

Abstract
All vertebrates contain two nonmuscle myosin II heavy chains, A and B, which differ in tissue expression and subcellular distributions. To understand how these distinct distributions are controlled and what role they play in cell migration, myosin IIA and IIB were examined during wound healing by bovine aortic endothelial cells. Immunofluorescence showed that myosin IIA skewed toward the front of migrating cells, coincident with actin assembly at the leading edge, whereas myosin IIB accumulated in the rear 15–30 min later. Inhibition of myosin light-chain kinase, protein kinases A, C, and G, tyrosine kinase, MAP kinase, and PIP3 kinase did not affect this asymmetric redistribution of myosin isoforms. However, posterior accumulation of myosin IIB, but not anterior distribution of myosin IIA, was inhibited by dominant-negative rhoA and by the rho-kinase inhibitor, Y-27632, which also inhibited myosin light-chain phosphorylation. This inhibition was overcome by transfecting cells with constitutively active myosin light-chain kinase. These observations indicate that asymmetry of myosin IIB, but not IIA, is regulated by light-chain phosphorylation mediated by rho-dependent kinase. Blocking this pathway inhibited tail constriction and retraction, but did not affect protrusion, suggesting that myosin IIB functions in pulling the rear of the cell forward.
INTRODUCTION
Nonmuscle myosin II is a ubiquitous motor protein that can form bipolar filaments, enabling it to contract actin-filament networks. Such contractility drives cortical constriction that divides cells during cytokinesis (Satterwhite and Pollard, 1992), creates pulling forces exerted by fibroblasts rearranging extracellular matrix fibers (Ehrlich et al., 1991; Kolodney and Wysolmerski, 1992), causes cytoplasm retraction when endothelial barriers open during inflammation (Dudek and Garcia, 2001), and plays a major role in generating traction forces during cell migration (Elson et al., 1999). In Dictyostelium amoebae, myosin II is also essential in orienting cell migration, as null mutants extend protrusions in multiple directions and fail to migrate with persistent orientation (Wessels and Soll, 1990).
In all vertebrates examined to date, including human (Simons et al., 1991), cow (Murakami and Elzinga, 1992), rat (Choi et al., 1996), rabbit (Murakami et al., 1998), mouse (Tullio et al., 1997), frog (Kelley et al., 1996), and chicken (Kawamoto and Adelstein, 1991), two different genes encode heavy chains of nonmuscle myosin II. Northern blotting and histochemistry with isoform-specific antibodies have shown that the two different heavy chains, A and B, are expressed in different proportions in different tissues and at different times during development (Kawamoto and Adelstein, 1991; Murakami and Elzinga, 1992; Murakami et al., 1993; Maupin et al., 1994; Phillips et al., 1995; Choi et al., 1996; Bhatia-Dey et al., 1998). When A and B heavy chains are expressed in the same cell, they have different subcellular distributions (Maupin et al., 1994; Rochlin et al., 1995; Kelley et al., 1996, Saitoh et al., 2001), suggesting that they perform different functions.
Myosin containing A-type heavy chains (myosin IIA) and myosin containing B-type heavy chain (myosin IIB) have slightly different enzymatic and assembly properties in vitro. Both isoforms possess actin-activated ATPase that is stimulated when myosin's regulatory light chains are phosphorylated by myosin light-chain kinase (MLCK). Light chain phosphorylation also stimulates filament assembly for both isoforms. However, fully activated myosin IIA and IIB move actin filaments at different rates in a molecular motility assay (Kelley et al., 1996), and filament assembly by myosin IIB, but not myosin IIA, is inhibited when heavy chains are phosphorylated by protein kinase C or casein kinase II (Murakami et al., 2000). Conversely, filament assembly by myosin IIA, but not myosin IIB, is disrupted by metastasis-associated protein, mts1 (Murakami et al., 2000).
The significance of these differences in vivo is not known. Despite extensive evidence for myosin II playing an essential part in moving amoebae (Fukui and Yumura, 1986; Wessels and Soll, 1990; Wessels et al., 1996; Elson et al., 1999; Zhang et al., 2002), the function of myosin II during migration of vertebrate nonmuscle cells is not well understood. Myosin IIB knockout mice have recently been generated, and these animals have severe cardiovascular and brain abnormalities, indicating that myosin IIB has an important and specific role in determining tissue organization (Tullio et al., 1996, 1997, 2001). In addition, neurons cultured from myosin IIB–deficient mice extend more slowly than cells from normal mice (Bridgman et al., 2001), suggesting a role in cell migration. The contributions of myosin IIA and IIB have not been examined in endothelial cells, which form the initial foundation for vascular structures that are defective in myosin IIB–deficient mice. Nor is it known how the behaviors of myosin IIA and IIB are regulated so that they distribute differently in vivo. Experiments described below address how myosin IIA and IIB act during endothelial migration and how their distinct distributions arise. Changes in the distribution of the two isoforms were observed as cells began to migrate at the edge of a wound and indicated that myosin IIA and IIB are involved in different aspects of migration. Myosin IIA was closely associated with protrusive activity, whereas myosin IIB appeared to be responsible for retracting the tails of migrating cells. Moreover, inhibition of protein kinases and the small G-protein, rhoA, revealed that the distribution of myosin IIB can be regulated in living cells independently from myosin IIA via rho-kinase–dependent phosphorylation of its regulatory light chains.
MATERIALS AND METHODS
Cell Culture and Wounding
All experiments were performed on primary cultures of bovine aortic endothelial cells (BAECs) between passage 13 and 18 after isolation. Cells were maintained in DMEM supplemented with 10% fetal calf serum as described previously (Kolega, 1999). Wounds were made within 24–48 h after cells reached confluence by dragging a plastic comb across the surface of the culture, creating a series of uniform, parallel wounds ∼0.5 μm wide and 0.5 μm apart.
Plasmids and Transfections
An expression vector for the dominant-negative form of rhoA, T19Nrho, was provided by Dr. Klaus Hahn (Scripps Research Institute, La Jolla, CA; Subauste et al., 2000). This vector is a pcDNA3 plasmid (Invitrogen, Carlsbad, CA) with T19Nrho cDNA inserted in tandem with a GFP S65T cDNA, which encodes enhanced green fluorescent protein (EGFP) as an indicator of expression levels. pCMV plasmids containing constitutively active and kinase-dead mutants of MLCK, each tagged with a FLAG epitope, were provided by Dr. Patricia Gallagher (Indiana University Purdue University at Indianapolis, IN). The kinase-dead MLCK consists of full-length rabbit smooth muscle MLCK (130 kDa) that has a deletion in the ATP-binding site, rendering it catalytically inactive while maintaining calmodulin and myosin light-chain binding activity (Gallagher et al., 1993). It has been shown to act as a dominant negative inhibitor of endogenous MLCK activity in fibroblasts (Klemke et al., 1997). In the constitutively active mutant, the N-terminal autoinhibitory domain has been deleted.
Adherent BAECs were transiently transfected with nonlinearized plasmids using Mirus TransIT-LT1 polyamine transfection reagent (PanVera Corp., Madison, WI). To study plasmid-expressing cells at wound edges, cultures were transfected when they were ∼70% confluent and then incubated for an additional 24–48 h to allow them to reach confluence before wounding.
Antibodies
Isoform-specific polyclonal rabbit antibodies against the A and B isoforms of myosin II heavy chain were supplied by Dr. Robert Adelstein (National Institutes of Health) or purchased from Covance (Richmond, CA). These antibodies do not cross-react with other myosin isoforms (Kelley et al., 1996) and recognize only myosin II on immunoblots of endothelial cell proteins (Kolega, 1998b). For staining both myosin heavy chain isoforms, polyclonal antibody against smooth- and nonmuscle myosin from Biomedical Technologies Inc. (Bedford, MA) was used. Antibodies against phosphorylated myosin regulatory light chains were obtained from Cell Signaling Technology (Beverly, MA). One antibody recognizes light chains that are phosphorylated at serine 19, and another recognizes light chains that are phosphorylated at both serine 19 and threonine 18. FLAG-tagged proteins were stained using M2 anti-FLAG mAb (Sigma, St. Louis, MO). All secondary antibodies and Fab fragments were from Jackson ImmunoResearch (West Grove, PA), except Alexa488-conjugated antibodies, which were from Molecular Probes (Eugene, OR).
Fluorescent Staining
Cells were fixed for 30 min in 3.7% freshly prepared formaldehyde in cytoskeletal buffer (CB; 137 mM NaCl, 5 mM KCl, 1.1 mM, Na2HPO4, 0.4 mM KH2PO4, NaHCO3, MgCl2, 2 mM EGTA, 50 mM d-glucose, 5 mM PIPES, pH 6.0) and permeabilized for 5 min with 0.1% Triton X-100 in CB. Protein was stained by incubating cells for 30 min in CB containing a 1:100 dilution of CyDye, a lysine-reactive succinimidyl ester of cy5 (Amersham Life Science, Pittsburgh, PA). Cells were washed three times with CB, and any remaining unreacted dye was blocked by treating the coverslips for 1 h with 10% goat serum in phosphate-buffered saline. Coverslips were then stained for F-actin, using rhodamine-conjugated phalloidin (Molecular Probes) and for myosin IIA or IIB by indirect immunofluorescence using Alexa488-conjugated secondary antibodies.
To stain myosin IIA and IIB in the same cell, specimens were prepared as described above except that phalloidin was omitted and a fluorescent Fab fragment was used for the secondary antibody. Specimens were then blocked for 1 h with 1 mg/ml unconjugated Fab fragments and subsequently stained with the second antimyosin II primary and another secondary Fab antibody fragment conjugated with a different fluorophore.
In some experiments, fluorescent dextran was used as an inert indicator of cytoplasmic volume. Dextran was introduced into cells by “scrape-loading”; i.e., including the dextran in the culture medium during wounding: scraping the monolayer produced sufficient transient damage to surviving cells at the wound edge that nearly all of them took up sufficient dextran to permit fluorescence imaging. Lysine-derivatized dextran (Molecular Probes), which is fixed by formaldehyde, was used in order to preserve dextran distribution when permeabilizing cells for cytoskeletal staining.
Image Acquisition and Analysis
Fluorescence staining was observed through a Zeiss Axiovert 135 microscope using a 100× Plan-NEOFLUAR oil-immersion objective (Carl Zeiss Microimaging, Thornwood, NY) and imaged with a Hamamatsu Orca-ER CCD camera (Micro Video Instruments, Avon, MA). Specimen illumination and camera gain were controlled so that the maximum pixel intensities in the images were within the linear range of the camera. For illustrations used in this article, image contrast was linearly stretched to enhance the visibility of certain features, such as thin edges of spreading cells. However, all calculations were performed on unadjusted images.
Cytoskeletal asymmetry was assessed using a vector measurement based on the method described by Coates et al. (1992). Briefly, specimens stained with multiple fluorophores were imaged, and background signal (as determined from an image of an empty field) was subtracted from each image. A single cell in the field was outlined manually in IPLab software (Scanalytics, Fairfax, VA) using the fluorescence image of total protein (i.e., CyDye staining), and then the center of mass of protein for that cell was calculated from the CyDye fluorescence intensity at each pixel within the outlined region, assuming protein mass to be proportional to fluorescence intensity. The center of mass of other components was determined within the same outline superimposed onto the respective fluorescence image. The distance and direction between the center of a particular component and the center of total protein gave a vector that measures asymmetry: the direction of the vector indicates the direction in which the distribution of the component is skewed, and the magnitude of the vector reflects how large the asymmetry is. Images of beads that fluoresce in all three fluorescent channels (Molecular Probes) were used to check the registration of images taken through different filter sets, and the correction was ≤1 pixel in both the x and y directions in our system.
Western Blotting
For determination of myosin light-chain phosphorylation, cultures were rinsed with serum-free medium, drained, and scraped into a minimal volume of 2% SDS, 1 mM DTT, 50 mM Tris, pH 6.8. This extract was heated to 100°C for 2 min, clarified by centrifugation for 5 min at 14000 × g, and loaded onto 20% polyacrylamide gels. Separated proteins were electrophoretically transferred to nitrocellulose paper and stained with antibody against phosphorylated myosin light chains and peroxidase-conjugated secondary antibody. Antibody binding was visualized by chemiluminescence using a stabilized luminol substrate (SuperSignal Westdura, Pierce Biotechnology, Rockford, IL) and Kodak XAR film (Rochester, NY).
RESULTS
Myosin IIA and Myosin IIB Distribute toward Opposite Ends of Migrating Endothelial Cells
In confluent monolayers of BAECs, the A and B isoforms of myosin II heavy chains were symmetrically distributed around the center of the cell and overlapped extensively. Some diffuse staining was observed throughout the cytoplasm for both isoforms, but most of the myosin IIA and myosin IIB was located in rows of dots that were coincident with actin stress fibers. These formed rings or polygons around the body of the cell a few micrometers proximal to the contacts between cells (Figure 1, C and E). When cells were induced to migrate by wounding the monolayer, myosin IIA and myosin IIB moved to different parts of the cells. Both myosins were still found predominantly in linear arrays of dots, but myosin IIA, which constitutes 70–90% of the myosin II in BAECs (Kolega, 1998a), distributed throughout the broad lamellar protrusions that extended toward the wound, except at the extreme distal edge (Figure 1D, arrowheads). Staining for myosin IIA in the front of the cell was equal to that in the main body of the cell, even though the front of the cell was much flatter and contained much less protein. Thus, on a concentration basis, myosin IIA was strongly enriched toward the front of the cell. In contrast, myosin IIB, which makes up 10–30% of the cells' myosin II, was largely excluded from the front of the cell and instead accumulated in large, dense bundles along the cells' trailing edges (Figure 1F, arrows). In migrating cells, the distributions of myosin IIA and IIB did continue to overlap somewhat, especially in the largest myosin bundles, but as cells became more polarized the two isoforms became more distinctly skewed in opposite directions.
Figure 1.

Asymmetric distribution of myosin IIA and myosin IIB in migrating endothelial cells. BAECs were fixed and stained 1 d after reaching confluence (A, C, and E) or 1 h after wounding the confluent monolayer (B, D, and F). The wound is at the right of the figure. Cells were stained with lysine-reactive cy5 to directly visualize total protein distribution (A and B) and with antibodies against myosin IIA (C and D) and myosin IIB (E and F), which were imaged by indirect immunofluorescence. Arrowheads in B, D, and F mark the location of the leading edges of one cell's lamellipodia. Note that the lamellar protrusions are very flat and contain much less protein mass compared with the cytoplasm around the nucleus, so the strong myosin IIA staining in the protrusions represents local concentration of myosin IIA. Myosin IIB is much less abundant toward the front of migrating cells, but is concentrated in cytoplasm at their trailing edges (F, arrows).
To determine if these opposite, asymmetric distributions might play a role in determining the polarity of protrusive activity, myosin asymmetries were compared with asymmetry in the assembly of actin filaments, which is responsible for formation of protrusions at a cell's leading edge. Asymmetry was measured by determining the extent to which myosin II or F-actin mass was shifted away from the center of mass of the cell (Figure 2). In migrating cells, F-actin and myosin IIA were both strongly skewed toward the front of the cell, whereas myosin IIB skewed away from the front (Figure 2C). In contrast, a 40-kDa fluorescent dextran, which does not assemble or bind to specific structures in the cell and which was introduced into cells by scrape loading, did not distribute asymmetrically. This demonstrated that the asymmetries measured for actin and myosin were not caused simply by changes in the shape of the cytoplasm. Actin and myosin must undergo specific changes that cause them to concentrate in particular regions relative to the direction of cell movement.
Figure 2.

Quantitation of the asymmetry of F-actin and myosin II in confluent and migrating cells. (A) Calculation of asymmetries: The cell was fixed, double-stained with lysine-reactive cy5 (to visualize protein distribution) and with rhodamine phalloidin (to show F-actin), and digital fluorescence images were acquired of both stains. The center of mass of protein in the cell (○) was taken as the center of mass of the cy5, calculated as described in MATERIALS AND METHODS. Similarly, the center of mass of F-actin (×) was calculated from the rhodamine phalloidin image. The displacement between the two centers gave the asymmetry vector for F-actin distribution in the cell. (B) F-actin asymmetries were measured as described in A for 25 cells in confluent monolayers (left) and for 25 cells migrating at the edge of a wound 1 h after wounding (right). Each arrow represents the asymmetry vector, in μm, for a single cell, and all vectors are plotted with the cell's protein center at the origin (0,0). Note the random orientation of asymmetries in confluent cells, and the larger, oriented asymmetries in migrating cells. (C) Fluorescence images were acquired of F-actin, myosin IIA, myosin IIB, and 40-kDa fluorescein dextran in confluent monolayers or at the edges of 1-h wounds. Asymmetry vectors were calculated for 30–40 cells in each case, and the mean projection onto the Y axis (i.e., in the direction of the wound) used to quantify polarization. Error bars indicate one SD from the mean; asterisks indicate values that are significantly different from 0 (p > 0.95). Only migrating cells displayed asymmetry in the distribution of actin and myosin II. Dextran, which acts as an inert volume indicator, did not polarize at all.
Polarization of Protrusive Activity Precedes Polarization of Myosin IIB, But Not Myosin IIA
Endothelial monolayers were fixed and stained at various times after wounding to examine the development of cytoskeletal polarity as endothelial cells begin to migrate. As shown in Figure 3, both F-actin and myosin IIA were asymmetrically distributed within 5 min of wounding. At this time, most cells at the wound edge had just begun to extend lamellipodia from their newly free borders. F-actin and myosin IIA asymmetry continued to increase for 30–60 min as lamellipodia enlarged and the cells elongated toward the wound. They reached maximal values 1–2 h after wounding, when virtually all cells at the wound edge were fully extended and migrating on the wound surface.
Figure 3.

Development of F-actin and myosin II asymmetry. Mean asymmetries of F-actin, myosin IIA, and myosin IIB were measured for cells along wound edges that were fixed at various times after wounding. To compare components more easily, values were normalized to the maximum mean asymmetry attained during the 2-h time course, which was observed at 120 min for F-actin and myosin IIA and at 60 min for myosin IIB. Each point represents the mean of 30–40 cells; error bars, 1 SD.
In contrast, there was no asymmetry in the distribution of myosin IIB during the first 15 min after wounding. Accumulation of myosin IIB toward the rear of the cell was not apparent until 15–30 min postwounding (Figure 3, dashed line), after which it rapidly skewed away from the wound edge, reaching maximal values at about the same time that asymmetries in F-actin and myosin IIA plateaued. Thus, myosin IIB was asymmetrically distributed in fully migrating cells, but this asymmetry was not required for the initial polarization of the cells' protrusions.
Inhibition of rho-dependent Protein Kinase Selectively Affects the Posterior Accumulation of Myosin IIB
We have previously shown that inhibiting phosphorylation of myosin II's regulatory light chains decreases stress fibers and increases the solubility and mobility of myosin II in fibroblasts (Kolega et al., 1993; Kolega and Taylor, 1993) and endothelial cells (Kolega, 1997; Kolega and Kumar, 1999). Therefore, kinase inhibitors known to affect light-chain phosphorylation were used to disrupt myosin II organization in order to determine the role of myosin II asymmetry in endothelial migration. BAECs were treated with ML-7, an inhibitor of myosin light-chain kinase, and Y-27632, which inhibits rho-dependent kinases, particularly p160ROCK and ROCK-II (Uehata et al., 1997; Davies et al., 2000) and which has been shown to block phosphorylation of myosin light chains in endothelial cells.
ML-7 had surprisingly little effect. Even in 10 μM ML-7, cells migrated normally and the distribution of both myosin IIA and IIB appeared unaffected (Figure 4, A–C). Both myosin IIA and IIB were found in linear, punctate patterns and in numerous stress fibers. Most significantly, myosin IIA still distributed preferentially toward the front of migrating cells, whereas myosin IIB still accumulated in the tail.
Figure 4.

Myosin II distribution in cells treated with ML-7 and Y-27632. BAECs were incubated for 15 min in 10 μM ML-7 to inhibit myosin light-chain kinase (A–C) or 2 μM Y-27632 to inhibit rho-dependent kinase (D and E). Cultures were wounded and cells were allowed to migrate in the continued presence of drug for 60 min before fixation and staining with lysine-reactive cy5 (A and D) and antibodies against myosin IIA (B and E) or myosin IIB (C and F). Arrowheads mark the leading edges of lamellipodia. Observe myosin IIB excluded from lamellar protrusions in ML-7–treated cells (C), but extending to the very edge of cells treated with Y-27632 (F). Myosin IIB was concentrated along the trailing edge in ML-7 (C, arrows), but not in Y-27632.
In contrast, Y-27632 significantly changed the organization of myosin II. Cells treated with 2 μM Y-27632, still extended large lamellar protrusions at the edge of a wound (Figure 4D), but stress fibers were less prominent, and large, dense bundles of myosin II were rare (Figure 4, E and F). Both myosin IIA and myosin IIB continued to localize in small, discrete aggregates, which tended to line up in rows throughout the cytoplasm (Figure 4, E and F). These rows colocalized with F-actin bundles, suggesting that myosin IIA and IIB were still bound to an actin cytoskeleton, albeit one with smaller actin-myosin II structures. Despite this more dispersed arrangement of myosin IIA, both F-actin and myosin IIA were still asymmetrically distributed relative to cell migration. In fact, the centers of mass of F-actin and of myosin IIA were more strongly skewed toward the front of cells migrating in 2 μM Y-27632 than in untreated controls (Figure 5). Y-27632 consistently increased the asymmetry of myosin IIA in five separate experiments, although this change did not prove statistically significant at 90% confidence levels. More strikingly, myosin IIB no longer accumulated in the rear of these cells. Instead, it was found throughout the cell body and also in the lamellar protrusions at the front (Figure 4F), from which it was largely excluded in control cells (compare Figure 1F). In Y-27632–treated cells, the overall distribution of myosin IIB actually became polarized toward, rather than away from, the wound (Figure 5). Thus, the signal that normally causes myosin IIB to localize in the tail of migrating cells was inhibited by Y-27632.
Figure 5.

Effects of kinase inhibitors on F-actin and myosin II asymmetry. BAECs were fixed and stained 60 min after wounding in the presence of: 0.5% dimethyl sulfoxide (control), 10 μM ML-7, 2 μM Y-27632, 1 μM K252b, 2 μM bisindolylmaleimide I, 1 μM KT5720, 10 μM PD98509, 10 μM genistein, 50 nM wortmannin, or 10 μM LY294002. Asymmetries were calculated for F-actin, myosin IIA, and myosin IIB from the images of 30–40 cells in each case; error bars, 1 SD. #Values that were moderately significantly different from controls (p > 0.9, but <0.95); *highly significant differences (p > 0.99).
A variety of other kinase inhibitors were also tested for their effects on myosin II asymmetry, but only Y-27632 had significant effects on the overall distribution of myosin IIB (Figure 5). The myosin light-chain kinase inhibitor, ML-7, did cause a modest reduction in F-actin asymmetry, but myosin IIA and myosin IIB still polarized normally as noted above. K252b, a cell-permeable inhibitor of myosin light-chain kinase as well as protein kinases A, C, and G (Kase et al., 1987), had little effect on protrusive polarity (as assessed by F-actin asymmetry), nor on the asymmetry of myosin IIA and myosin IIB. No effect was observed at a concentration of 1 μM, which is a 10-fold excess over the concentration of K252b required to inhibit myosin light-chain kinase and protein kinases A, C, and G in vitro (Nakanishi et al., 1990). Bis-indolylmaleimide I, a very selective inhibitor of protein kinase C (Toullec et al., 1991; Davies et al., 2000), and KT5720, which preferentially inhibits protein kinase A (Kase et al., 1987; Davies et al., 2000), also failed to alter asymmetry of F-actin, myosin IIA or myosin IIB, at concentrations well above their in vitro Kis. Likewise, inhibitors of MAP kinase (PD98509), tyrosine kinases (genistein), and PIP3 kinase (wortmannin and LY294002; Davies et al., 2000), all failed to disrupt the development of F-actin and myosin II asymmetry in migrating BAECs. Thus, only Y-27632 changed the direction of asymmetry so that myosin IIB became skewed toward—rather than away from—the wound.
If the posterior accumulation of myosin IIB is dependent on rho-dependent kinase, it should also be dependent on rho activity. To test this, signaling by rho was blocked using a dominant-negative mutant of rhoA. Endothelial cells were transiently transfected with a plasmid carrying genes for T19Nrho, which behaves as a dominant negative mutation of rhoA (Pan et al., 1998), and for enhanced green fluorescence protein (EGFP), which serves as an indicator of plasmid expression. Fluorescence microscopy of cells expressing T19Nrho showed that myosin IIB did not accumulate along the trailing edge and was no longer excluded from anterior protrusions, even while immediately adjacent cells displayed normal polarization (Figure 6, A–C). Measurements of asymmetry confirmed that myosin IIB distributed toward the front of cells expressing T19Nrho (Figure 6D) and that this effect was apparent even at the minimum detectable levels of plasmid expression (Figure 6E). This occurred despite the fact that cells expressing dominant negative rhoA did not elongate as much as untransfected cells, possibly because rhoA is involved in regulating turnover of cellsubstratum adhesions (Ridley, 2001). Myosin IIA asymmetry was reduced by dominant-negative rhoA (Figure 6, D and E), but the direction of asymmetry was unchanged, with myosin IIA always remaining skewed toward the wound. Thus, rhoA's influence on the direction of asymmetry was specific to myosin IIB.
Figure 6.

Reversal of myosin IIB asymmetry by dominant-negative rhoA. BAEC cultures that had been transiently transfected with EGFP-T19Nrho plasmids were fixed 1 h after wounding and then stained with lysine-reactive cy5 (A) and antibody against myosin IIB (C). EGFP fluorescence of the same field reveals plasmid expression (B). Arrowheads mark leading edges of migrating cells. The cell on the right was expressing high levels of plasmid encoding dominant-negative rhoA and displayed myosin IIB extending far into its lamellar protrusions, whereas its untransfected neighbor to the left excluded myosin IIB from the front and accumulated in the rear. Myosin IIA and myosin IIB asymmetries were calculated for 35 transfected and 35 untransfected cells, and mean values ± 1 SD are shown in D. (E) The asymmetry of myosin IIA (left) and myosin IIB (right) in individual cells plotted as a function of the expression levels of dominant-negative rhoA, as measured by GFP fluorescence. Dotted lines indicates the mean myosin II asymmetry in 20 untransfected cells in the same fields. Dominant-negative rhoA decreased myosin IIA asymmetry, but in no instance was it observed to cause negative asymmetry (i.e., posterior accumulation) of myosin IIA. For myosin IIB, even the lowest levels of plasmid expression (minimum detected = three times background fluorescence), inhibited the development of negative myosin IIB asymmetry, and myosin IIB became positively polarized (i.e., skewed toward the wound).
Posterior Accumulation of Myosin IIB Is Induced by Myosin Light-chain Phosphorylation
Rho-dependent kinase can regulate the activity of myosin II in vitro by phosphorylating the myosin II regulatory light chains at the same sight that is recognized by myosin light-chain kinase (Amano et al., 1996). To determine if this is how myosin IIB behavior is regulated in migrating BAECs, the effect of Y-27632 on light-chain phosphorylation was examined. Western blots of endothelial cell extracts were probed with antibodies that recognize myosin II regulatory light chains that have been either singly phosphorylated at serine 19 or doubly phosphorylated at serine 19 and threonine 18. These sites can be phosphorylated by rho-dependent kinase and myosin light-chain kinase in vitro. In cells treated with 2 μM Y-27632, levels of both mono- and di-phosphorylated light chains were markedly reduced (Figure 7). Quantitative measurements of Western blot chemiluminescence indicated that Y-27632 reduced monophosphorylated light chains to 35 ± 8% (n = 5) of control levels and diphosphorylated light chains to 34 ± 5% (n = 5) of controls. In contrast, the myosin light-chain kinase inhibitor, ML-7, which did not affect myosin II asymmetry, had little or no effect on phosphorylation at these sites (light-chain phosphorylation was 97 ± 15% and 110 ± 28% of controls for mono- and di-phosphorylated light chains, respectively).
Figure 7.
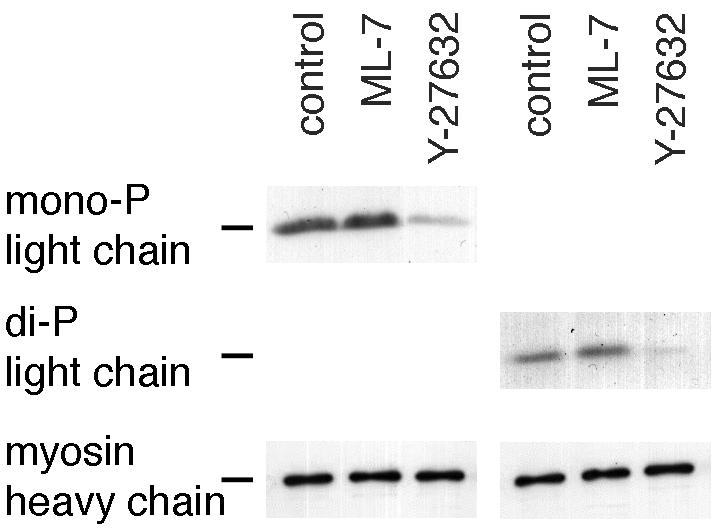
Inhibition of myosin light-chain phosphorylation by Y-27632. Confluent cultures of BAECs were treated for 15 min with 0.5% dimethyl sulfoxide (control), 10 μM ML-7, or 2 μM Y-27632 and then wounded in continued presence of drugs by raking with a flea comb to create a high density of wound edges. After 1 h, cell lysates were separated by SDS-PAGE and transferred to nitrocellulose, and the bottom half was stained either with an antibody that specifically recognizes 20-kDa regulatory light chains of myosin II that have been phosphorylated at serine 19 (monophosphorylated, left) or an antibody against light chains phosphorylated at both serine 19 and threonine 18 (diphosphorylated, right). The top half of the blot was stained for myosin heavy chains (using a broadspecificity antibody against smooth- and nonmuscle heavy chains) to verify equal loading between lanes and is shown at the bottom of the panel. Light chain phosphorylation was markedly reduced in cells treated with Y-27632, but not ML-7.
The distribution of phosphorylated light chains in migrating cells was determined by immunofluorescence with the phosphorylation-specific antibodies. Like myosin IIB, both mono- and di-phosphorylated light chains were particularly concentrated along the posterior and lateral edges of migrating cells (Figure 8). This suggests that light chains were preferentially phosphorylated on myosin IIB over those on myosin IIA. However, some patches of light-chain phosphorylation were also detected toward the front of migrating cells, where there was very little myosin IIB, indicating that phosphorylated light chains were not exclusively associated with myosin IIB. It should be noted that when phosphorylated light chains were seen toward the front of a cell, they were found in regions where lamellipodia were absent or retracting and the cell margin had a concave contour (arrows in Figure 8, C and F). Thus, there was a strong correlation between light-chain phosphorylation and the absence of protrusion.
Figure 8.

Distribution of phosphorylated regulatory light chains in migrating cells. BAECs were fixed and stained 1 h after wounding a confluent monolayer, with the wound at the top of the figure. Cells were triple-stained with antibodies against myosin II heavy chain A (A and D), myosin II heavy chain B (B and E), and myosin II regulatory light chains phosphorylated at either serine 19 (C) or serine 19 and threonine 18 (F). Both the mono- and di-phosphorylated light chains accumulated predominantly in bundles forming a U-shaped band along the posterio-lateral margins of migrating cells. This corresponds to accumulations of the B heavy chain. Smaller patches of phosphorylated light chains can be seen in nonspreading regions nearer to the front of the cells (arrows in C and F)
If phosphorylation of myosin IIB is indeed responsible for the normal distribution of myosin IIB in the tails of migrating cells, then artificially phosphorylating the regulatory light chains should cause posterior accumulation of myosin IIB even when rho-kinase activity is blocked. This was tested by transiently transfecting cells with a FLAG-tagged, constitutively active mutant of MLCK (Figure 9). In the presence of 2 μM Y-27632, staining of phosphorylated light chains was dramatically reduced (compare untransfected cells in Figure 9C with Figure 8C), but in cells expressing the mutant MLCK, phosphorylation of myosin regulatory light chains at serine 19 was restored (Figure 9C). A similar result was observed for di-phosphorylated light chains. Furthermore, myosin IIB accumulated in the tails of MLCK-transfected, Y-27632–treated cells, but not in untransfected cells in the same wound (Figure 10, A–C). Myosin IIB asymmetry was “rescued” in all cells that expressed high levels of constitutively active MLCK and in most cells expressing even minimally detectable amounts (Figure 10E, right). Note, however, that the magnitude of the asymmetry in highly expressing cells was not greater than in the cells with lower levels. Nor did constitutively active MLCK increase the posterior distribution of myosin IIB in control wounds (Figure 10D). This suggests that there is a threshold of kinase activity, above which myosin IIB is caused to accumulate posteriorly. A dominant-negative mutant of MLCK did not affect the loss of posterior accumulation of myosin IIB in Y-27632–treated wounds, nor did it disrupt the polarization of myosin IIB in control wounds (Figure 10D).
Figure 9.

Constitutively active MLCK can phosphorylate myosin II regulatory light chains in Y-27632–treated cells. BAEC cultures that had been transiently transfected with plasmid encoding FLAG-tagged, constitutively active MLCK were fixed 1 h after wounding in 2 μM Y-27632, then stained with lysine-reactive cy5 (A), antibody against the FLAG epitope on constitutively active MLCK (B), and antibody against myosin II regulatory light chains phosphorylated at serine 19 (C). The transfected cell (*) stained much more strongly for phosphorylated light chains than its untransfected neighbors. Note the strong localization of phosphorylated light chains along the lateral trailing edges of the cell.
Figure 10.

Constitutively active MLCK restores myosin IIB polarity in Y-27632–treated cells. BAEC cultures that had been transiently transfected with plasmid encoding FLAG-tagged, constitutively active MLCK were fixed 1 h after wounding in 2 μM Y-27632 and then stained with lysine-reactive cy5 (A), antibody against the FLAG epitope on constitutively active MLCK (B), and antibody against myosin IIB (C). Arrowheads mark leading edges of migrating cells. In untransfected cells, myosin IIB extended far into the lamellar protrusions, but it was excluded from the front of the cell at the upper right, which contained high levels of constitutively active MLCK. Note the heavy accumulation of myosin IIB along the rear of this cell (C, arrow), whereas myosin IIB was more generally distributed through the cytoplasm of untransfected neighboring cells. (D) Transfected cultures were fixed and stained after cells had migrated for 1 h in the presence or absence of Y-27632. Myosin IIB asymmetries were then calculated for cells transfected with constitutively active MLCK (caM-LCK), for cells transfected with dominant-negative MLCK (dnMLCK) and for untransfected cells. Mutant MLCKs did not affect asymmetry in control wounds, but constitutively active MLCK prevented the reversal of myosin IIB asymmetry caused by Y-27632. Error bars, 1 SD; n = 26 for each mean. (E) Asymmetries of myosin IIA (left) and myosin IIB (right) plotted as a function of the expression levels of constitutively active MLCK, as measured by the fluorescence intensity of FLAG staining. Dotted lines indicate the mean myosin II asymmetry in 20 untransfected cells in the same fields. At low levels of MLCK expression, myosin IIA asymmetry in transfected cells was less than the untransfected average, but it did not become negative in most cells until expression levels reached ∼1 U. Myosin IIB asymmetry was negative; i.e., skewed toward the tail even at very low levels of MLCK expression (∼0.2 U).
Constitutively active MLCK also affected myosin IIA. Low levels of expression inhibited the anterior polarization of myosin IIA, but did not change the direction of asymmetry. However, at very high levels of MLCK expression, myosin IIA began to skew toward the posterior of the cell like myosin IIB (Figure 10E). Dominant-negative MLCK had no effect on myosin IIA distribution (our unpublished results). Thus, increasing phosphorylation of myosin II regulatory light chains generally promoted posterior accumulation of myosin II, with myosin IIA being more resistant to this effect than myosin IIB.
Inhibition of Posterior Accumulation of Myosin IIB Affects Tail Retraction But Not the Forward Extension of Lamellipodia
To understand better the function of myosin IIB accumulation in the tail, the morphology and dynamics of cell migration were examined in the presence and absence of Y-27632. Immediately after wounding, BAECs rapidly extended lamellipodia into the wound, with the cells' leading edges advancing at ∼0.5 μm/min. Y-27632 had little or no effect on the rate of protrusion during the first 30 min after wounding (Figure 11). This indicated that myosin IIB asymmetry was not necessary for the development of polarized protrusive activity. However, just when cells in control wounds became fully polarized 20–40 min after wounding, differences in behavior were seen in Y-27632–treated cells. In control cells, spreading of the leading edge slowed to about half of its original speed, and the trailing edge began to move forward. Thereafter, the leading and trailing edges advanced, on average, at the same rate (Figure 11, solid symbols). In contrast, the trailing edge did not begin to move until 50–60 min after wounding in Y-27632, even though the leading edge continued to spread until 90 min after wounding and did so more rapidly than controls (Figure 11, open symbols). Because of the long delay in moving the rear of the cell, Y-27632–treated cells became more elongated before the bodies of the cells began to translocate.
Figure 11.

Movements of the leading and trailing edges of migrating BAECs. Time-lapse movies were made of BAECs migrating at the edge of a wound in the presence (dotted lines, open symbols) or absence (solid lines, closed symbols) of 2 μM Y-27632. Each point represents the average distance moved toward the wound by the leading edges (circles) and trailing tails (triangles) of six cells migrating in the same experiment (error bars, 1 SD). The results shown are representative of three separate experiments. Note the constant distance between the leading and trailing edges of control cells after 50 min and the much larger separation between the front and rear of cells in Y-27632.
These observations suggested that Y-27632 might inhibit contractility in the rear of the cell. This idea was supported by the morphology of migrating cells. In control cells, the cytoplasm posterior to the nucleus was typically sharply constricted, so that the cells had tapering, phase-dense tails with concave contours (Figure 12A). Gaps developed not only behind cells as they moved into the wound, but also between adjacent cells along the edge, indicating that the cells contracted laterally as they elongated. The tails retracted rapidly toward the cell body when cell-substratum attachments broke, as if under contractile tension. But in Y-27632, the shapes of the trailing edges did not change much while the leading edges of the cells spread into the wound (Figure 12B). Gaps between cells were fewer, smaller, and developed later than in control cells. The cell bodies did narrow slightly when the cells were fully elongated, but even 60 min after wounding the tails of drugtreated cells were wider than their untreated counterparts (Figure 12C). In untransfected cells, the average width of the cell immediately posterior to the nucleus was 14 ± 3 μm in Y-27632 compared with 9 ± 2 μm in controls, which is significantly larger (n = 48; p > 95%). Similarly broad posteriors were observed for cells in which rho activity was inhibited by transient transfection with T19Nrho (see Figure 6). In contrast, the tails of Y-27632–treated cells that were transfected with constitutively active MLCK did constrict and taper (see Figures 9 and 10). Thus, tail morphology was regulated in parallel with myosin IIB asymmetry.
Figure 12.

Effects of Y-27632 on tail morphology. (A) Phase contrast micrographs of live, untreated cells 60 min after wounding illustrate the narrow, phase-dense tails (arrows) that developed as cells began to pull away from the edge of the monolayer. (B) Parallel cultures were wounded and imaged at the same time, but in the presence of 2 μM Y-27632. Bracket indicates a broad, flat tail on a cell that was just beginning to separate from its neighbors. (C) The width of the tail was measured 60 min after wounding in 48 untreated cells (▪) or 48 cells in 2 μM Y-27632 (□). Tail width was taken as the distance across the cell body along a line drawn parallel to the wound edge and located immediately posterior to the nucleus (e.g., dotted lines in A and B).
DISCUSSION
Because myosin IIA and IIB redistributed toward opposite ends of an endothelial cell as it began to migrate (Figure 1) and because they did so at different times (Figure 3), they must perform very different functions during cell migration. Saitoh et al. (2001) reported a similarly polarized distribution of myosin IIA and IIB in human fetal lung fibroblasts and proposed that myosin IIA was generally involved in cell migration, with myosin IIB playing a supporting role in the rear. In both endothelial cells and fibroblasts, the distribution of myosin IIB is very similar to myosin II in Dictyostelium amoebae, where only a single myosin II isoform is present. Genetic knockouts of the Dictyostelium myosin II indicate that it contributes to the formation of a contractile cortex around the tail: the tails of myosin II–deficient cells are less rigid (Pasternak et al., 1989), exert less traction force (Elson et al., 1999; Fukui et al., 2000), and produce less hydrostatic pressure for squeezing cytoplasm toward the front of the cell (Pasternak et al., 1989; Elson et al., 1999; Fukui et al., 2000). Myosin IIB appears to have a similar role in endothelial cells, because disruption of myosin IIB by inhibiting rhoA or rho-kinase resulted in delayed tail retraction and diminished constriction of the tail. Although inhibition of rho and rho-kinase undoubtedly affects other proteins in the cell besides myosin IIB, the posterior accumulation of myosin IIB, phosphorylation of myosin II regulatory light chains, and the narrow, tapered morphology characteristic of contracting tails were all specifically restored by constitutively active MLCK. This demonstrates that the altered tail contractility is specific to rho-kinase's effects on phosphorylation of myosin II regulatory light chains. Bridgman and colleagues have also shown correlations between myosin IIB and cytoplasmic retraction: in cultured nerve growth cones, myosin IIB is concentrated behind edges that are retracting rather than extending (Rochlin et al., 1995), and neurons from myosin IIB–deficient mice exert less traction force on the substratum (Bridgman et al., 2001). Thus, the role of myosin IIB in migrating cells may be to contract the locomotive cytoskeleton after protrusions have extended. Such contractility can pull the rear of the cell forward (or retract a protrusion from the front or side of the cell), increase traction forces between the front and rear of the cell, and create hydrostatic pressure to drive flow of cytoplasm toward the front of the cell.
Contraction of the cortical cytoplasm by myosin IIB might also constrain protrusive activity in the tail, because myosin II–deficient amoebae display excessive protrusive activity from their posterior and lateral edges (Wessels et al., 1988). However, we did not observe any new protrusions along the rear or side edges of endothelial cells migrating without posterior accumulation of myosin IIB (i.e., in Y-27632 or after transfection with T19Nrho). Furthermore, myosin IIB was not asymmetrically distributed until >15 min after wounding, even though BAECs already had new protrusions restricted to the wound edge within 5 min of wounding. Thus, myosin IIB apparently does not dictate protrusive polarity at the beginning of wound healing. It is likely that inhibitory signals from cell-cell contacts prevent lateral and posterior protrusion in the wound-healing scenario, obviating a role for myosin IIB early on. However, myosin IIB and tail retraction may become more important when cell-cell contacts are absent. Experiments are underway to test the effects of disrupting myosin IIB on protrusive polarity and locomotive efficiency in isolated endothelial cells migrating toward a chemotactic signal or during random-walk migration.
The role of myosin IIA in endothelial migration remains unclear. The timing with which myosin IIA becomes asymmetrically redistributed so closely parallels that of F-actin that it is tempting to postulate a structural role for myosin IIA in forming protrusions. Although myosin II is initially excluded from the tips of new protrusions in fibroblasts and endothelial cells (DeBiasio et al., 1988; Kolega, 1998a) and is not required at all for protrusive activity in Dictyostelium (Wessels et al., 1988), disrupting the assembly of myosin IIA with truncated fragments of the heavy chain caused retraction of protrusions and rounding up of HeLa cells (Wei and Adelstein, 2000). Verkhovsky et al. (1995) have shown that the myosin II toward the front of keratinocytes is organized into short, bipolar ribbons that interconnect actin filaments and accumulate in large actin-myosin bundles, suggesting that myosin II acts as a cross-linker more than a contractile motor in this region. This is supported by the behavior of microinjected, fluorescently labeled, chicken-gizzard myosin II, which we have shown colocalizes with myosin IIA in endothelial cells (Kolega, 1998b). In fibroblasts and endothelial cells, this fluorescent myosin II analog accumulates into actin-containing bundles parallel to the leading edge, which associate with cell-substratum adhesions but do not contract in the direction of movement as the cell moves forward (DeBiasio et al., 1988, and our own unpublished observations). Thus, the function of myosin IIA may be to stabilize and organize the actin cytoskeleton in protrusions at the front, whereas myosin IIB contracts it in the rear. The present demonstration that the distributions of myosin IIA and IIB can be independently regulated may lead to agents that selectively disrupt myosin IIA, which will permit more definitive tests of its function in cell migration.
How are the distributions of myosin IIA and IIB controlled in the cell? The posterior accumulation of myosin IIB was blocked by inhibition of either rho-dependent kinase or rhoA itself and was restored by artificially activating MLCK, indicating a pathway in which rhoA activates rho-dependent kinase, leading to myosin light-chain phosphorylation. Because phosphorylation of myosin II by rho-kinase in vitro stimulates myosin II filament assembly and motor activity (Amano et al., 1997), direct phosphorylation of myosin IIB by rho-kinase could induce contraction of the cortical cytoskeleton in the tail. Rho-kinase also inhibits myosin phosphatase (Kimura et al., 1996), decreasing the rate of light-chain dephosphorylation, which could further enhance myosin II assembly and activation.
Surprisingly, it appears that MLCK is not the primary regulator of posterior accumulation of myosin IIB in BAECs, because neither the pharmacological inhibitor, ML-7, nor the dominant negative mutant, kinase-dead MLCK, had a significant effect on myosin IIB asymmetry. The ability of constitutively active MLCK to induce posterior accumulation of myosin IIB when rho-kinase is inhibited, suggests that endogenous MLCK could perform this function, but is not sufficiently active during the normal wound response or is not present in sufficient amounts in BAECs to produce myosin IIB rearrangement. The activity of rho-kinase, on the other hand, may be elevated after wounding, because loss of cell-cell contacts is accompanied by activation of rhoA in MDCK and HEK293 epithelial cells (Noren et al., 2001). A similar response in endothelial cells could provide the necessary cue for rho-kinase–mediated phosphorylation of myosin IIB.
The ability of constitutively active MLCK to produce posterior accumulation of myosin IIB in Y-27632–treated cells also indicates that the asymmetry in myosin IIB distribution does not require asymmetric distribution of the kinase. Myosin IIB may get phosphorylated throughout the cytoplasm, causing it to assemble and contract, so that it accumulates in the rear simply because it remains in place while the rest of the cell advances. However, the punctate, linear arrays of myosin IIB in Y-27632–treated cells (Figure 4F) indicate that myosin IIB remains assembled in some cytoskeletal framework even when rho-kinase is inhibited. This is further demonstrated by the skewing of myosin IIB toward the front during rho-kinase inhibition. Fumoto et al. (2003) showed that myosin II containing nonphosphorylatable light chains (in which serine 19 and threonine 18 are replaced with alanines) still distributes with F-actin networks in fibroblasts, demonstrating that assembly of actin-myosin structures can occur without light-chain phosphorylation. Therefore, the posterior accumulation of myosin IIB during normal migration must involve an active contractile event or motor activity that is turned on by light-chain phosphorylation. Contraction of actin cytoskeleton by myosin IIB would tend to pull both the actin and myosin IIB toward the cells' strongest attachments, which early in migration are toward the rear. As new cell-substratum adhesions develop toward the leading edge and posterior attachments weaken, this contractility would also lead to tail retraction later in migration.
Why is myosin IIA not affected in the same way as myosin IIB? Several lines of evidence suggest that myosin IIA is not phosphorylated in the same manner as myosin IIB in BAECs. First, regulatory light chains were most heavily phosphorylated in the posterior cytoplasm where myosin IIB accumulated (Figure 8). Second, little phosphorylation was detected in the anterior cytoplasm of migrating cells, where myosin IIA is abundant and myosin IIB is absent. Third, changes in light-chain phosphorylation (Figure 7) correlated with changes in asymmetry of myosin IIB, but not myosin IIA (Figure 5). Fourth, much higher levels of constitutively active MLCK were required to cause posterior accumulation of myosin IIA in Y-27632–treated cells than were required to skew myosin IIB rearward (Figure 10E). Two alternative mechanisms could account for different levels of light-chain phosphorylation between myosin IIA and IIB: In one scenario, the relevant kinase(s) could distinguish between myosin IIA and IIB. Little is known about the relative affinity of rho-dependent kinases for different isoforms of nonmuscle myosin II in vitro or in vivo. If a rho-dependent kinase preferentially phosphorylates myosin IIB over myosin IIA, it could cause myosin IIB to contract F-actin structures, pulling them toward stable anchor points that become the tail of the cell. Meanwhile, the less phosphorylated myosin IIA would be free to enter the cell's newly formed protrusions and incorporate into the F-actin cytoskeleton at the leading edge. Alternatively, both myosin IIA and IIB may get phosphorylated, but myosin IIA may be preferentially dephosphorylated. We previously showed that the phosphate on myosin regulatory light chains turns over more rapidly in migrating BAECs than in unwounded monolayers (Kolega, 1999). If the responsible phosphatases have a preference for myosin IIA, myosin IIA would get dephosphorylated to a greater extent than myosin IIB, perhaps contributing to myosin IIA's ability to move forward into actin networks in new protrusions, whereas myosin IIB lags behinds in large contractile structures. Preferential dephosphorylation of myosin IIA could also explain why constitutively active MLCK more readily affects myosin IIB— more kinase activity would be required to outpace the phosphatase.
The preponderance of light-chain phosphorylation in the posterior cytoplasm in BAECs is somewhat different from what has been observed in fibroblasts, where phosphorylation of the regulatory light chains at serine 19 and/or threonine 18 is observed at both the front and the rear of migrating cells (Post et al., 1995; Matsumura et al., 1998; Saitoh et al., 2001; Fumoto et al., 2003). Because fibroblasts also display anterior skewing of myosin IIA and posterior accumulation of myosin IIB (Saitoh et al., 2001), such anterior phosphorylation of regulatory light chains most likely occurs on myosin IIA in fibroblasts. Why this was not observed in BAECs is not clear; however, BAECs were notably insensitive to the MLCK inhibitor, ML-7 (Figures 4, 5, and 7), suggesting that they may have lower MLCK activity or less MLCK than fibroblasts. A higher level of endogenous MLCK in fibroblasts might be sufficient to cause anterior phosphorylation of myosin IIA in fibroblasts, but not enough to induce posterior accumulation, much as moderate levels of constitutively active MLCK in BAECs decrease myosin IIA asymmetry but do not reverse it (Figure 10E, left panel). It would be interesting to determine if migrating BAECs exert less contractile tension in the anterior portions of the cell when compared with fibroblasts and whether constitutively active MLCK changes the distribution of traction forces along the anterior-posterior axis.
Acknowledgments
I thank Dr. Chris Cohan for valuable criticism of the manuscript of this work and Marian Pazik and Jason Myers for their excellent assistance in the laboratory. This work was supported by a grant from the American Heart Association.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E03–04–0205. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E03-04-0205.
References
- Amano, M., Chihara, K., Kimura, K., Fukata, Y., Nakamura, N., Matsuura, Y., and Kaibuchi, K. (1997). Formation of actin stress fibers and focal adhesions enhanced by rho-kinase. Science 275, 1308–1311. [DOI] [PubMed] [Google Scholar]
- Amano, M., Itoh, M., Kimura, K., Fukata, Y., Chihara, K., Nakano, T., Matsuura, Y., and Kaibuchi, K. (1996). Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 271, 20246–20249. [DOI] [PubMed] [Google Scholar]
- Bhatia-Dey, N., Taira, M., Conti, M.A., Nooruddin, H., and Adelstein, R.S. (1998). Differential expression of non-muscle myosin heavy chain genes during Xenopus embryogenesis. Mech. Dev. 78, 33–36. [DOI] [PubMed] [Google Scholar]
- Bridgman, P.C., Dave, S., Asnes, C.F., Tullio, A.N., and Adelstein, R.S. (2001). Myosin IIB is required for growth cone motility. J. Neurosci. 21, 6159–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, O.H., Park, C.S., Itoh, K., Adelstein, R.S., and Beaven, M.A. (1996). Cloning of the cDNA encoding rat myosin heavy chain-A and evidence for the absence of myosin heavy chain-B in cultured rat mast (RBL-2H3) cells. J Muscle Res. Cell Motil. 17, 69–77. [DOI] [PubMed] [Google Scholar]
- Coates, T.D., Watts, R.G., Hartman, R., and Howard, T.H. (1992). Relationship of F-actin distribution to development of polar shape in human polymorphonuclear neutrophils. J. Cell Biol. 117, 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, S.P., Reddy, H., Caivano, M., and Cohen, P. (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBiasio, R.L., Wang, L.-L., Fisher, G.W., and Taylor, D.L. (1988). The dynamic distribution of fluorescent analogues of actin and myosin in protrusions at the leading edge of migrating Swiss 3T3 fibroblasts. J. Cell Biol. 107, 2631–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek, S.M., and Garcia, J.G. (2001). Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 91, 1487–1500. [DOI] [PubMed] [Google Scholar]
- Ehrlich, H.P., Rockwell, W.B., Cornwell, T.L., and Rajaratnam, J.B.M. (1991). Demonstration of a direct role for myosin light chain kinase in fibroblastpopulated collagen lattice contraction. J. Cell. Physiol. 146, 1–7. [DOI] [PubMed] [Google Scholar]
- Elson, E.L., Felder, S.F., Jay, P.Y., Kolodney, M.S., and Pasternak, C. (1999). Forces in cell locomotion. Biochem. Soc. Symp. 65, 299–314. [PubMed] [Google Scholar]
- Fumoto, K., Uchimura, T., Iwasaki, T., Ueda, K., and Hosoya, H. (2003). Phosphorylation of myosin II regulatory light chain is necessary for migration of HeLa cells but not for localization of myosin II at the leading edge. Biochem. J. 370, 551–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui, Y., Uyeda, T.Q., Kitayama, C., and Inoue, S. (2000). How well can an amoeba climb? Proc. Natl. Acad. Sci. USA 97, 10020–10025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui, Y., and Yumura, S. (1986). Actomyosin dynamics in chemotactic amoeboid movement of Dictyostelium. Cell Motil. Cytoskel. 6, 662–673. [Google Scholar]
- Gallagher, P.J., Herring, B.P., Tafny, A., Sowadski, J., and Stull, J.T. (1993). A molecular mechanism for autoinhibition of myosin light chain kinases. J. Biol. Chem. 268, 26578–26582. [PMC free article] [PubMed] [Google Scholar]
- Kase, H., Iwahashi, K., Nakanishi, S., Matsuda, Y., Yamada, K., Takahashi, M., Murakata, C., Sato, A., and Kaneko, M. (1987). K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem. Biophys. Res. Commun. 142, 436–440. [DOI] [PubMed] [Google Scholar]
- Kawamoto, S., and Adelstein, R.S. (1991). Chicken nonmuscle myosin heavy chains: differential expression of two mRNAs and evidence for two different polypeptides. J. Cell Biol. 112, 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley, C.A., Sellers, J.R., Gard, D.L., Bui, D., Adelstein, R.S., and Baines, I.C. (1996). Xenopus nonmuscle myosin heavy chain isoforms have different subcellular localizations and enzymatic activities. J. Cell Biol. 134, 675–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, K. et al. (1996). Regulation of myosin phosphatase by rho and rho-associated kinase (rho-kinase). Science 273, 245–248. [DOI] [PubMed] [Google Scholar]
- Klemke, R.L., Cai, S., Giannini, A.L., Gallagher, P.J., de Lanerolle, P., and Cheresh, D.A. (1997). Regulation of cell motility by mitogen-activated protein kinase J. Cell Biol. 137, 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolega, J. (1997). Asymmetry in the distribution of free versus cytoskeletal myosin II in locomoting microcapillary endothelial cells. Exp. Cell Res. 231, 66–82. [DOI] [PubMed] [Google Scholar]
- Kolega, J. (1998a). Cytoplasmic dynamics of myosin IIA and IIB: spatial `sorting' of isoforms in locomoting endothelial cells. J. Cell Sci. 111, 2085–2095. [DOI] [PubMed] [Google Scholar]
- Kolega, J. (1998b). Fluorescent analogues of myosin II for tracking the behavior of different myosin isoforms in living cells. J. Cell. Biochem. 68, 389–401. [DOI] [PubMed] [Google Scholar]
- Kolega, J. (1999). Turnover rates at regulatory phosphorylation sites on myosin II in endothelial cells. J. Cell. Biochem. 75, 629–639. [PubMed] [Google Scholar]
- Kolega, J., and Kumar, S. (1999). Regulatory light chain phosphorylation and the assembly of myosin II into the cytoskeleton of microcapillary endothelial cells. Cell Motil. Cytoskel. 43, 255–268. [DOI] [PubMed] [Google Scholar]
- Kolega, J., Nederlof, M.A., and Taylor, D.L. (1993). Quantitation of cytoskeletal fibers in fluorescence images: stress fiber disassembly accompanies dephosphorylation of the regulatory light chains of myosin II. Bioimaging 1, 136–150. [Google Scholar]
- Kolega, J., and Taylor, D.L. (1993). Gradients in the concentration and assembly of myosin II in living fibroblasts during locomotion and fiber transport. Mol. Biol. Cell 4, 819–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodney, M.S., and Wysolmerski, R.B. (1992). Isometric contraction by fibroblasts and endothelial cells in tissue culture: a quantitative study. J. Cell Biol. 117, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura, F., Ono, S., Yamakita, Y., Totsukawa, G., and Yamashiro, S. (1998). Specific localization of serine 19 phosphorylated myosin II during cell locomotion and mitosis of culture cells. J. Cell Biol. 140, 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maupin, P., Phillips, C.L., Adelstein, R.S., and Pollard, T.D. (1994). Differential localization of myosin-II isozymes in human cultured cells and blood cells. J. Cell Sci. 107(Pt 11), 3077–3090. [DOI] [PubMed] [Google Scholar]
- Murakami, N., Chauhan, V.P., and Elzinga, M. (1998). Two nonmuscle myosin II heavy chain isoforms expressed in rabbit brains: filament forming properties, the effects of phosphorylation by protein kinase C and casein kinase II, and location of the phosphorylation sites. Biochemistry 37, 1989–2003. [DOI] [PubMed] [Google Scholar]
- Murakami, N., and Elzinga, M. (1992). Immunohistochemical studies on the distribution of cellular myosin II isoforms in brain and aorta. Cell Motil. Cytoskel. 22, 281–295. [DOI] [PubMed] [Google Scholar]
- Murakami, N., Kotula, L., and Hwang, Y.W. (2000). Two distinct mechanisms for regulation of nonmuscle myosin assembly via the heavy chain: phosphorylation for MIIB and mts 1 binding for MIIA. Biochemistry 39, 11441–11451. [DOI] [PubMed] [Google Scholar]
- Murakami, N., Trenkner, E., and Elzinga, M. (1993). Changes in expression of nonmuscle myosin heavy chain isoforms during muscle and nonmuscle tissue development. Dev. Biol. 157, 19–27. [DOI] [PubMed] [Google Scholar]
- Nakanishi, S., Yamada, K., Iwahashi, K., Kuroda, K., and Kase, H. (1990). KT5926, a potent and selective inhibitor of myosin light chain kinase. Mol. Pharmacol. 37, 482–488. [PubMed] [Google Scholar]
- Noren, N.K., Niessen, C.M., Gumbiner, B.M., and Burridge, K. (2001). Cadherin engagement regulates Rho family GTPases. J. Biol. Chem. 276, 33305–33308. [DOI] [PubMed] [Google Scholar]
- Pan, Z.K., Ye, R.D., Christiansen, S.C., Jagels, M.A., Bokoch, G.M., and Zuraw, B.L. (1998). Role of the Rho GTPase in bradykinin-stimulated nuclear factorkappaB activation and IL-1beta gene expression in cultured human epithelial cells. J. Immunol. 160, 3038–3045. [PubMed] [Google Scholar]
- Pasternak, C., Spudich, J.A., and Elson, E.L. (1989). Capping of surface receptors and concomitant cortical tension are generated by conventional myosin. Nature 341, 549–551. [DOI] [PubMed] [Google Scholar]
- Phillips, C.L., Yamakawa, K., and Adelstein, R.S. (1995). Cloning of the cDNA encoding human nonmuscle myosin heavy chain-B and analysis of human tissues with isoform-specific antibodies. J Muscle Res. Cell Motil. 16, 379–389. [DOI] [PubMed] [Google Scholar]
- Post, P.L., DeBiasio, R.L., and Taylor, D.L. (1995). A fluorescent protein biosensor of myosin II regulatory light chain phosphorylation reports a gradient of phosphorylated myosin II in migrating cells. Mol. Biol. Cell 6, 1755–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley, A.J. (2001). Rho GTPases and cell migration. J. Cell Sci. 114, 2713–2722. [DOI] [PubMed] [Google Scholar]
- Rochlin, M.W., Itoh, K., Adelstein, R.S., and Bridgman, P.C. (1995). Localization of myosin II A and B isoforms in cultured neurons. J. Cell Sci. 108, 3661–3670. [DOI] [PubMed] [Google Scholar]
- Saitoh, T., Takemura, S., Ueda, K., Hosoya, H., Nagayama, M., Haga, H., Kawabata, K., Yamagishi, A., and Takahashi, M. (2001). Differential localization of non-muscle myosin II isoforms and phosphorylated regulatory light chains in human MRC-5 fibroblasts. FEBS Lett. 509, 365–369. [DOI] [PubMed] [Google Scholar]
- Satterwhite, L.L., and Pollard, T.D. (1992). Cytokinesis. Curr. Opin. Cell Biol. 4, 43–52. [DOI] [PubMed] [Google Scholar]
- Simons, M., Wang, M., McBride, O.W., Kawamoto, S., Yamakawa, K., Gdula, D., Adelstein, R.S., and Weir, L. (1991). Human nonmuscle myosin heavy chains are encoded by two genes located on different chromosomes. Circ. Res. 69, 530–539. [DOI] [PubMed] [Google Scholar]
- Subauste, M.C., Herrath, M.V., Benard, V., Chamberlain, C.E., Chuang, T.-H., Chu, K., Bokoch, G.M., and Hahn, K.M. (2000). Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and fas. J. Biol. Chem. 275, 9725–9733. [DOI] [PubMed] [Google Scholar]
- Toullec, D. et al. (1991). The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 266, 15771–15781. [PubMed] [Google Scholar]
- Tullio, A.N., Accili, D., Ferrans, V.J., Yu, Z.X., Takeda, K., Grinberg, A., Westphal, H., Preston, Y.A., and Adelstein, R.S. (1997). Nonmuscle myosin II-B is required for normal development of the mouse heart. Proc. Natl. Acad. Sci. USA 94, 12407–12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullio, A.N., Bridgman, P.C., Tresser, N.J., Chan, C.C., Conti, M.A., Adelstein, R.S., and Hara, Y. (2001). Structural abnormalities develop in the brain after ablation of the gene encoding nonmuscle myosin II-B heavy chain. J. Comp. Neurol. 433, 62–74. [DOI] [PubMed] [Google Scholar]
- Tullio, A.N., Ferrans, V.J., Yu, Z.-X., Ward, J.M., Chan, C.-C., Zimmer, A., Westphal, H., Preston, Y.A., and Adelstein, R.S. (1996). Targeted disruption of the gene encoding nonmuscle myosin heavy chain II-B results in structural abnormalities in the mouse heart, brain, and retina. Mol. Biol. Cell 7, 195a. [Google Scholar]
- Uehata, M. et al. (1997). Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389, 990–994. [DOI] [PubMed] [Google Scholar]
- Verkhovsky, A.B., Svitkina, T.M., and Borisy, G.G. (1995). Myosin II filament assemblies in the active lamella of fibroblasts: their morphogenesis and role in the formation of actin filament bundles. J. Cell Biol. 131, 989–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, Q., and Adelstein, R.S. (2000). Conditional expression of a truncated fragment of nonmuscle myosin II-A alters cell shape but not cytokinesis in HeLa cells. Mol. Biol. Cell 11, 3617–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels, D., and Soll, D.R. (1990). Myosin II heavy chain null mutant of Dictyostelium exhibits defective intracellular particle movement. J. Cell Biol. 111, 1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels, D., Soll, D.R., Knecht, D., Loomis, W.F., De Lozanne, A., and Spudich, J. (1988). Cell motility and chemotaxis in Dictyostelium amoebae lacking myosin heavy chain. Dev. Biol. 128, 164–177. [DOI] [PubMed] [Google Scholar]
- Wessels, D., Titus, M., and Soll, D.R. (1996). A Dictyostelium myosin I plays a crucial role in regulating the frequency of pseudopods formed on the substratum. Cell Motil. Cytoskel. 33, 64–79. [DOI] [PubMed] [Google Scholar]
- Zhang, H., Wessels, D., Fey, P., Daniels, K., Chisholm, R.L., and Soll, D.R. (2002). Phosphorylation of the myosin regulatory light chain plays a role in motility and polarity during Dictyostelium chemotaxis. J. Cell Sci. 115, 1733–1747. [DOI] [PubMed] [Google Scholar]