum (1:1000 in blocking solution) (45) was incubated with 10 μl protein A–Sepharose beads (Amersham Bioscience) for 30 min at 4°C. Three hundred and fifty microgram protein were incubated with the antibodies–bead complexes for 1.5 h at 4°C and washed three times with RIPA washing buffer for 10 min. For immunoblotting, the total protein was separated in SDS–PAGE and blotted onto Hybond ECL membrane (Amersham Bioscience). After blocking with 5% milk powder in washing buffer (8.5 mM Tris–HCl, 1.7 mM Tris-base, 50 mM NaCl, 0.1% Tween-20) for 1 h at room temperature, a 1 h incubation was performed with rabbit Cx43 antibodies (1:2000 in blocking solution) and a 45 min incubation with goat anti-rabbit horseradish peroxidase (HRP)-conjugated antibodies (1:25000 Dianova, Hamburg, Germany), with three wash steps in-between and afterwards. The membranes were incubated with enhanced chemiluminescence reagents (Amersham Bioscience) and developed on X-ray films.
Immunohistochemistry
Hearts were frozen in liquid nitrogen and embedded in Tissue Tec (Miles Inc., OH, USA) for cryostat sectioning. The sections (9 μm) were dried on air, fixed with absolute ethanol for 5 min at −20°C, washed three times with PBS−, blocked in 4% bovine serum albumin (Sigma Aldrich, Steinheim, Germany) for 30 min and incubated with rabbit anti-Cx43 serum (45) in blocking solution for 1 h. After washing with blocking solution, a 50 min incubation with goat anti-rabbit antibodies conjugated with Alexa 594 (1:2000 in blocking solution; MoBiTec, Göttingen, Germany) followed. After an additional washing step, the sections were embedded in Permaflour and analyzed with a Laser Scanning Microscope (LSM Axioplan 2; Zeiss, Germany).
Histological studies
Hearts investigated in the electrophysiological studies were fixed in 4% paraformaldehyde for 24 h at 4°C, dehydrated and embedded in paraffin. The 3 μm thick sections were dried, deparaffinized with xylene, hydrated in a dilution series of ethanol, stained with hematoxylin/eosin, treated with xylene and mounted in Entellan (Merck, Hohenbrunn, Germany).
For bone analyses, tibiae were dissected and fixed in 10% formalin, decalcified in 14% EDTA for 14 days and embedded in paraffin. Sections were stained with hematoxylin/eosin. Quantitative histomorphometry was performed in an area 175–875 μm distal to the growth plate using the OsteoMeasure software program (Osteometrix, Atlanta, GA, USA) in an epifluorescence microscopic system, as detailed (46) elsewhere. The following parameters of bone remodeling were estimated: trabecular bone volume as a percentage of total tissue volume (BV/TV), trabecular separation (Trabec.Sp. in micrometers) and osteoblast perimeter per bone surface (Ob.Pm/BS. in percent).
Cell culture experiments
HeLa cells were stably transfected with a vector containing the Cx43G138R-IRES-eGFP cassettes or the Cx43-coding DNA driven by a CMV promoter for overexpression. Double transfectants were generated by stable transfection of Cx43-expressing cells (47) with the Cx43G138R vector. The recombinant ES cells were generated by homologous recombination, as described earlier. To express the Cx43G138R protein, the ES cells were transduced with Cre protein and subcloned, as previously described (48). The cardiomyocytes were derived from Cx43+/floxG138R:alphaMyHC-Cre and for controls from Cx43+/floxG138R ventricles of embryonic hearts on ED 12.5 and ED 16.5, as described previously (49). Each heart was dissociated and plated separately; its genotype was identified by green fluorescent protein and proven by PCR of the remaining embryonic tissue.
ATP release and dye uptake studies
To measure the activity of ATP-releasing channels, cells were incubated in 500 μl modified Hank’s balanced salt solution (HBSS; Sigma Aldrich) with or without Ca2+, Mg2+ and additional 1 mm EGTA for 20 min at 37°C. For hypoxic stimulation, the cells were incubated for 2 h in a hypoxic incubator (Herolab, Wiesloch, Germany).
For ATP release experiments, 100 μl of the supernatant was collected and incubated with 100 μl nucleotide releasing reagent (ViaLight HS kit, Cambrex, Rockland, ME, USA) for 20 min. The luciferase activity indicating ATP concentration was measured for 10 s with a Berthold Microplate LB96V luminometer after addition of 20 μl ATP monitoring reagent. The luminometric results were normalized to whole protein amount. In studies with HeLa transfected cells, the expression level of the mutant and the wild-type Cx43 protein were determined by the immunoblot analysis. Expression of Cx43G138R was normalized by GFP fluorescence and protein amount.
For dye uptake studies, ED 16.5 cardiomyocytes were incubated for 20 min in HBSS solutions with 10 μm propidium iodide, washed twice after bathing, fixed in 4% paraformaldehyde and mounted in Permaflour mounting medium (Immunotech, Marseille, France). The propidium iodide fluorescence was detected with a Zeiss microscope. To exclude all dead cells showing a weak fluorescence, only the number of pixels above a fixed threshold was used for further evaluation.
Microinjection analysis and patch clamp analysis
All tested cells were microinjected by iontophoresis with neurobiotin at confluency. In the case of embryonic cardiomyocytes, the peripheral cells of beating cell clusters were injected. The cultures were fixed in a fresh 1% glutaraldehyde in PBS− for 5 min, washed twice with PBS− and permeabilized with 2% Triton X-100 for 2 h at room temperature. After washing with PBS−, the cells were incubated with HRP-conjugated avidinD (Vector Laboratories, Burlingame, CA, USA) solution for 90 min, washed again and stained with the HistoGreen POD substrate kit (Linaris, Wertheim-Bettingen, Germany), following manufacturer’s instructions.
To study whether heterotypic Cx43G138R-Cx43WT channels are functional, Cx43G138R expressing HeLa cells and HeLa cells expressing Cx43WT-eGFP as a fusion protein were co-cultured. Cell pairs showing visible eGFP fluorescent plaques were identified and measured. Patch clamp protocols were used, as described previously (24).
Comparative analysis of skull bones
Skulls from mutant and control mice of 3 weeks and 6 months of age were prepared. The skin, the brain and most of the flesh were removed. Then, the skulls were incubated with maggots cultures over night. The remaining tissue was removed carefully, and the skull bones were cleared in a 13% H2O2 solution for 5–10 min.
Application of inhalation anesthetics induces ventricular arrhythmias in cardiac-specific ODDD mutants
Ubiquitous and cardiac-specific mutants showed in contrast to wild-type mice a high lethality during isoflurane or sevoflurane anesthesia following standard protocols. In order to investigate the reason for the obvious incompatibility toward inhalation anesthetics, we administered ketamine (10 mg/kg weight) with disoprivan (0.05 mg/kg weight) for sedation and increasingly applied isoflurane (0.3% till exitus) during echocardiographic recording. Between 2 and 5% isoflurane, all cardiac-specific Cx43+/floxG138R:alphaMyHC-Cre mutant mice investigated developed tachycardias and ventricular fibrillation, whereas wild-type mice tolerated these concentrations. Moreover, mutated mice developed apnea at normal sinus-rhythm electrograms, indicating possible hemodynamical complications because of electromechanical uncoupling suggesting severe electrical and electromechanical disturbances as the cause of death. However, ECG recordings could be successfully performed in several mice before these alterations occurred.
Electrophysiological and Langendorff studies
All electrophysiological in vivo measurements were carried out as described previously (35). For evaluation of ex vivo myocardial conduction velocities and characteristics, hearts were Langendorff-perfused, and epicardial activation mapping (EAM) was performed using a 128-electrode array. Hearts were excorporated, dissected from surrounding tissue in ice-cold Krebs–Henseleit buffer and retrogradely perfused in a Langendorff apparatus (Radnoti Technologies Inc., Monrovia, CA, USA) at constant pressure (80 mmHg). The perfusate composition was (in millimolar): NaCl 110, KCl 4.6, MgSO4 1.2, CaCl2, NaH2PO4 2, NaHCO3 25, glucose 8.3, Na-pyruvate 2 and gassed with carbogen (O2 95%, CO2 5%), pH, 7.3–7.45 at constant 37°C. Under normal perfusion conditions, all hearts started beating spontaneously. Hundred twenty-eight unipolar extracellular electrograms were recorded from the epicardial surface of the left ventricle using a custom-made electrode array. Interelectrode distance was 300 ±7 μm. Unipolar electrograms were recorded using a 128 channel computer-assisted recording system (Multi Channel Systems, Reutlingen, Germany), with a sampling rate of up to 25 kHz. Data were bandpass-filtered (50 Hz), digitized with 12 bit and a range of 20 mV.
Activation maps were calculated from these data using custom-made software (Labview 7.1, National Instruments, Austin, TX, USA). The first derivative of each unipolar electrogram was evaluated, and the maximal negative dV/dt activation was defined as time point of local activation in these mappings, as described before (35,50,51). Color-coded mapping indicates with red earliest activated and with blue latest activated areas at given beat.
For in vivo ECG recordings, each animal was placed in a small apparatus (ECG tube, QRS Phenotyping Inc., Calgary, Canada), where the feet made contact with gel electrode pads (52). ECG waves and intervals were measured with a PowerLab 26T device (ADInstruments, Colorado Springs) and tabulated using a pattern recognition program (Chart 5 Pro ADInstruments). For hypoxic stimulation in vivo, the animals were placed into the ECG tube, anesthetized with 0.5% isoflurane and exposed to hypoxic (hypoxia = gas mixture of 5–10% O2 with 95–90% argon) for <2 min or normoxic conditions.
Contractions of cultured embryonic cardiomyocytes
The beating frequency of cultured embryonic cardiomyocytes was measured after application of different drugs. Cells were exposed to FFA (50 μm, Sigma Aldrich) or the mimetic peptide Gap26 (300 μm, Genscript, USA) for 30 min at 37°C. Movies of beating cardiomyocytes were recorded with a Sony XCD-X710 (Koeln, Germany) camera at 30 frames/s, and beating areas were analyzed off-line with a custom written software (Labview 7.1 and IMAQ, National Instruments). The number of pixels, which change their intensity above a defined threshold in consecutive frames, increased with each beat. This was used to calculate interspike intervals that were averaged over at least 20 s to obtain the average frequency.
Supplementary Material
Acknowledgments
We thank Luc Leybaert and Elke De Vuyst (Gent, Belgium) for testing the function of the mimetic peptide Gap26, used in our studies, in their cell systems. Furthermore, we want to thank Frank Holst and Christine Siegmund for their excellent technical assistance.
FUNDING This work was supported by grants from the German Research Association (SFB 645, project B2 to K.W. and SFB 656, project C3 to C.T., A.G. and K.T.); Bonn University Grant BONFOR O-109.0008 to T.L.; NIH Grant AR41255 to M.W. and R.C. and NIH Grant RO1 NS036706 to F.B.
Footnotes
Conflict of Interest statement. None declared.
Supplementary Material is available at HMG Online.
References
- 1.Judisch GF, Martin-Casals A, Hanson JW, Olin WH. Oculodentodigital dysplasia: four new reports and a literature review. Arch Ophthal. 1979;97:878–884. doi: 10.1001/archopht.1979.01020010436007. [DOI] [PubMed] [Google Scholar]
- 2.Gillepsie FD. A hereditary syndrome: ‘dysplasia oculodentodigitalis’. Arch Ophthalmol. 1964;71:187–192. doi: 10.1001/archopht.1964.00970010203009. [DOI] [PubMed] [Google Scholar]
- 3.Weintraub DM, Baum JL, Pashayan HM. A family with oculodentodigital dysplasia. Cleft Palate J. 1975;12:323–329. [PubMed] [Google Scholar]
- 4.Fara M, Horak I, Hrivnakova J, Kapras J, Nova M, Stloukalova M. Oculodentodigital dysplasia. Acta Chir Plast. 1977;19:110–122. [PubMed] [Google Scholar]
- 5.Nivelon-Chevallier A, Audry D, Audry F, Dumas R. Dysplasie oculodento-digital: a propos d’un cas avec paraplégie spasmodique. J Genet Hum. 1981;29:171–179. [PubMed] [Google Scholar]
- 6.van Steensel MAM, Spruijt L, van der Burgt I, Bladergroen RS, Vermeer M, Steijlen PM, vanGeel M. A 2-bp deletion in the GJA1 gene is associated with oculo-dento-digital dysplasia with palmoplantar keratoderma. Am J Med Genet A. 2005;132:171–174. doi: 10.1002/ajmg.a.30412. [DOI] [PubMed] [Google Scholar]
- 7.Loddenkemper T, Grote K, Evers S, Oelerich M, Stogbauer F. Neurological manifestations of the oculodentodigital dysplasia syndrome. J Neurol. 2002;249:584–595. doi: 10.1007/s004150200068. [DOI] [PubMed] [Google Scholar]
- 8.Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, Innis JW, Dinulos MB, Christian C, Hannibal MC, et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72:408–418. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Söhl G, Willecke K. An update on connexin genes and their nomenclature in mouse and man. Cell Commun Adhes. 2003;10:173–180. doi: 10.1080/cac.10.4-6.173.180. [DOI] [PubMed] [Google Scholar]
- 10.Bennett MVL, Verselis VK. Biophysics of gap junctions. Semin Cell Biol. 1992;3:29–47. doi: 10.1016/s1043-4682(10)80006-6. [DOI] [PubMed] [Google Scholar]
- 11.Bruzzone S, Guida L, Zocchi E, Franco L, De Flora A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 2001;15:10–12. doi: 10.1096/fj.00-0566fje. [DOI] [PubMed] [Google Scholar]
- 12.Evans WH, De Vuyst E, Leybaert L. The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem J. 2006;397:1–14. doi: 10.1042/BJ20060175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stout CE, Costantin JL, Naus CCG, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;12:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- 14.Spray DC, Ye ZC, Ransom BR. Functional connexin “hemichannels”: a critical appraisal. Glia. 2006;54:758–773. doi: 10.1002/glia.20429. [DOI] [PubMed] [Google Scholar]
- 15.Seki A, Coombs W, Taffet SM, Delmar M. Loss of electrical communication, but not plaque formation, after mutations in the cytoplasmic loop of connexin43. Heart Rhythm. 2004;1:227–233. doi: 10.1016/j.hrthm.2004.03.066. [DOI] [PubMed] [Google Scholar]
- 16.Roscoe W, Veitch GI, Gong XQ, Pellegrino E, Bai D, McLachlan E, Shao Q, Kidder GM, Laird DW. Oculodentodigital dysplasia-causing connexin43 mutants are non-functional and exhibit dominant effects on wild-type connexin43. J Biol Chem. 2005;280:11458–11466. doi: 10.1074/jbc.M409564200. [DOI] [PubMed] [Google Scholar]
- 17.Shibayama J, Paznekas W, Seki A, Taffet S, Jabs EW, Delmar M, Musa H. Functional characterization of connexin43 mutations found in patients with oculodentodigital dysplasia. Circ Res. 2005;96:83–91. doi: 10.1161/01.RES.0000168369.79972.d2. [DOI] [PubMed] [Google Scholar]
- 18.Lai A, Le D-N, Paznekas WA, Gifford WS, Wang Jabs E, Charles AC. Oculodentodigital dysplasia connexin43 mutations result in non-functional connexin hemichannels and gap junctions in C6 glioma cells. J Cell Sci. 2006;119:532–541. doi: 10.1242/jcs.02770. [DOI] [PubMed] [Google Scholar]
- 19.Dobrowolski R, Sommershof A, Willecke K. Some oculodentodigital dysplasia-associated Cx43 mutations cause increased hemichannel activity in addition to deficient gap junction channels. J Membr Biol. 2007 doi: 10.1007/s00232-007-9055-7. [DOI] [PubMed] [Google Scholar]
- 20.Gong XQ, Shao Q, Langlois S, Bai D, Laird DW. Differential potency of dominant negative connexin43 mutants in oculodentodigital dysplasia. J Biol Chem. 2007;26:19190–19202. doi: 10.1074/jbc.M609653200. [DOI] [PubMed] [Google Scholar]
- 21.Flenniken AM, Osborne LR, Anderson N, Ciliberti N, Fleming C, Gittens JE, Gong XQ, Kelsey LB, Lounsbury C, Moreno L, et al. A Gja1 missense mutation in a mouse model of oculodentodigital dysplasia. Development. 2005;132:4375–4386. doi: 10.1242/dev.02011. [DOI] [PubMed] [Google Scholar]
- 22.Lallemand Y, Luria V, Haffner-Krausz R, Lonai P. Maternally expressed PGK-Cre transgene as a tool for early and uniform activation of the Cre site-specific recombinase. Transgenic Res. 1998;7:105–112. doi: 10.1023/a:1008868325009. [DOI] [PubMed] [Google Scholar]
- 23.Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–1834. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- 24.Kreuzberg MM, Söhl G, Kim JS, Verselis VK, Willecke K, Bukauskas FF. Functional properties of mouse connexin30.2 expressed in the conduction system of the heart. Circ Res. 2005;11:1169–1177. doi: 10.1161/01.RES.0000169271.33675.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rackauskas M, Kreuzberg MM, Pranevicius M, Willecke K, Verselis VK, Bukauskas FF. Gating properties of heterotypic gap junction channels formed of connexins 40, 43 and 45. Biophys J. 2007;92:1952–1965. doi: 10.1529/biophysj.106.099358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bukauskas FF, Verselis VK. Gap junction channel gating. Biochim Biophys Acta. 2004;1662:42–60. doi: 10.1016/j.bbamem.2004.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bukauskas FF, Angele AB, Verselis VK, Bennett MV. Coupling asymmetry of heterotypic connexin 45/connexin 43-EGFP gap junctions: properties of fast and slow gating mechanisms. Proc Natl Acad Sci USA. 2002;99:7113–7118. doi: 10.1073/pnas.032062099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delmar M, Coombs W, Sorgen P, Duffy HS, Taffet SM. Structural bases for the chemical regulation of connexin43 channels. Cardiovasc Res. 2004;62:268–275. doi: 10.1016/j.cardiores.2003.12.030. [DOI] [PubMed] [Google Scholar]
- 29.Oh SY, Dupont E, Madhukar BV, Briand JP, Chang CC, Beyer E, Trosko JE. Characterization of gap junctional communication-deficient mutants of a rat liver epithelial cell line. Eur J Cell Biol. 1993;60:250–255. [PubMed] [Google Scholar]
- 30.Solan JL, Lampe PD. Key connexin 43 phosphorylation events regulate the gap junction life cycle. J Membr Biol. 2007 doi: 10.1007/s00232-007-9035-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duffy HS, Sorgen PL, Girvin ME, O’Donnell P, Coombs W, Taffet SM, Delmar M, Spray DC. pH-dependent intramolecular binding and structure involving Cx43 cytoplasmic domains. J Biol Chem. 2002;277:36706–36714. doi: 10.1074/jbc.M207016200. [DOI] [PubMed] [Google Scholar]
- 32.McLachlan E, Manias JL, Gong XQ, Lounsbury CS, Shao Q, Bernier SM, Bai D, Laird DW. Functional characterization of oculodentodigital dysplasia-associated Cx43 mutants. Cell Commun Adhes. 2005;5-6:279–292. doi: 10.1080/15419060500514143. [DOI] [PubMed] [Google Scholar]
- 33.Yamada KA, Rogers JG, Sundset R, Steinberg TH, Saffitz JE. Up-regulation of connexin45 in heart failure. J Cardiovasc Electrophysiol. 2003;11:1205–1212. doi: 10.1046/j.1540-8167.2003.03276.x. [DOI] [PubMed] [Google Scholar]
- 34.Eckardt D, Kirchhoff S, Kim JS, Degen J, Theis M, Ott T, Wiesmann F, Doevendans PA, Lamers WH, de Bakker JM, et al. Cardiomyocyte-restricted deletion of connexin43 during mouse development. J Mol Cell Cardiol. 2006;41:963–971. doi: 10.1016/j.yjmcc.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 35.van Rijen HV, Eckardt D, Degen J, Theis M, Ott T, Willecke K, Jongsma HJ, Opthof T, de Bakker JM. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation. 2004;109:1048–1055. doi: 10.1161/01.CIR.0000117402.70689.75. [DOI] [PubMed] [Google Scholar]
- 36.Bukauskas FF, Kreuzberg MM, Rackauskas M, Bukauskiene A, Bennett MVL, Verselis VK, Willecke K. Properties of mouse connexin 30.2 and human connexin 31.9 hemichannels: implications for atrioventricular conduction in the heart. Proc Natl Acad Sci USA. 2006;103:9726–9731. doi: 10.1073/pnas.0603372103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen YH, Xu SJ, Bendahhou S, Wang Y, Xu WY, Jin HW, Sun H, Zhuang QN, Yang YQ, Li YB, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–254. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- 38.Degen J, Meier C, Van Der Giessen RS, Söhl G, Petrasch-Parwez E, Urschel S, Dermietzel R, Schilling K, De Zeeuw CI, Willecke K. Expression pattern of lacZ reporter gene representing connexin36 in transgenic mice. J Comp Neurol. 2004;473:511–525. doi: 10.1002/cne.20085. [DOI] [PubMed] [Google Scholar]
- 39.Buchholz F, Angrand PO, Stewart AF. A simple assay to determine the functionality of Cre or FLP recombination targets in genomic manipulation constructs. Nucleic Acids Res. 1996;24:3118–3119. doi: 10.1093/nar/24.15.3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magin TM, McWhir J, Melton DW. A new mouse embryonic stem cell line with good germ line contribution and gene targeting frequency. Nucleic Acids Res. 1992;20:3795–3796. doi: 10.1093/nar/20.14.3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Theis M, Magin TM, Plum A, Willecke K. General or cell type-specific deletion and replacement of connexin-coding DNA in the mouse. Methods. 2000;20:205–218. doi: 10.1006/meth.1999.0938. [DOI] [PubMed] [Google Scholar]
- 42.Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–179. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- 44.Hennemann H, Schwarz HJ, Willecke K. Characterization of gap junction genes expressed in F9 embryonic carcinoma cells: molecular cloning of mouse connexin31 and -45 cDNAs. Eur J Cell Biol. 1992;57:51–58. [PubMed] [Google Scholar]
- 45.Wilgenbus KK, Kirkpatrick CJ, Knuechel R, Willecke K, Traub O. Expression of Cx26, Cx32 and Cx43 gap junction proteins in normal and neoplastic human tissues. Int J Cancer. 1992;51:522–529. doi: 10.1002/ijc.2910510404. [DOI] [PubMed] [Google Scholar]
- 46.Castro CH, Shin CS, Stains JP, Cheng SL, Sheikh S, Mbalaviele G, Szejnfeld VL, Civitelli R. Targeted expression of a dominant-negative N-cadherin in vivo delays peak bone mass and increases adipogenesis. J Cell Sci. 2004;117:2853–2864. doi: 10.1242/jcs.01133. [DOI] [PubMed] [Google Scholar]
- 47.Traub O, Eckert R, Lichtenberg-Frate H, Elfgang C, Bastide B, Scheidtmann KH, Hülser DF, Willecke K. Immunochemical and electrophysiological characterization of murine connexin40 and -43 in mouse tissues and transfected human cells. Eur J Cell Biol. 1994;64:101–112. [PubMed] [Google Scholar]
- 48.Nolden L, Edenhofer F, Haupt S, Koch P, Wunderlich FT, Siemen H, Brüstle O. Site-specific recombination in human embryonic stem cells induced by cell-permeant Cre recombinase. Nat Methods. 2006;3:461–467. doi: 10.1038/nmeth884. [DOI] [PubMed] [Google Scholar]
- 49.Herr C, Smyth N, Ullrich S, Yun F, Sasse P, Hescheler J, Fleischmann B, Lasek K, Brixius K, Schwinger RH, et al. Loss of annexin A7 leads to alterations in frequency-induced shortening of isolated murine cardiomyocytes. Mol Cell Biol. 2001;21:4119–4128. doi: 10.1128/MCB.21.13.4119-4128.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lammers WJ, Schalij MJ, Kirchhof CJ, Allessie MA. Quantification of spatial inhomogeneity in conduction and initiation of reentrant atrial arrhythmias. Am J Physiol. 1990;259:1254–1263. doi: 10.1152/ajpheart.1990.259.4.H1254. [DOI] [PubMed] [Google Scholar]
- 51.Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation. 1999;100:87–95. doi: 10.1161/01.cir.100.1.87. [DOI] [PubMed] [Google Scholar]
- 52.Hesse M, Kondo CS, Clark RB, Su L, Allen FL, Geary-Joo CT, Kunnathu S, Severson DL, Nygren A, Giles WR, Cross JC. Dilated cardiomyopathy is associated with reduced expression of the cardiac sodium channel Scn5a. Cardiovasc Res. 2007;75:498–509. doi: 10.1016/j.cardiores.2007.04.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.