

Abstract
The past decade has seen an explosion in the use of DNA-based microarrays. These techniques permit to assess RNA abundance on a genome-wide scale. Medical applications emerged in the field of cancer, with studies of both solid tumors and hematological malignancies leading to the development of tests that are now used to personalize therapeutic options. Microarrays have also been used to analyze the blood transcriptome in a wide range of diseases. In human autoimmune diseases, these studies are showing potential for identifying therapeutic targets as well as biomarkers for diagnosis, assessment of disease activity and response to treatment. More quantitative and sensitive high throughput RNA profiling methods are starting to be available and will be necessary for transcriptome analyses to become routine tests in the clinical setting. We expect this to crystallize within the coming decade, as they become part of the personalized medicine armamentarium.
Introduction
The immune system is a powerful defense operation. Protective immunity results from the interplay between two cardinal systems: non-specific innate immunity and antigen-specific adaptive immunity. Dysfunctions of the immune system lie at the center of a wide variety of diseases, including autoimmunity, allergy, infections, cancer, and even some cardiovascular diseases.
As most diseases of the immune system, autoimmune diseases arise from interactions between environmental, epigenetic and genetic factors that result in downstream perturbations of complex and interactive biological networks. Attempts to identify single causative factors (i.e. genes or cytokines) with the use of classic genetic approaches, or in vitro studies focusing on a limited number of genes and/or cell types have for the most part not succeeded. Furthermore, in vivo studies using animal models of human immune-mediated diseases have been of limited value in the identification of relevant therapeutic targets (1). For example, the many existing murine lupus models have not yet led to the development of specific treatments for human lupus (2). Likewise, animal models of rheumatoid arthritis (RA) predicted IL1B to be an appropriate target in human RA (3). Blocking IL-1 was indeed effective only in a minor fraction of patients (4), which unfortunately cannot be identified using currently available disease markers. The successful blockade of TNFα in RA patients represents considerable progress (5), but many autoimmune diseases continue to be treated with non-specific medications such as corticosteroids and chemotherapeutic drugs. The use of these later medications is unfortunately associated with considerable adverse events. Additional challenges in the field of autoimmunity include the lack of specific biomarkers that can be used for diagnosis, assessment of disease activity and prediction of flares. These problems are especially significant as these diseases are life-long with a relapsing and remitting course.
An integrative evaluation of the complex network of alterations underlying the pathogenesis of autoimmune diseases was until recently difficult to conceive. Technological advances in the past 10 years, however, now permit us to analyze DNA, RNA or protein in patient samples on a genome-wide scale. These techniques, combined with bioinformatics, are changing the face of clinical research and opening the path for novel approaches to patient care. DNA microarrays can assess in a single sample the activity of the entire transcriptome (6, 7). Current techniques detect mRNA species from known genes as immunofluorescent labeled cRNA hybridized to arrays of either cDNA or oligonucleotide fragments, but novel approaches are rapidly emerging (8). Tissue samples, blood, purified cells and even saliva can be tested in these assays.
The first hint that genomic studies could have clinical applications came in 1999, when microarray-based transcriptional profiling was proposed for the differential diagnosis of acute myeloid and lymphocytic leukemias (9). These studies have since resulted in the identification of gene expression signatures correlating with clinical outcomes both in hematological and solid tumors (10, 11). In breast cancer, for example, microarray studies have helped identifying subgroups of patients that may benefit from adjuvant therapy (12). When applied to diseases of the immune system in humans, restricted access to sampling relevant tissue(s), such as the brain in multiple sclerosis or the joints in rheumatoid arthritis, becomes a major limitation. Cells of the immune system, however, get educated and implement their functions by recirculating between central and peripheral lymphoid organs as well as by migrating to and from sites of injury via the blood. The blood therefore represents the pipeline of the immune system. Indeed, it is the preferred route for immune cells to reach the lymph nodes. After exiting through outgoing lymphatic vessels, these cells reach again the bloodstream to be transported to tissues throughout the body. Upon patrolling these tissues, they gradually drift back into the lymphatic system to begin the cycle all over again. The complex patterns of recirculation depend on the state of cell activation, the adhesion molecules expressed by immune and endothelial cells and the presence of chemotactic molecules which selectively attract particular populations of blood cells. Sampling the blood therefore provides a “snap shot” of the complex immune networks that operate throughout the entire body. Indeed, profiling blood sample RNA from patients with both systemic as well as organ-specific autoimmune diseases has proved to be a valid approach to find clues about pathogenesis as well as to identify potential biomarkers (Figure 1) [reviewed in (13, 14)].
Figure 1.
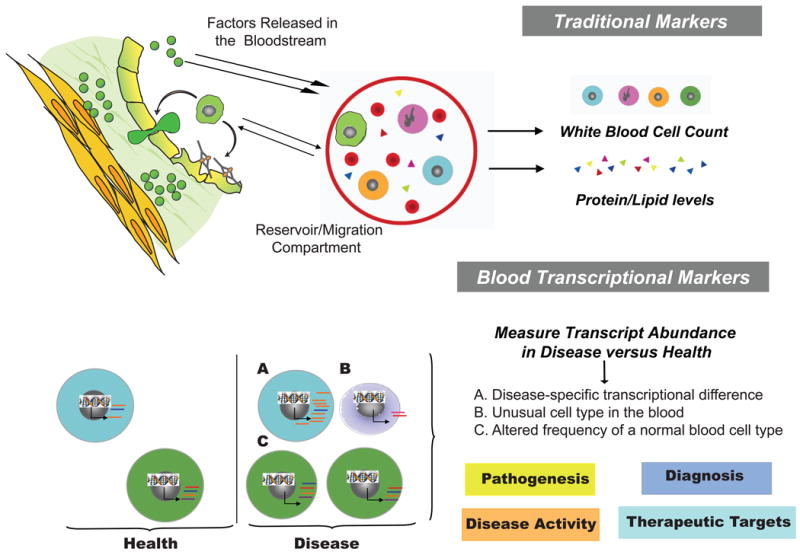
The blood can be used as a source of transcriptional biomarkers to understand the pathogenesis, diagnose, assess severity and find therapeutic targets for human autoimmune diseases.
Furthermore, in addition to providing a static view of the transcriptional activity of blood cells in the steady state, the transcriptome of whole blood, PBMCs or purified blood cell populations can be assessed with DNA microarray technology after exposure in vivo (15, 16) or in vitro (17) to a variey of immune stimuli. This approach, also known as “exercising the genome” [reviewed in (18)] has the potential to reveal alterations in gene pathways that are only visible after specific types of activation. The transcriptional effects of patient serum factors on healthy blood cells can also be studied using this approach. Indeed, we and others have successfully applied this “reverse proteomics” or reporter assay approach to understand autoimmune disease pathogenesis (19, 20).
About 10 years ago we surmised that analyzing the blood transcriptome of patients with autoimmune diseases would help us understand their pathogenesis and ultimately provide better care to these patients. Here we will describe our experience and that of others using this powerful yet challenging approach. We will especially focus on Systemic Lupus Erythematosus (SLE) and Systemic onset Juvenile Idiopathic Arthritis (SoJIA), two diseases in which microarray technology has led to the identification of pathogenic pathways, therapeutic targets as well as potential biomarkers to diagnose and follow patient disease activity and response to therapy.
Blood DNA Microarrays to Understand Autoimmune Disease Pathogenesis
The first reports on blood gene expression profiling in autoimmune/inflammatory diseases date back to 2002-2003. Systemic Lupus Erythematosus (21-24), Juvenile Idiopathic Arthritis (19, 25-28), multiple sclerosis (29, 30), rheumatoid arthritis (31-34), Sjogren's syndrome (35), diabetes (36, 37), inflammatory bowel disease (38), psoriasis and psoriatic arthritis (39, 40), inflammatory myopathies (41, 42), scleroderma (43, 44), vasculitis (45) and anti-phospholipid syndrome (46) have been studied using this approach. Overall, these studies have shown that diseases with diverse pathogenesis and clinical manifestations may share common mediators, which represent therapeutic targets for intervention. Blood RNA profiling has also rapidly extended to other diseases involving the immune system, especially infections caused by viruses such as HIV (47), adenovirus (48, 49), influenza (50), or dengue (51); bacteria such as Staphylococcus aureus, Streptococcus pneumoniae or Escherichia coli (52); Mycobacterium tuberculosis (53); parasites (Plasmodium) (54); as well as sepsis (55). The blood of transplant recipients has also been profiled, in kidney (56), liver (57), heart (58), and hematopoeitic cell transplant recipients (59). Furthermore, disease signatures have been detected in the blood cells of patients with cardio vascular diseases (60), and non-hematological malignancies (61, 62).
Systemic Lupus Erythematosus (SLE)
SLE and Type I IFN
Box 1.
Systemic Lupus Erythematosus (SLE) occurs in 17–48/100,000 people. There is a spectrum of human lupus ranging from solely skin involvement to systemic disease, which is characterized by a relapsing and remitting course with flares of high morbidity. SLE patients are predominantly women who present with chronic non-specific symptoms such as fever, weight loss and fatigue often associated to lymphadenopathy and especially lymphopenia. Some patients may present with severe acute illness characterized by seizures, psychosis, renal failure, profound anemia, pulmonary hemorrhage, or sepsis. Confirming the diagnosis of SLE requires the fulfillment of 4 out of 11 criteria, one of those being the presence of anti-nuclear antibodies (ANA), which are detected in >95% of patients. Loss of tolerance to nuclear antigens focuses on chromatin components (such as dsDNA, histones, and nucleosomes) and U-rich ribonucleoproteins (such as RNP and Sm), which are Toll-like receptor 9 (TLR9) and TLR7 ligands, respectively [reviewed in (193)]. Antibodies against phospholipids (complexed to β2-glycoprotein) exposed on the surface of dying cells are also frequently detected in SLE patients and correlate with the development of thromboembolic complications. Autoantibodies directed to cell surface molecules, especially those expressed on cells of hemopoietic origin, cause hemolytic anemia, neutropenia and thrombocytopenia. Antibodies against antigens expressed in target organs like the kidney (glomerular extracts) are also found in patients with severe kidney disease. In addition to the direct damage caused by cellular and/or tissue antigen-antibody interactions, many lupus symptoms result from indirect damage through the deposition of IC on tissues (i.e., nephritis, arthritis, and vasculitis).
Multiple cell types and soluble mediators, including IL10 (63, 64) and IFNγ (65-67), but not Type I IFN, have been proposed to be at the center of lupus pathogenesis. Yet, in the late 60s, polyI:C, a known inducer of type I IFN, was reported to accelerate disease in young NZBxNZW (F1) female mice (68). Then, 10 years later, increased interferon levels were detected in the sera of patients with SLE (69). Intriguingly, treatment with recombinant IFN-α resulted in autoantibody formation in up to 20% of patients and in the appearance of SLE-specific symptoms in a fraction of them (reviewed in (70)). In the late 90's, the number of natural IFN producing cells (IPCs), later identified as plasmacytoid dendritic cells (pDCs), were reported decreased in lupus blood (71) but increased at sites of injury (i.e. the skin) (72, 73). Furthermore, DNA and/or nucleic acid-containing immune complexes could induce these cells to secrete type I IFN (74, 75). At this time, we described that monocytes from some children with SLE displayed DC function, and that the serum of these patients was able to induce the differentiation of healthy monocytes into mature dendritic cells (DCs) in an IFNα-dependent manner (76). These studies, together with those describing the numerous effects of type I IFN on adaptive immunity (77-80), now converged to place this family of cytokines at the center of lupus pathogenesis. Yet, several observations did not support the hypothesis: first, not every SLE patient has detectable serum type I IFN levels (81); second, dysregulation of type-I IFN production is not obvious in most murine SLE-models (2); and third, genetic linkage and association studies had not identified candidate lupus susceptibility genes within the IFN-pathway (82). Gene expression profiling helped addressing some of these questions.
Because serum levels of type I IFN family members are difficult to measure, we explored whether leukocyte gene expression profiling might help us assess the prevalence of lupus patients exposure to these cytokines. Thirty children with SLE (representing four ethnicities, 60% female), 9 healthy children and a group of 12 patients with juvenile chronic arthritis (JCA) were recruited. Peripheral blood mononuclear cells (PBMCs) from the pediatric SLE patients displayed striking transcriptional differences compared to healthy controls (375 transcripts) (Figure 2). After correction for multiple testing-related errors, the very conservative Bonferroni correction (Box 7) selected 15 genes, 14 of which were IFN-inducible. A less conservative correction (Benjamini and Hochberg) selected 33 genes, 26 of which were IFN-regulated (21).
Figure 2. SLE signature.

Hierarchical clustering of gene expression from blood leukocytes of 9 healthy children, 30 with SLE and 12 with juvenile chronic arthritis. SLE patients were ranked according to their SLEDAI at the time of blood draw. Each row represents a separate gene and each column a separate patient. 374 transcript sequences were selected as being differentially expressed in SLE compared to healthy patients. The normalized expression index for each transcript sequence (rows) in each sample (columns) is indicated by a color code. Red, yellow and blue squares indicate that expression of the gene is greater than, equal to or less than the mean level of expression across 9 healthy controls. The scale extends from fluorescence ratios of 0.25 to 4.0. (Original figure published in J. Exp. Med. 2003 Mar 17;197(6):711-23)
Box 7. Analysis Primer.
Per-Chip Normalization
This step allows controlling array-wide variations in intensity across multiple samples that form a given dataset. After background subtraction, a normalization algorithm is used to rescale the difference in overall intensity to a fixed intensity level for all samples across multiple arrays.
Data filtering
Typically more than half of the probes present on a microarray do not detect a signal for any of the samples in a given analysis. Thus, a detection filter is applied to remove such probes. This step avoids the introduction of unnecessary noise in downstream analyses.
Unsupervised analysis
The aim of this step is to group samples on the basis of their molecular profiles without a priori knowledge of their phenotypic classification. The first step consists of selecting transcripts that are expressed in the dataset (detection filter), and display some degree of variability (which will facilitate sample clustering). For instance, this filter could select transcripts with expression levels that deviate by at least 2-fold from the median intensity calculated across all samples. It is important though to apply this filter independently of any knowledge of sample grouping or phenotype (which makes this type of analysis “unsupervised”). Next, a pattern discovery algorithm is applied to identify molecular phenotypes or trends in the data.
Clustering
Clustering is commonly used for the discovery of expression patterns in large datasets. Hierarchical clustering is an iterative agglomerative clustering method that can be used to produce gene trees and condition trees. Condition tree clustering groups samples based on the similarity of their expression profiles across a specified gene list. Other commonly employed clustering algorithms include k-means clustering and self-organizing maps.
Class Comparison
Class comparison analyses identify genes differentially expressed among groups and/or time points. The methods for analysis are chosen based on the study design. For studies with independent observations and two or more groups, t-tests, ANOVA, Mann-Whitney U tests, or Kruskal-Wallis tests are used. For more complex studies (e.g. longitudinal) appropriate linear mixed model analyses are chosen.
Multiple Testing Correction
Multiple testing correction (MTC) methods provide a means to mitigate the level of noise in sets of transcripts identified by class comparison (in order to lower permissiveness to false positives). While it reduces noise, MTC promotes higher false negative rate as a result of dampening the signal (). The methods available are characterized by varying degrees of stringency, and therefore they produce gene lists with different levels of robustness.
Benjamini and Hochberg false discovery rate is the least stringent MTC applied and provides a good balance between discovery of statistically significant genes while limiting false positives. By using this procedure with an alpha of 0.01, 1% of the statistically significant transcripts might be identified as significant by chance alone (false positives).
Bonferroni correction is the most stringent method used and can drastically reduce false positive rates. Conversely, it increases the probability of having false negatives.
Class Prediction
Class prediction analyses assess the classification capability of gene expression data for a study subject or sample. K-nearest neighbors is a commonly used technique for this task. Using Euclidian distance, this method identifies the user defined “k” number of closest observations for an unclassified sample. Class prediction is then determined by the lowest p-value, which is calculated for each group. The p-values are based on the likelihood of obtaining the observed number of neighbors for a specific class given the overall class proportion in the data set. Other available class prediction procedures include, but are not limited to, Discriminant Analysis, General Linear Model Selection, Logistic Regression, Distance Scoring, Partial Least Squares, Partition Trees, and Radial Basis Machine (refs???).
Sample Size
The number of samples necessary for the identification of a robust signature is variable. Indeed, sample size requirements will depend on the amplitude of the difference between and the variability within study groups.
A number of approaches have been devised for the calculation of sample size for microarray experiments, but to date little consensus exists. Hence, best practices in the field consist in the utilization of independent sets of samples for the purpose of validating candidate signatures. Thus, the robustness of the signature identified will rely on a statistically significant association between the predicted and true phenotypic class in the first and the second test sets.
All but one of the pediatric patients in our study exhibited upregulation of IFN-inducible genes, and the only patient lacking this signature had been in remission for over 2 years (21). Similar studies performed with adult SLE PBMCs showed an IFN signature in approximately 50% of patients (22-24). The higher prevalence of the signature in children might have been due to sample selection, as more patients with newly diagnosed, untreated disease were included in the pediatric study. It might also reflect a higher degree of disease severity and/or a unique genetic background responsible for the earlier appearance of disease manifestations in the pediatric cohort. Larger population studies will be required to resolve the source of this difference.
All nucleated cells express the type I IFN receptor (IFNAR) and respond to type I IFN with phosphorylation of STAT1 and STAT2, which ultimately leads to the transcription of interferon-responsive genes (83). A notable exception is represented by immature neutrophils, which do not phosphorylate STAT1 in response to receptor ligation (84). All type I IFNs signal through this receptor and give rise to similar signaling and transcriptional cascades. Sequence differences between type I IFNs result, however, in different binding affinities with each IFNAR chain and consequent biological activities (85). Hundreds of genes are induced or repressed [reviewed in (86)], including among others classical anti-viral/proliferative transcripts (i.e. OAS, MX1 etc), interferon regulatory factors (IRF5 and IRF7), pro-apoptotic molecules (i.e. FAS and TRAIL), B cell differentiation factors (i.e. BLyS/BAFF), chemokines and chemokine receptors (MCP1 and IP10) and, interestingly, even lupus autoantigens such as Ro/SSA (21). Many IFN-inducible proteins might indeed play a significant role in disease pathogenesis. Fas and TRAIL can for example contribute to nucleosome overload and therefore to increased nuclear antigen presentation. BLyS/BAFF induces the proliferation and differentiation of mature B cells into antibody secreting cells. Indeed, a neutralizing anti-BAFF/BLyS antibody being tested in clinical trials appears to reduce SLE disease activity and prevent flares (87).
Treating SLE patients with high dose IV steroids, which are used to control disease flares, results in the silencing of the IFN signature (21) (Figure 3). This is associated with pDC depletion (88), although both the blood pDCs and the IFN signature return in less than 1 week after IV steroid administration (our own unpublished data). Altogether, these observations support the central role of pDCs as producers of type I IFN in lupus pathogenesis.
Figure 3. High dose steroid intravenous pulse extinguishes the type I IFN signature in SLE blood.

Analysis of PBMCs from 3 pediatric SLE patients before and after treatment with high dose i.v. Methylprednisolone (1g/day for 3 days). All patients show down-regulation of IFN-regulated transcripts (upper panel) while expression of non-type I IFN-inducible transcripts (lower panel) does not change significantly. Patient #5 did not display granulopoiesis signature before high dose GC therapy. Original figure published in J. Exp. Med. 2003 Mar 17;197(6):711-23
A surprise from these initial studies was the absence of type I IFN gene transcripts in the face of an abundance of IFN-inducible ones in SLE PBMCs. A likely explanation is that the cells producing type-I IFN, and therefore transcribing these genes, migrate to sites of injury. Indeed, IFN-inducible genes and/or proteins can be detected in lupus kidney (89), skin (73) and synovial biopsies from SLE patients (90). These tissues are all infiltrated with pDCs (72, 91), further supporting them as being the source of type I IFN in SLE. Increased levels of Flt3L, a pDC growth factor, in SLE serum (92) suggest that this cytokine might contribute to the increased levels of these cells at the sites of inflammation.
Type I IFN and Murine Lupus
A number of murine lupus models have been available for decades (2). Yet, they did not predict that type-I Interferon might be an important mediator of human SLE. However, in line with the human studies, two independent groups showed that the crossing of NZB and B6 lpr/lpr mice respectively with a type I IFN receptor KO strain significantly decreased morbidity and prolonged the survival of these animals (93, 94). Additionally, delivering type I IFN to young (6-8 weeks old) preautoimmune NZB/W F(1) mice resulted in the rapid development of severe SLE, possibly explaining the deleterious effect of poly I:C reported decades ago (95). Conversely, administration of a type I IFN immunogen that induced transient specific neutralizing antibodies (Abs) delayed/prevented lupus manifestations, including proteinuria, histological renal lesions and death in these mice (96).
Introduction of the Yaa locus into lupus-prone mice accelerates the development of autoimmune manifestations (97). The Yaa phenotype results from a duplication of the TLR7 gene (98, 99). Engagement of TLR7, which can be triggered by viruses or immune complexes containing RNA and by RNA-associated autoantigens, induces pDCs to secrete type-I IFN (100-102). B cells, which also express this receptor, respond to the same triggers with activation and pro-inflammatory cytokine production (103). A role for TLR7 in the induction of autoimmunity is further demonstrated in transgenic mice, which spontaneously develop autoimmunity. Whereas a modest increase in TLR7 gene dosage promotes autoreactive lymphocytes with RNA specificities and myeloid cell proliferation, a substantial increase in TLR7 expression causes fatal acute inflammatory pathology and profound dendritic cell dysregulation (104). Using the Unc93b1 3d mutation that selectively abolishes nucleic acid-binding TLR3, -7, and -9 signaling, endosomal TLR signaling has been shown to be required for optimal production of IgG autoAbs, IgM rheumatoid factor, and other clinical parameters of disease in lupus-prone mice as well (105). Although there is currently no evidence that TLR7 mutations contribute to human SLE, polymorphisms in TLR8 have been recently reported associated to SLE in pediatric and adult populations (106).
Genome in the Footsteps of Transcriptome Analyses
Traditional genetic approaches had not pointed out type I IFN to be a contributor to lupus pathogenesis. Instead, the HLA locus, Fcγ receptors, complement components etc. were among the candidates to explain disease susceptibility (82). Nevertheless, the evidence accumulated through some of the studies described above led geneticists to perform a “genome-focused” search for polymorphisms in 13 Type I IFN-related genes (107). This study revealed a strong association between SLE and certain variants of the interferon regulatory factor 5 (IRF5), which has since been confirmed in several genome-wide association (GWA) studies (108, 109). Indeed, the IRF5 SLE risk haplotype is associated with higher serum type I IFN activity in patients (110). A correlation between serum IFN-alpha activity in patients and their first degree healthy relatives has also been described (111). In addition to IRF5, polymorphisms in other genes connecting with the IFN signaling pathway, such as STAT4 and IRAK 1, have emerged as conferring susceptibility to SLE (112-114). More recently, mutations in the human TREX1 gene, which cause Aicardi-Goutieres syndrome (AGS) and chilblain lupus (115), have also been linked to SLE (116). TREX1 is a negative regulator of the IFN stimulatory DNA response, and single-stranded DNA derived from endogenous retroelements accumulates in Trex1-deficient cells (117). The persistent presence of dsDNA from dying cells could also lead to an aberrant immune response in patients carrying Trex 1 mutations (118). Finally, polymorphisms in the autophagy-related gene ATG5 have been described in SLE (109). ATG deficient pDCs seem to be less able to produce type I IFN in response to certain viruses (119). The specific mechanisms through which these lupus-related polymorphisms affect IFN production in response to environmental and/or endogenous triggers remain for the most part to be elucidated.
The IFN pathway is not the only genetic pathway predisposing to SLE. Genes within the HLA locus, TLR signaling, B cell and myeloid lineage-specific, FcRs etc. have been confirmed to be associated with disease. All together, however, IFN-related genetic alterations rank among the highest, closely behind the HLA locus, in the lupus susceptibility scale [reviewed in (120)] (Figure 4).
Figure 4. A unified view of SLE pathogenesis.

Left: HLA and non-HLA gene polymorphisms contribute to SLE susceptibility. Among them, TLR/IFN signaling pathway-related genes have recently been described. Right: Both environmental triggers (i.e. viral infections) and chromatin-containing immune complexes might contribute to the unabated production of type I IFN by pDCs in SLE patients. Increased bioavailability of type I IFN induces and maintains the generation of mature DCs, tilting the fate of autoreactive T lymphocytes which have escaped central tolerance from deletion to activation. These mature DCs activate cytotoxic CD8+ T cells to generate nucleosomes which can be captured and presented by IFN-DCs. Together with IL-6, type I IFN promotes the differentiation of mature B cells into plasma cells, which secrete autoantibodies. A direct effect of IFN-alpha on endothelial cells could also contribute to premature atherosclerosis in SLE patients.
Drug-induced Lupus and Type I IFN
Microarray analyses of PBMCs have also helped us shed some light on the pathogenesis of drug-induced lupus. Upon treatment with TNF antagonists, up to 20% of patients with rheumatoid arthritis (RA) and Crohn's disease develop anti-dsDNA antibodies, and a fraction of them even progress to reversible SLE [reviewed in (70)]. In line with those clinical observations, the PBMCs of juvenile arthritis patients treated with anti-TNF antibodies display an overt IFN signature. In vitro studies revealed that addition of TNF antagonists induced pDCs exposed to viruses to secrete higher levels of Type I IFN (121). This is best explained by the inhibition of TNF-dependent pDC maturation, which normally shuts down Type I IFN secretion. Further studies will be however necessary to understand if this mechanism extends to other drugs, such as hydralazine, anti-epileptic medications etc., that can induce lupus-like syndromes.
Non IFN-related signatures in SLE blood
Type I IFN-inducible transcripts represent only a fraction of the transcriptional alterations observed in SLE patient PBMCs (21, 22). A different set of signatures reflect changes in blood cell composition. Thus, the second most prevalent signature in our study (21) consisted of genes expressed by granulocytes at varying stages of development (e.g. elastase, MPO, and bactericidal proteins such as defensins). This finding was surprising, as Ficoll-purified PBMCs, which are normally freed from neutrophils, were the source of RNA for these arrays. However, phenotypic examination of the isolated PBMCs revealed a large population of low density neutrophils in the mononuclear cell fraction from patients but not from controls. The expression of neutrophil-specific transcripts (e.g. the defensin DEF3) correlated with the frequency of low density neutrophils in the patients' samples (Figure 5). Next, and as expected, the well known lupus-associated lymphopenia resulted in a relative decrease in B cell, T cell and NK cell-specific transcripts (e.g. TCRα/β, Lck, and CD3γ). Transcripts encoding variable and constant regions of immunoglobulins (Ig) together with plasma cell-lineage markers were increased in a number of patients, a finding correlating with the expansion of CD19low CD20-CD38++ CD27+/++plasma cell precursors (PCPs) (21). These cells, which are oligoclonal and highly proliferative, are not specific to SLE but can be detected in situations where there is immune activation, i.e. vaccinations, infections, and in other autoimmune diseases (122-124). Although Type I IFN induces in vitro the differentiation of memory B cells into PCPs very similar to those found in SLE patients (77), the IFN and plasma cell precursor signatures do not always correlate in individual patients. This discordance might be explained by the fact that some of the medications used to treat SLE, such as mycophenolate mofetil, target proliferative cells and suppress the PCP signature while having little effects on the overall IFN signature (Xu et al. unpublished data).
Figure 5. A non-type I IFN induced signature in SLE PBMCs (Granulopoiesis signature).

a) Genes are divided into three categories: enzymes and their inhibitors, bactericidal proteins and others. Median expression and the number of patients who display more than 2-fold increase (red) in gene expression. ** Significant after Bonferroni correction, * significant after Benjamini and Hochberg correction. b) presence of granular cells in leukocytes that display granulopoiesis – related RNA. Flow cytometry analysis (forward scatter vs. side scatter) of Ficoll-separated mononuclear cells. The gated cells are immature neutrophils. c) Correlation between the defensin alpha (DEF3) levels and the numbers of cells gated as shown in b.
Proof of Concept: clinical trials with IFNα antagonists in human SLE
A phase Ia trial to evaluate the safety, pharmacokinetics, and immunogenicity of an anti-IFNα monoclonal antibody (mAb) therapy in adult SLE patients was recently conducted (125). The antibody elicited a specific and dose-dependent inhibition of expression of type I IFN-inducible genes in both whole blood and skin lesions from SLE patients, at both the transcript and the protein levels. As expected, overexpression of BLyS/BAFF, a type I IFN inducible gene, decreased as well with treatment. Interestingly however, TNFα, IL-10, IL-1B, GM-CSF, and their respective downstream signatures were also suppressed upon treatment with anti-IFNα mAb (125). These data suggest that, beyond the IFN signature, many of the transcriptional alterations detected in SLE might be connected to the type I IFN pathway. Thus, this first trial supports the proposed central role of type I IFN in human SLE.
Juvenile Idiopathic Arthritis (JIA)
Box 3.
JIA is the most common rheumatic disease in childhood and an important cause of short and long-term disability. The term JIA includes a heterogeneous group of diseases characterized by the development of chronic peripheral arthritis starting in the first 16 years of life. JIA is classified according to three major types of disease presentation: oligoarthritis, polyarthritis and systemic onset (SoJIA). Each of these groups is defined by a constellation of clinical signs and symptoms during the first six months of illness. Systemic Onset Juvenile Idiopathic Arthritis (SoJIA) represents up to 20% of all the cases of JIA. This disease is unique in terms of clinical manifestations, prognosis and lack of response to conventional therapies. High spiking fever, a salmon-color rash that follows the fever spikes, anemia, leukocytosis and elevated erythrocyte sedimentation rate (ESR) are the main initial features of the disease, sometimes lasting several months before the diagnosis can be established at the time arthritis appears. Most SoJIA patients lack classical autoantibodies. As the symptoms are non specific and found in other diseases such as infections and malignancies, patients undergo a series of very costly diagnostic tests and prolonged hospitalizations. The systemic manifestations may last from weeks to months, and eventually subside, to be followed by chronic arthritis. About 50% of patients will present oligoarticular involvement and will recover. The other half will evolve into a polyarticular pattern. Almost half of the children with SoJIA will have active arthritis 10 years after diagnosis is made. These patients display an increased risk of developing hemophagocytic syndrome, also known as macrophage activation syndrome (MAS), a disorder associated with serious morbidity and/or death.
Systemic onset JIA (SoJIA) is one of the most challenging pediatric rheumatic diseases. Its pathogenesis had been for years linked to pro-inflammatory cytokines, as elevated serum levels of IL-6 and TNF had been found in active patients (126, 127). IL6 antagonists however are not yet available in the USA, and TNF antagonists did not prove to be efficacious in treating these patients. To try to understand SoJIA pathogenesis, we developed two complementary approaches based on microarray technology: 1) analysis of transcriptional patterns in active patient PBMCs (25); 2) analysis of the transcriptional changes induced by serum from active patients in healthy PBMCs (19). The development of biomarkers for disease diagnosis and patient follow up will be described in a later section of this review.
Transcriptional analysis of SoJIA PBMCs revealed alterations in the expression of >800 transcripts. The majority of these changes, however, were also found in patients suffering from systemic inflammation caused by bacterial and viral infections and did not point towards any specific molecular pathway (25). The clue came from in vitro experiments designed to test the effect of active patient sera on the transcriptional profile of healthy PBMCs. These “reverse proteomics” experiments revealed that SoJIA serum up-regulates the transcription and secretion of IL-1B in healthy leukocytes (Figure 6). SoJIA serum also upregulates the expression of genes known to be induced by IL-1b and/or involved in innate immunity pathways (19).
Figure 6. Gene expression profiling led to the identification of IL-1 b in the pathogenesis of SoJIA.

(A) Incubation of healthy PBMCs with autologous sera (AS) or sera from four patients with active SoJIA. (B) Transcriptional changes induced by SoJIA sera included the up-regulation of IL1b and its receptors. (C) Treatment with daily injections of anakinra induced the resolution of systemic symptoms (fever) and arthritis in 9/9 and 7/9 patients respectively (adapted from Curr Opin Immunol. 2007 Dec;19(6):623-32).
This unexpected finding led us to consider the role of IL1B in disease pathogenesis. An IL1 receptor antagonist (anakinra) had been developed to treat patients with rheumatoid arthritis (RA) but had shown limited clinical efficacy [reviewed in (4)]. We thus treated two SoJIA patients who were refractory to all conventional therapies and observed remarkable clinical and hematological responses. Fever and rash disappeared within 48 hr. Laboratory parameters of inflammation soon followed and, most importantly, refractory arthritis responded within a few weeks to anakinra treatment. Seven additional patients were similarly treated and 5/7 quickly went into complete remission (19) (Figure 6). Since then, 30 patients have been treated in our clinic with 72% of them having attained full and long-lasting remission (our own unpublished data). The efficacy of anakinra to treat SoJIA patients has been recently confirmed in a randomized clinical trial (Quartier et al, submitted). Rapid and sustained responses to IL1B blockade have also been described in patients with refractory adult-onset Still's disease (128), which is the adult equivalent of SoJIA. These patients display a blood microarray signature almost identical to that of SoJIA patients (our own unpublished data).
The origin of dysregulated IL1B production in SoJIA remains unknown. A common infectious or inflammatory trigger could lead to an excessive production of IL1 in patients with underlying mutations in genes controlling the regulation of this cytokine. In favor of this hypothesis, non-specific activation of PBMCs from a group of SoJIA patients in vitro results in excessive IL1B secretion (19). Since IL1B can upregulate its own transcription, IL1B itself could also be responsible for the serum effects described above. This cytokine, however, is difficult to detect in the serum because it binds to large proteins such as b-2-macroglobulin, complement, and the soluble type II IL1 receptor [reviewed in (129)]. Alternatively, some members of the S100A family (S100A8 and S100A9) have been described elevated in SoJIA serum and postulated to induce IL1B secretion (130, 131).
Thus, simple experiments performed with active patient sera helped focus our attention on the IL1 pathway. This pathway had been obscured by the many transcriptional alterations found in the blood of patients.
Other Rheumatic Diseases
Sjogren's Syndrome (SS)
SS is an autoimmune disease characterized by lymphocytic infiltration and progressive functional disruption of lacrimal and salivary glands. Extraglandular manifestations, however, are also common. The production of autoantibodies is a prominent feature, as antinuclear antibodies are found in about 80% of patients, and anti-SSA/Ro and anti-SSB/La autoantibodies in 65% and 40% respectively. While the etiology of Sjögren's syndrome is unknown, a genetic predisposition has long been recognized. Indeed, the major polymorphisms in Type I IFN-related genes (IRF5 and STAT4) found associated with SLE are also associated with primary SS patients (132, 133). Transcriptome analyses performed on blood (35), salivary gland tissue (134) and even saliva (135) of patients have also revealed an upregulation of IFN-inducible genes remarkably similar to those described in SLE.
Inflammatory Myopathies
Box 4.
The inflammatory myopathies include dermatomyositis (DM), polymyositis (PM) and inclusion body myositis (IBM). DM muscle pathology is quite distinct, with perivascular inflammatory cells and abnormal capillaries and perimysial perifascicular myofibers. Both PM and IBM have larger infiltrates of inflammatory cells into muscle that surround, displace, and even invade the myofibers. DM and PM are treated with corticosteroids and non specific immunosuppressive drugs. IBM is refractory to these therapies. A challenge common to these diseases is the lack of simple and specific tests, other than muscle biopsy and magnetic resonance imaging, for the diagnosis and disease activity assessment of patients.
Gene expression profiling of muscle and skin from patients with inflammatory myopathies have revealed unexpected signatures and led to the identification of previously unrecognized cell types and signaling pathways in involved tissues. The vast majority of upregulated transcripts in samples from dermatomyositis (DM) and polymyositis (PM) patients are type I interferon inducible [reviewed in (136)]. Indeed, pDCs account for most of the CD4+ cells present in DM muscle (41). These cells, which had been previously interpreted as T-helper cells (137), are also present in DM skin (138). Juvenile DM (JDM), the most common pediatric inflammatory myopathy, is strongly associated with HLA class II region genes (139). A type I interferon signature is also found in JDM muscle biopsies (140). Microarray analyses of PM and Inclusion Body Myositis (IBM) muscle revealed an overexpression of immunoglobulin gene transcripts. Consequently, abundant CD20-CD138+ plasma cells were identified by immunohistochemistry as the source of this signature (41).
Blood samples from JDM, DM and PM patients reflect the main signatures found in muscle with a distinct overexpression of type I IFN-inducible genes. Accordingly, clinical improvement during immunosuppressive treatment is generally associated with the disappearance of this signature (42, 141). Thus, microarray analyses have revealed new molecular and cellular alterations in inflammatory myopathies. These might enable the identification of novel therapeutic targets and open the door for biomarker development in these diseases. The prominent IFN signature in DM and PM patients advocates for the conduction of clinical trials with type I IFN antagonists.
Psoriasis and Psoriatic Arthritis (PsA)
Psoriasis vulgaris is a chronic inflammatory condition characterized by hyperproliferation of keratinocytes, dermal infiltration of activated CD4+ T cells, and lesional production of pro-inflammatory cytokines (142). The disease has a strong genetic component and inflammatory arthritis develops in as many as 30% of patients. Microarray studies revealed signatures corresponding to cytokines and chemokines within involved skin, and markers of dendritic cell (DC) activation in uninvolved skin. Several members of the IL-1 cytokine family as well as IFN-inducible genes were upregulated in psoriatic lesions of patients (143-146).
PBMC gene expression profiling of PsA patients identified a signature that differentiated these patients from healthy individuals and RA patients. Downregulated transcripts clustered to certain chromosomal regions, including those containing the psoriasis susceptibility loci PSORS1 and PSORS2. Genes involved in downregulation or suppression of innate and acquired immune responses were most downregulated, possibly explaining the pro-inflammatory responses associated with this disease. One gene, nucleoporin 62 kDa, could correctly classify all controls and 94.7% of the PsA patients. The combination of two genes, MAP3K3 and CACNA1S, correctly classified all RA and PsA patients (40). Larger studies are necessary to confirm these tantalizing data. This is particularly relevant to PsA, as diagnosing early and/or predicting who will develop this complication might lead to early therapeutic interventions, thus preventing the development of erosive arthritis.
Rheumatoid Arthritis (RA)
RA is a systemic autoimmune disorder characterized by chronic inflammation and destruction of bone and cartilage in diarthrodial joints. There is clearly a need for improved clinical laboratory evaluation and classification of patients with RA, as diagnosis is based on clinical observations and a combination of serological markers (i.e. rheumatoid factor and anti-cyclic citrullinated peptide antibodies) which are not present in every patient [reviewed in (147)]. Furthermore, although effective treatments are now available, their efficacy is not universal. Indeed, more that 1/3 of RA patients do not respond to current biological agents, including TNF antagonists, and there are no available biomarkers to predict response to therapy.
Gene expression profiling has been used to study both synovial tissues of affected joints and peripheral blood. Initial studies comparing RA synovial tissue to osteoarthritis (OA) revealed upregulation in RA of transcripts specific to antigen-presenting cells, T-, and B-cell genes (148). STAT1-inducible genes were found upregulated in another study (149), suggesting the involvement of the IFN pathway in the disease process of at least a subgroup of patients. Synovial tissue microarrays have also been used to predict responsiveness to anti-TNF therapy in a limited number of patients (150). Overall, patients with high level of tissue inflammation were more likely to benefit from anti-TNF treatment.
The transcriptome analysis of RA PBMCs revealed a signature significantly correlated with the percentage of circulating monocytes, in line with the known monocytosis and monocyte activation often seen in RA patients (34). Other studies support the results described in synovial tissue and reveal the presence of a type I IFN signature in a subgroup of patients (32). More studies will be necessary to determine whether this information might help understand the heterogeneity of this disease and better classify and/or predict treatment success in subsets of RA patients.
Systemic Sclerosis (SSc)
SSc is a clinically heterogeneous disease characterized by vascular dysfunction, tissue fibrosis and internal organ damage. Initial symptoms are difficult to recognize and patients are often diagnosed late in the disease process, when irreversible damage has occurred. Anti-nuclear antibodies are present in most patients. In a significant number of them, however, these antibodies do not display specificities that help with the diagnosis. There are no effective therapies for this disease (151).
Skin biopsies from patients with early diffuse SSc show nearly identical patterns of gene expression at clinically affected and unaffected sites. Upregulation of genes from endothelial cells, B lymphocytes, fibroblasts, cytotoxic T cells, macrophages and DCs have been described. Identified gene pathways include those of TGFβ, and a “cell proliferation signature” (152). Patient PBMCs showed increased expression of a cluster of IFN-regulated genes (43, 153), including Siglec-1 (CD169, sialoadhesin), which was confirmed by finding increased Siglec-1 surface expression on CD14+ monocytes (44). Risk alleles in the IRF5 and STAT4 genes have been reported in SSc patients (154). These observations indicate a potential role for type I IFN and call for testing IFN antagonists in this disease.
Other Autoimmune Diseases
Multiple Sclerosis (MS)
MS is a chronic autoimmune disease characterized by demyelination of the CNS white matter (155). Microarray studies in MS have focused on identifying gene expression differences in CNS lesions of patients to assess pathogenesis, and in blood to assess response to treatment. Genes showing differential expression in MS lesions include genes involved in immunological and inflammatory pathways, stress-response and antioxidant processes, as well as metabolic and central nervous system markers (156, 157). Of particular interest are a number of genes localized to susceptible loci previously shown to be in linkage with MS (158). Transcriptome analyses of purified blood T cells from MS patients showed altered expression of genes involved in apoptosis (159). Overall, due to the heterogeneity of the samples and the small scale of these expression studies, it is still difficult to define which of the identified gene pathways are directly associated to the pathogenesis of this disease.
As IFNβ represents an effective therapy in MS, attempts have been made to identify predictors of response to therapy using blood transcriptome analyses. As expected, IFN-regulated transcripts could be identified in treated patients (160-162). Larger studies will be necessary to establish definitive gene expression profiles that will be predictive of disease onset, flare, or response to therapy.
Diabetes
Following the identification of a major signature in SLE, we wondered whether an organ-specific autoimmune disease such as diabetes would be associated to alterations in the blood transcriptome. We even hypothesized that these patients might display a type I IFN signature because of previous reports of elevated IFNα in pancreatic tissue from patients (163). Thus, expression profiles of PBMCs from children with newly diagnosed Type I diabetes (T1D) were compared to those from healthy controls, and 1 and 4 month follow-up samples were obtained from a subgroup of patients. PBMCs from children with new onset Type 2 diabetes (T2D) were also analyzed (36). These studies identified changes in expression of IL1B, early growth response gene 3, and prostaglandin-endoperoxide synthase 2, which resolved within 4 months of insulin therapy in both T1D and T2D. IL1B was highly overexpressed in both diseases, suggesting that they likely share a final common pathway for beta-cell dysfunction that includes secretion of IL1B, which exacerbates existing beta-cell dysfunction and causes further hyperglycemia.
The presence of proinflammatory factors in serum of T1D patients has also been investigated using the genomic strategy of “reverse proteomics” described above for SoJIA patients (Hessner, 2008, JI). Thus, sera from recent onset diabetes patients, long-standing diabetes patients, “at risk” siblings of diabetes patients, and healthy controls were tested for the induction of gene expression patterns in unrelated, healthy PBMC (20). All recent onset sera induced an expression signature that included IL-1 cytokine family members and chemokines involved in monocyte/macrophage and neutrophil chemotaxis. This molecular signature was not induced with the sera of healthy controls or long standing diabetes patients. Furthermore, longitudinal analyses of “at risk” siblings before and after onset support the hypothesis that the signature emerges years before onset.
Overall, these results identify IL1B as a target for disease-modifying therapy of diabetes. Indeed, clinical trials using IL1 antagonists have already proven some efficacy in adults with T2D (164, 165) and are underway for patients with T1D. Additionally, these studies support that proinflammatory serum markers may be used as inclusion criteria or endpoint measures in clinical trials aimed at preventing T1DM.
Blood Microarrays and Biomarker Discovery
In addition to the identification of disease signatures that may yield critical insight regarding underlying immune dysfunctions and identify potential therapeutic targets in autoimmune diseases, blood transcriptional studies have been used to identify biomarkers for diagnosis, monitoring of disease activity and prediction of clinical outcomes. We have applied this approach to SoJIA and SLE because there are no available diagnostic tests or disease activity measures (SoJIA) or because current measures are too burdensome to apply in the clinical setting (SLE). In order to select specific biomarkers, we had to devise in both cases novel analysis strategies.
Analysis of Significance Patterns
The diagnosis of SoJIA takes weeks to months, as it is based on clinical criteria which lack specificity (166). Indeed, initial symptoms mimic infections or malignancies and it is only when arthritis appears that the disease can be recognized. We surmised that the blood transcriptome of these children could be a source of diagnostic biomarkers. The profiles that differentiate SoJIA patients from healthy controls, however, were highly similar to those of children with febrile infectious diseases of both bacterial and viral origin (25). Because SoJIA can present at any age during childhood, matching the control groups for this disease and for infections that predominate at earlier or later times during childhood was however a challenge. Thus, we devised a custom meta-analysis strategy for biomarker selection relying on the analysis of patterns of significance (Box 5) (167). This approach can be used to compare diseases across multiple datasets, each being analyzed in relation to its own set of healthy controls. First, statistical comparisons were performed between each group of patients (SoJIA, S. aureus, S. pneumoniae, E. coli, influenza A, and SLE) and their respective control groups composed of age- and gender-matched healthy donors. The P-values obtained from each comparison were then subjected to selection criteria. This permitted us to identify genes significantly changed in SoJIA patients versus their control group, and not in any of the other disease versus their own control groups. The SoJIA-specific signature that we obtained using this algorithm was composed of 88 transcripts (Figure 7). Treatment with IL1 antagonists was able to extinguish this signature in the majority of patients. Using a more stringent analysis, 12/88 transcripts seemed to be enough to correctly classify an independent group of patients during the systemic phase of the disease against healthy and febrile disease controls. These 12 genes were not dysregulated however in SoJIA patients who had resolved the systemic phase and were left with chronic arthritis, suggesting that they are specifically dysregulated in the initial phase of the disease, probably the time of greatest sensitivity to IL1 blockade. The specificity of 7/12 genes has been recently validated in an independent study of PBMC transcriptional profiles including different types of JIA patients (28). The same type of analysis has allowed us to identify blood disease-specific transcriptional markers differentiating SLE patients from patients with diseases that also display a Type I IFN signature such as Influenza infection (167).
Box 5. Analysis of Significance Patterns (25, 167).
The strategy is based on the comparison of P-values obtained by generating gene expression data from well defined groups of patients and their corresponding healthy controls, which are well matched for age, sex, ethnicity etc. Genes significantly changed (P < .01) in the “study group” are divided in two sets: overexpressed versus control and underexpressed versus control. P-Values of the genes forming the overexpressed set are obtained for the “reference groups”. P-Values of the reference groups are set to 1 when genes are underexpressed. The same procedure is used in the set of genes underexpressed in the study group, only this time P-values of the reference group are set to 1 when genes are overexpressed. P-Value data are then processed to perform hierarchical clustering and groups of genes based on significance patterns.
Figure 7. Analysis of significance across diseases identifies 88 SoJIA-specific transcripts.

(A) Eight healthy and eight SoJIA samples were used as training set to generate a list of 50 classifier genes displaying the best ability to discriminate SoJIA patients from healthy controls. Those classifier genes were hierarchically clustered in a test set composed of 35 healthy controls, 16 SoJIA, 31 S. aureus, 12 S. pneumoniae, 31 E. coli, 18 Influenza A and 38 SLE patients. (B) Genes expressed at statistically different levels in SoJIA patients compared to healthy volunteers (p<0.01, Wilcoxon-Mann-Whitney test) were selected (4311 probe sets). Out of those, 88 were found expressed at statistically different levels in SoJIA patients compared to healthy volunteers (p<0.01, Wilcoxon-Mann-Whitney test) but not in all the other groups (p>0.5, Wilcoxon-Mann-Whitney test). The 88 genes are hierarchically clustered in the 107 samples from different disease groups used in (A). Expression values or the genes are normalized per-gene to the healthy group (Published in Curr Opin Immunol. 2007 Dec;19(6):623-32)
Modular Analysis Framework for Blood Genomic Studies
The exploitation of large-scale data can be hindered by our limited ability to separate signal from noise and to grasp the overall biological significance of the results. Numerous approaches have been developed (168-171), including our modular analysis framework (172) that reduces the dimension of blood transcriptional data in order: a) to facilitate functional interpretation; b) enable comparative analyses across multiple datasets and diseases; c) minimize noise and improve robustness of biomarker signatures; and d) derive multivariate metrics that can be used at the bedside. The steps involved in the development of this framework are depicted in Box 6.
Box 6. Module-based analysis of gene expression(172).
Transcriptionally co-regulated transcripts emerging from the blood profiles of patients with 8 different diseases were identified using a clustering algorithm. Once patterns were identified for each disease, the cluster membership of individual transcripts was compared across all other diseases. A module is formed of transcripts found to always belong to the same clusters across all diseases (8 out of 8 in our example). The stringency of this requirement was progressively relaxed during the subsequent rounds of selection so that modules are formed when transcripts fall in the same clusters in any combination of 7 (round 2) or combination of 6 diseases (round 3). This stepwise reduction of the stringency of filtering criteria accounts for the fact that transcripts may not be “turned on” in all diseases. Indeed, modules linked to interferon or inflammation (M3.1 and M3.2) were for instance not formed until the third round of selection. As should be expected of transcriptional modules, literature profiling of genes forming each one of them revealed significant functional convergence, with half of the modules associated with clearly identifiable functional themes (Figure 8). By including profiles from a wide range of diseases, we identified a “universal” set of modules that could be used as a stable framework for subsequent analysis of any PBMC dataset.
Once the framework has been established, transcriptional changes can then be mapped for individual diseases on a modular basis (Figure 8). Briefly, differences in expression levels between study groups are displayed on a grid. Each position on the grid is assigned to a given module; a red spot indicates an increase and a blue spot a decrease in transcript abundance. The spot intensity is determined by the proportion of transcripts reaching significance for a given module. A posteriori, biological interpretation has linked several modules to immune cells or pathways as indicated by a color code on the figure legend (Figure 8).
Figure 8. Modular fingerprints of human immune-mediated diseases.

A. Expression levels of 4 groups of transcriptionally co-regulated genes in PBMCs of healthy children and children with SLE are shown in the upper panel. Transcripts within modules 2.2 (neutrophil-related transcripts) and 3.1 (type I IFN-inducible transcripts) are upregulated while 2.4 (ribosomal protein-encoding transcripts) and 2.8 (T cell –related transcripts) are downregulated in the majority of SLE patients. These differentially expressed modules and 7 additional ones are represented on a greed as red (over expressed) or blue (under expressed) circles. The intensity of the colour reflects the percentage of transcripts within each module that are significantly differentially expressed. Statistical comparisons between patient and healthy control groups were performed independently on a module-by-module basis (Mann-Whitney rank test, p<0.05). B. Modular analysis of PBMC differential gene expression in patients with Melanoma, liver transplant under immunosuppression, and S. pneumoniae infection. The colour code of modules containing transcripts with annotated function is depicted in the lower right quadrant. (Adapted from Immunity. 2008 July 18; 29(1): 150–164).
We have explored the use of transcriptional modules as a basis for biomarker discovery in SLE (172), a disease that presents with a wide range of clinical and laboratory abnormalities. Objectively assessing disease activity across patients or longitudinally in individual patients can therefore be challenging. At least 6 composite measures of SLE global disease activity have been developed and have been used to assess disease progression during clinical trials (173). These measures, however, rely on many clinical and laboratory findings and are cumbersome to obtain. Additionally, given the heterogeneous nature of the clinical disease, not all SLE manifestations are computed within these measures, making the overall assessment of the patient sometimes difficult. The SLEDAI (174), one of the simplest measures, considers 24 different attributes that need to be obtained at every clinic visit. Thus, establishment of a simple and objective disease activity index would be beneficial. We engaged to assess whether such activity index could be generated from blood leukocyte microarray transcriptional data applying the modular interpretation described above. The idea was that this approach would permit us of focus on biologically relevant transcripts while reducing the dimension of microarray data from over 44,000 variables to about 5000 distributed in 28 modules. Comparisons carried out on a module-by-module basis identified 11 sub-modules with a minimum of 15% of transcripts over- or under-expressed compared to healthy children. We next transformed the expression values of the transcripts within these modules into 11 composite values (or expression vectors) and summarized the results as one single multivariate score. These multivariate transcriptional scores were correlated to clinical disease activity indices in both cross-sectional and longitudinal sets of samples. Indeed, upon longitudinal follow up of patients, severity of disease was in some cases more accurately assessed, and even predicted, by the transcriptional score than by the SLEDAI. A striking example is represented in Figure 9, which displays the discordance between a very low SLEDAI, suggestive of low disease activity, and a high transcriptional score in a 7 year old patient with SLE (SLE 78). This patient was subsequently diagnosed with severe pulmonary hypertension, an uncommon but serious complication of SLE that is not recorded within the classic SLEDAI. Although this approach needs to be validated in large multi-centric studies, it might prove useful for the discovery of diagnostic/prognostic markers and the monitoring of disease progression and response to treatment in other diseases.
Figure 9. Development of transcriptome-based SLE disease activity biomarkers.
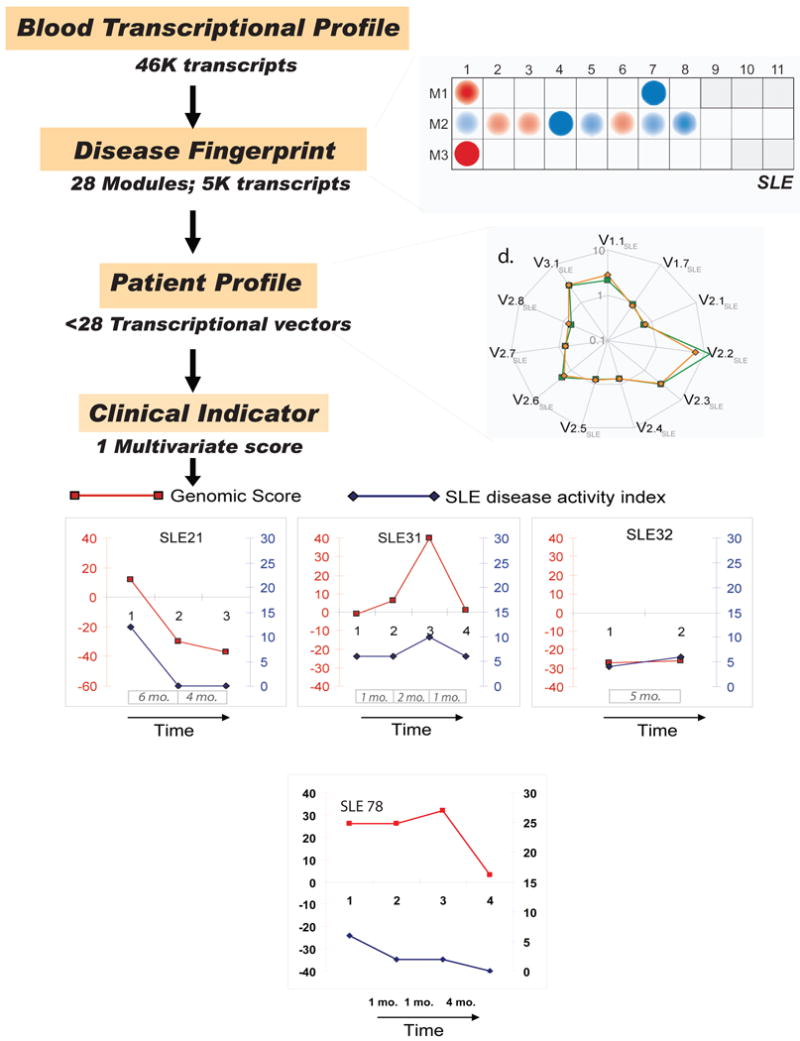
Starting from a full set of 28 modules, 11 modules for which a minimum proportion of transcripts (>15%) are significantly changed (p<0.05) between the study groups (SLE and healthy) are selected. Next, composite values for each sample are generated by calculating the arithmetic average of normalized expression values across significantly over-expressed or under-expressed genes selected from each module. Each resulting “transcriptional vector” recapitulates the expression of a given module (or select set of genes within a module) in a given patient. A spider graph connecting all the vector values in untreated (average in orange) and treated (average in green) patients is shown. The values are normalized per-gene using the median expression value of healthy and are represented on a logarithmic scale. A non-parametric method for analyzing multivariate ordinal data was then used to score the patients based on 5/11 vectors that best correlated individually with SLE disease activity according to the SLEDAI. The correlation achieved by this score was superior to that of its individual components. Upon longitudinal follow-up of patients, parallel trends were observed between transcriptional scores and SLEDAI longitudinal measures in a majority of patients. Disease flaring and subsequent recovery was detected in one patient (SLE31) upon longitudinal follow up using both SLEDAI and transcriptional score. Interestingly, however, the amplitude of change observed in the case of the transcriptional U-score appears not only to be much greater (0 to 40 vs. 6 to 10 for SLEDAI), but an increase could already be detected at the second time point, 2 months before the worsening of the clinical condition of this patient was detected by SLEDAI. One of the patients (SLE78) showing discrepancy between the SLEDAI (low) and transcriptional score (high) was diagnosed during the follow-up period with a life-threatening complication (pulmonary hypertension) which is not computed within the SLEDAI. Thus, severity of disease was more accurately assessed by the transcriptional score.
Challenges in Transcriptome-Based Biomarker Discovery
To date, few diseases besides cancer have received the level of attention required to provide robust assays that could be used in clinical practice. Most published data on autoimmune diseases, including our own, have resulted from proof of principle studies that need to be validated. Inter-individual variability, whether driven by genetic or environmental factors, and inherent disease heterogeneity require these studies to be performed on large datasets and in multiple centers. Indeed, progress in this complex field is more likely to be achieved through concerted, focused consortium-type efforts.
Disease heterogeneity is an important initial limiting step in autoimmune disease biomarker discovery. Thus, patient clinical characteristics and disease stages should be taken into account and carefully recorded at the time of selecting the samples to study. The importance of implementation of common methods and standards for the collection of this type of information, samples and measurements cannot be stressed enough (175, 176).
Drug treatments and co-morbidities may impact blood transcriptional signatures and those variables cannot always be isolated, as patients cannot be taken off treatments. These factors also pose significant challenges in terms of study design and downstream data analysis. We have found that including samples from recently diagnosed and untreated patients is useful when selecting biomarkers related to disease pathogenesis. Selective inclusion criteria can be subsequently relaxed to broaden the scope and potential clinical impact of a study. Working with pediatric populations is attractive because it is easier to accrue patients in the initial phases of disease and because significantly less co-morbidities and modalities of treatments will be encountered.
Sample collection / Logistics
High quality RNA can be easily isolated from PBMCs. However, cell isolation procedures represent a well-documented source of variability (176, 177). As a result, most transcriptional studies are now carried out using whole blood. Indeed it has become possible to preserve intact RNA from small volumes of blood collected in tubes under vacuum that can be stored frozen for extended periods of time without the need for further processing (178, 179). A drawback of using whole blood as a source of RNA is its high content in globin transcripts. Although globin reduction techniques increase sensitivity and data reproducibility (178-180), there is some controversy regarding whether globin removal may interfere with biomarker discovery (179, 181).
Sensitivity
The sensitivity of microarray-based technologies is relatively low when compared to real-time PCR. Furthermore, it is important to keep in mind that the measurement of transcript abundance using microarrays is relative rather than absolute. As a result, data may not be directly compared due to systematic variations introduced for instance between manufacturing, sample, or array processing batches. This fact has implications for data reproducibility as well.
Reproducibility
Standardization efforts have been found to improve comparability, but the reproducibility of microarray data remains a legitimate concern (182-184). Indeed no standards have been adopted by the community and, given the need to keep up with the rapid technology developments that characterize this field, they would be difficult to implement. It is likely, for instance, that microarray technologies will be superseded in the near future by more robust digital expression technologies (discussed below), therefore obviating efforts to standardize microarray-based transcriptome analyses.
Interpretation
The factors affecting data reproducibility across microarray platforms and laboratories can also impact the quality or interpretability of a dataset. It is important to avoid confounding the analysis with technical variables. For example, reuse of pre-existing data for direct group comparison should be avoided. Samples should be run if possible in one single batch. If this is not possible, case and control samples should be randomized across the different runs. Overall, the availability of cost-effective commercial platforms has reduced the number of formats used for analysis and contributes to enhance data quality and reproducibility when compared to early cDNA arrays. Meta-analysis of data obtained using different platforms and from different laboratories is possible, but one must proceed with caution. A common strategy consists in the use of a control group that is common to all datasets under study (e.g. non stimulated or healthy controls) (167, 185, 186).
Finally, when interpreting results from blood transcriptome analyses it is important to consider that changes in transcript abundance might reflect two phenomena: 1) transcriptional regulation; 2) relative changes in cell composition (Figure 1). The later can also provide reliable and valid clinical correlate/biomarker signatures, as shown in patients with SLE who display plasma cell precursor and/or immature neutrophils signatures (21). The analysis of purified cell subpopulations might represent a useful way to identify low frequency biomarkers. Subpopulation frequencies (i.e. shifts in naïve, memory and/or effector populations of T and/or B cells) may still be responsible however for transcriptional differences between patients and controls. Cell purification techniques might also be a source of biased results. Positive selection might for example alter the steady state transcriptome by delivering signals through surface receptors, while enrichment purification methods tend to be more prone to contamination with other cell types.
Data analysis primer
While data acquisition brings its own set of challenges, data analysis might represent the main bottleneck for blood transcriptional studies. It is beyond the scope of this article to review in detail microarray data analysis methods, but generalizations are provided for the main analysis steps (see Box 7).
New analytical techniques, including network and global analytical tools, are being devised to facilitate the interpretation of genomics data in the real context of interactive networks where all molecular players influence each other (187). Many computational tools are also being created to help interpreting this type of complex data (188). A detailed account of this topic is beyond the scope of this review.
The Future of Blood Genomics
Microarray technologies are limited by several factors, such as hybridization noise (background signal, non-specific binding) and lack of sensitivity for transcripts expressed at very low or very high levels (dynamic range). Additional limitations derive from the fact that they rely on existing sequence knowledge based on the human genome and lack the capacity to quantify alternative messages, such as splice variants of a given gene. When considering human studies with potential clinical applications, perhaps the main limitation, however, is that direct comparability of data across batches and platforms is sometimes impossible.
Real-time PCR technology is currently considered the gold standard for measurement of gene expression in clinical settings. However, it can be used to measure transcript abundance for only a small number of genes. Thus the interest for other technologies such as the one developed by Nanostring, which detects transcripts abundance for up to 500 transcripts with high sensitivity (189). The approach is “digital” since it consists of counting individual RNA molecules, and therefore there is no need for “normalization” to a healthy control population. But a distinct advantage of this technology, which like microarrays is hybridization-based, is that sample preparation needs are reduced to a minimum – for instance none of the steps involving enzymatic reactions. Also, given its high sensitivity, fast turnaround time, sample throughput and intermediate multiplexing ability this approach seems particularly promising for bedside applications.
The important limitations of current microarray-based technologies might also be lifted by methods relying on high-throughput sequencing for genome-wide measurement of RNA abundance (190). RNA-seq (RNA sequencing) (191) starts with a population of RNA (total or fractionated, such as poly(A)+) that is converted to a library of cDNA fragments with adaptors attached to one or both ends. Each molecule, with or without amplification, is then sequenced in a high-throughput manner to obtain short sequences from one end (single-end sequencing) or both ends (pair-end sequencing). The reads are typically 30–400 bp, depending on the DNA-sequencing technology used. Thus, this approach does not rely on probe design and is uses instead tag-based sequencing. The obtained sequences are then uniquely mapped against a reference genome, which basically eliminates background signals. A detailed genome-wide transcription map for a given sample can thus be obtained by sequencing several tens of millions of tags. From this map it is possible to obtain several types of information, including not only transcript abundance but also transcriptome structure (splice variants), non-coding RNA species such as miRNA, and genetic polymorphisms. Because it uses high throughput sequencer, with offering from companies such as Applied Biosystems, Helicos Biosciences, Illumina or Roche/454, RNA-seq is also cost-effective and is expected to supersede microarray technologies. Another advantage of RNA-Seq compared to DNA microarrays is that RNA-Seq does not have an upper limit for quantification, and therefore it can measure a large dynamic range of expression levels. The data generated should also be reproducible across different platforms. The use of RNA-seq has not reached the mainstream, with most publications in the field reporting results for a small number of samples and limited for the most part to model organisms. Indeed, challenges ahead are multiple, including sample preparation (laborious, and potentially a source of bias), storage of massive amounts of data and sequence alignment. The powerful advantage of measuring transcriptome dynamics across different tissues or conditions without sophisticated normalization of data sets confers however this technology with a tremendous advantage for understanding transcriptomic dynamics in health and disease (8) (Table I).
Table I.
| Microarrays | RNA-seq | PCR | Nanostring | |
|---|---|---|---|---|
| Principle | Hybridization | Sequencing | Hybridization | Hybridization |
| Necessitates Probe design | Yes | No | Yes | Yes |
| Sensitivity | Low | High | High | High |
| Amplification | Yes | Yes | Yes | No |
| Scale | Genome-wide | Genome-wide | Low | Intermediate |
| Detect Genetic Polymorphisms | No | Yes | No | No |
| Output | Spot intensity | Sequence Tag Counts | Cycles of Amplification | Molecule Counts |
 |
 |
 |
Transcriptome analyses have recently started to be combined with genome-wide association studies to identify networks of genes involved in disease pathogenesis. The abundance of a gene transcript can in fact be directly modified by polymorphisms in regulatory elements. Consequently, expression levels of most genes have been found to have statistically significant heritability, and transcript abundance might therefore be considered as a quantitative trait. These have been named expression QTLs (eQTLs) [reviewed in (18)]. By combining genome-wide assays of gene expression and genetic variation, the genetic factors responsible for individual differences in quantitative levels of expression can be mapped. This information could provide significant insight into the biological basis for disease associations identified through GWA studies.
In addition to DNA sequence variants, gene transcription is also modulated by epigenetic modifications [reviewed in (192)]. Post-translational modifications of histones, for example, modulate DNA accessibility and chromatin stability to provide an enormous variety of alternative interaction surfaces for trans-acting factors [reviewed in Ref. (18)]. Epigenetic studies will undoubtedly complement transcriptome studies by providing additional clues to understand gene expression alterations in disease.
We therefore expect that rapid technology developments currently occurring in the genomic field, including genetic, epigenetic and proteomic studies, will directly impact transcriptome research. Keeping up with these advances will be a challenge for the teams currently engaged in this line of work. Especially, it increases the demands on already strained bioinformatics resources. At the same time, these recent developments hold tremendous promise for understanding and better treating autoimmunity and beyond.
Conclusion
Basic immunologists have made great progresses in the past 80 years in understanding the cellular and molecular components of the immune system. The 00's have started to see translation of this knowledge into the human, but, as stated by Mark Davis, you can go to the most prestigious medical center in the world and ask “How is my immune system?”, and, after a short period of eye rolling and looks of amused incomprehension, you might, if they don't throw you out, be offered a white blood cell count…(1). We contend that the quantitative analysis of the transcriptome, together with other approaches such as polychromatic flow cytometry and advanced proteomics, will provide physicians with a comprehensive picture of their patient immune system. The successful identification of pathogenic pathways and therapeutic targets in some human autoimmune diseases through the use of this technology supports that it could be applied to other types of diseases. Furthermore, blood transcriptome studies will facilitate the development of diagnostic biomarkers and personalized treatment options, thus leading to more efficient and cost/effective therapies. Eventually, this added knowledge will transform the practice of Medicine into a true Health Care System focusing on monitoring health and preventing disease, rather that the “Sick Care System” that we have today.
Summary Points.
Blood transcriptome analyses can be performed using cost-effective, commercially available DNA microarrays.
Blood transcriptome analyses in health and disease reflect not only transcriptional changes but also cell composition, including altered balances of leukocyte subsets normally present in the blood as well as presence of unusual cell types such as plasma cell precursors, immature neutrophils, etc.
Blood transcriptome analyses reveal cytokine signatures reflecting activation of molecular pathways that can be targeted in patients with autoimmune diseases.
Blood transcriptome analyses reveal cytokine signatures shared among different autoimmune diseases.
Signatures identified through blood transcriptome analyses can help genetic studies pinpoint autoimmune disease-specific susceptibility loci.
Blood transcriptome analyses reveal disease-specific alterations that might be used to develop diagnostic biomarkers.
Blood transcriptome analyses enable the measurement of disease activity and might permit clinicians to assess treatment efficacy in patients with autoimmune diseases.
Future Issues.
Novel bioinformatics tools and data analyses remain limiting steps.
DNA microarrays allow a genome-wide assessment of gene expression, but they have limited sensitivity, are relatively slow and not fully quantitative.
Improved data mining tools and expanded data sets will facilitate the identification of both novel therapeutic targets and predictors of autoimmune diseases outcomes.
Biomarkers identified through blood transcriptome studies need to be fully validated in large multi-centric trials.
The mechanisms leading to the expression of disease-specific genomic signatures need to be understood. Information generated through transcriptome analyses must be integrated with genome-wide genetic association and epigenetic studies. Such assimilation will allow the determination of whether or not variation in gene expression underlies susceptibility to complex autoimmune diseases.
Autoimmune disease-specific genomic signatures will be used as diagnostic tests using rapid and quantitative methods such as real-time PCR or Nanostring.
Blood genomic analyses will become routine assays to assess health status and, as such, represent an essential component of personalized medicine.
Box 2.
Different genetic alterations may lead to type I IFN overproduction in human SLE. The increased bioavailability of type I IFN contributes to peripheral tolerance breakdown through the activation of immature myeloid DCs. While immature DCs contribute to peripheral tolerance through their effects on autoreactive T cells that escape negative selection in the thymus, IFN-matured DCs activate autoreactive T cells. These cells, together with pDCs, help expand autoreactive B cells. IFN-matured DCs also activate cytotoxic CD8+ T cells, possibly increasing the apoptotic cell load. The capture of apoptotic cells by mDCs and of nucleic acid-containing immune complexes by pDCs and B cells amplifies the autoimmune reaction by increasing the antigenic load, type I IFN production and B cell activation respectively. Endogenous proteins such as HMGB1 (194) and LL37 (195))(196) have been recently shown to alter the conformation and/or trafficking and subsequently increase the interferogenic capacity of immune complex-bound endogenous DNA and RNA.
Acknowledgments
This study was partially supported by U19-A1057234, RO1-CA078846, AR054083-01 (J.B.) and Baylor Health Care foundation (J.B). J.B. holds the W.W. Caruth, Jr. Chair for Transplantation Immunology Research. R01 AR050770-01, ARO54083-01CORT, NIH ARO55503-01CORT, U19- AI082715-01, Alliance for Lupus Research and the Mary Kirkland Foundation to VP. U01AI082110 and the DANA Foundation to DC.
The authors want to thank Florence Allantaz, Lynda Bennett, Quynh-Ahn Nguyen, Charlie Quinn, Pinakeen Patel and Dorothee Stichweh for their contributions through the past 10 years to autoimmune disease microarray studies. Jeanine Baisch, Romain Banchereau, Patrice Mannoni, Karolina Palucka and Octavio Ramilo for critical reading of the manuscript. And Barbara and Jerry Nepom for their insightful comments.
Contributor Information
Virginia Pascual, Email: Virginip@baylorhealth.edu.
Damien Chaussabel, Email: Damienc@baylorhealth.edu.
Jacques Banchereau, Email: Jacquesb@baylorhealth.edu.
References
- 1.Davis MM. A prescription for human immunology. Immunity. 2008;29:835–8. doi: 10.1016/j.immuni.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu K, Mohan C. What do mouse models teach us about human SLE? Clin Immunol. 2006;119:123–30. doi: 10.1016/j.clim.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 3.Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, Ikuse T, Asano M, Iwakura Y. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000;191:313–20. doi: 10.1084/jem.191.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mertens M, Singh JA. Anakinra for rheumatoid arthritis: a systematic review. J Rheumatol. 2009;36:1118–25. doi: 10.3899/jrheum.090074. [DOI] [PubMed] [Google Scholar]
- 5.Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19:163–96. doi: 10.1146/annurev.immunol.19.1.163. [DOI] [PubMed] [Google Scholar]
- 6.Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–70. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 7.Shalon D, Smith SJ, Brown PO. A DNA microarray system for analyzing complex DNA samples using two-color fluorescent probe hybridization. Genome Res. 1996;6:639–45. doi: 10.1101/gr.6.7.639. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–7. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 10.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Jr, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W, Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–11. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 11.Winnepenninckx V, Lazar V, Michiels S, Dessen P, Stas M, Alonso SR, Avril MF, Ortiz Romero PL, Robert T, Balacescu O, Eggermont AM, Lenoir G, Sarasin A, Tursz T, van den Oord JJ, Spatz A. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J Natl Cancer Inst. 2006;98:472–82. doi: 10.1093/jnci/djj103. [DOI] [PubMed] [Google Scholar]
- 12.Sotiriou C, Pusztai L. Gene-expression signatures in breast cancer. N Engl J Med. 2009;360:790–800. doi: 10.1056/NEJMra0801289. [DOI] [PubMed] [Google Scholar]
- 13.Pascual V, Allantaz F, Patel P, Palucka AK, Chaussabel D, Banchereau J. How the study of children with rheumatic diseases identified interferon-alpha and interleukin-1 as novel therapeutic targets. Immunol Rev. 2008;223:39–59. doi: 10.1111/j.1600-065X.2008.00643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bauer JW, Bilgic H, Baechler EC. Gene-expression profiling in rheumatic disease: tools and therapeutic potential. Nat Rev Rheumatol. 2009;5:257–65. doi: 10.1038/nrrheum.2009.50. [DOI] [PubMed] [Google Scholar]
- 15.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–7. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 16.Zaas AK, Chen M, Varkey J, Veldman T, Hero AO, 3rd, Lucas J, Huang Y, Turner R, Gilbert A, Lambkin-Williams R, Oien NC, Nicholson B, Kingsmore S, Carin L, Woods CW, Ginsburg GS. Gene Expression Signatures Diagnose Influenza and Other Symptomatic Respiratory Viral Infections in Humans. Cell Host Microbe. 2009 doi: 10.1016/j.chom.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boldrick JC, Alizadeh AA, Diehn M, Dudoit S, Liu CL, Belcher CE, Botstein D, Staudt LM, Brown PO, Relman DA. Stereotyped and specific gene expression programs in human innate immune responses to bacteria. Proc Natl Acad Sci U S A. 2002;99:972–7. doi: 10.1073/pnas.231625398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10:184–94. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005 doi: 10.1084/jem.20050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Jia S, Geoffrey R, Alemzadeh R, Ghosh S, Hessner MJ. Identification of a molecular signature in human type 1 diabetes mellitus using serum and functional genomics. J Immunol. 2008;180:1929–37. doi: 10.4049/jimmunol.180.3.1929. [DOI] [PubMed] [Google Scholar]
- 21.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–23. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–5. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crow MK, Wohlgemuth J. Microarray analysis of gene expression in lupus. Arthritis Res Ther. 2003;5:279–87. doi: 10.1186/ar1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han GM, Chen SL, Shen N, Ye S, Bao CD, Gu YY. Analysis of gene expression profiles in human systemic lupus erythematosus using oligonucleotide microarray. Genes Immun. 2003;4:177–86. doi: 10.1038/sj.gene.6363966. [DOI] [PubMed] [Google Scholar]
- 25.Allantaz F, Chaussabel D, Stichweh D, Bennett L, Allman W, Mejias A, Ardura M, Chung W, Wise C, Palucka K, Ramilo O, Punaro M, Banchereau J, Pascual V. Blood leukocyte microarrays to diagnose systemic onset juvenile idiopathic arthritis and follow the response to IL-1 blockade. J Exp Med. 2007;204:2131–44. doi: 10.1084/jem.20070070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogilvie EM, Khan A, Hubank M, Kellam P, Woo P. Specific gene expression profiles in systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007;56:1954–65. doi: 10.1002/art.22644. [DOI] [PubMed] [Google Scholar]
- 27.Fall N, Barnes M, Thornton S, Luyrink L, Olson J, Ilowite NT, Gottlieb BS, Griffin T, Sherry DD, Thompson S, Glass DN, Colbert RA, Grom AA. Gene expression profiling of peripheral blood from patients with untreated new-onset systemic juvenile idiopathic arthritis reveals molecular heterogeneity that may predict macrophage activation syndrome. Arthritis Rheum. 2007;56:3793–804. doi: 10.1002/art.22981. [DOI] [PubMed] [Google Scholar]
- 28.Barnes MG, Grom AA, Thompson SD, Griffin TA, Pavlidis P, Itert L, Fall N, Sowders DP, Hinze CH, Aronow BJ, Luyrink LK, Srivastava S, Ilowite NT, Gottlieb BS, Olson JC, Sherry DD, Glass DN, Colbert RA. Subtype-specific peripheral blood gene expression profiles in recent-onset juvenile idiopathic arthritis. Arthritis Rheum. 2009;60:2102–12. doi: 10.1002/art.24601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Achiron A, Feldman A, Mandel M, Gurevich M. Impaired expression of peripheral blood apoptotic-related gene transcripts in acute multiple sclerosis relapse. Ann N Y Acad Sci. 2007;1107:155–67. doi: 10.1196/annals.1381.017. [DOI] [PubMed] [Google Scholar]
- 30.Singh MK, Scott TF, LaFramboise WA, Hu FZ, Post JC, Ehrlich GD. Gene expression changes in peripheral blood mononuclear cells from multiple sclerosis patients undergoing beta-interferon therapy. J Neurol Sci. 2007;258:52–9. doi: 10.1016/j.jns.2007.02.034. [DOI] [PubMed] [Google Scholar]
- 31.Edwards CJ, Feldman JL, Beech J, Shields KM, Stover JA, Trepicchio WL, Larsen G, Foxwell BM, Brennan FM, Feldmann M, Pittman DD. Molecular profile of peripheral blood mononuclear cells from patients with rheumatoid arthritis. Mol Med. 2007;13:40–58. doi: 10.2119/2006-000056.Edwards. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Pouw Kraan TC, Wijbrandts CA, van Baarsen LG, Voskuyl AE, Rustenburg F, Baggen JM, Ibrahim SM, Fero M, Dijkmans BA, Tak PP, Verweij CL. Rheumatoid arthritis subtypes identified by genomic profiling of peripheral blood cells: assignment of a type I interferon signature in a subpopulation of patients. Ann Rheum Dis. 2007;66:1008–14. doi: 10.1136/ard.2006.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lequerre T, Gauthier-Jauneau AC, Bansard C, Derambure C, Hiron M, Vittecoq O, Daveau M, Mejjad O, Daragon A, Tron F, Le Loet X, Salier JP. Gene profiling in white blood cells predicts infliximab responsiveness in rheumatoid arthritis. Arthritis Res Ther. 2006;8:R105. doi: 10.1186/ar1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Batliwalla FM, Baechler EC, Xiao X, Li W, Balasubramanian S, Khalili H, Damle A, Ortmann WA, Perrone A, Kantor AB, Gulko PS, Kern M, Furie R, Behrens TW, Gregersen PK. Peripheral blood gene expression profiling in rheumatoid arthritis. Genes Immun. 2005;6:388–97. doi: 10.1038/sj.gene.6364209. [DOI] [PubMed] [Google Scholar]
- 35.Emamian ES, Leon JM, Lessard CJ, Grandits M, Baechler EC, Gaffney PM, Segal B, Rhodus NL, Moser KL. Peripheral blood gene expression profiling in Sjogren's syndrome. Genes Immun. 2009;10:285–96. doi: 10.1038/gene.2009.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaizer EC, Glaser CL, Chaussabel D, Banchereau J, Pascual V, White PC. Gene expression in peripheral blood mononuclear cells from children with diabetes. J Clin Endocrinol Metab. 2007;92:3705–11. doi: 10.1210/jc.2007-0979. [DOI] [PubMed] [Google Scholar]
- 37.Takamura T, Honda M, Sakai Y, Ando H, Shimizu A, Ota T, Sakurai M, Misu H, Kurita S, Matsuzawa-Nagata N, Uchikata M, Nakamura S, Matoba R, Tanino M, Matsubara K, Kaneko S. Gene expression profiles in peripheral blood mononuclear cells reflect the pathophysiology of type 2 diabetes. Biochem Biophys Res Commun. 2007;361:379–84. doi: 10.1016/j.bbrc.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 38.Burczynski ME, Peterson RL, Twine NC, Zuberek KA, Brodeur BJ, Casciotti L, Maganti V, Reddy PS, Strahs A, Immermann F, Spinelli W, Schwertschlag U, Slager AM, Cotreau MM, Dorner AJ. Molecular classification of Crohn's disease and ulcerative colitis patients using transcriptional profiles in peripheral blood mononuclear cells. J Mol Diagn. 2006;8:51–61. doi: 10.2353/jmoldx.2006.050079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stoeckman AK, Baechler EC, Ortmann WA, Behrens TW, Michet CJ, Peterson EJ. A distinct inflammatory gene expression profile in patients with psoriatic arthritis. Genes Immun. 2006;7:583–91. doi: 10.1038/sj.gene.6364334. [DOI] [PubMed] [Google Scholar]
- 40.Batliwalla FM, Li W, Ritchlin CT, Xiao X, Brenner M, Laragione T, Shao T, Durham R, Kemshetti S, Schwarz E, Coe R, Kern M, Baechler EC, Behrens TW, Gregersen PK, Gulko PS. Microarray analyses of peripheral blood cells identifies unique gene expression signature in psoriatic arthritis. Mol Med. 2005;11:21–9. doi: 10.2119/2006-00003.Gulko. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, Barohn RJ, Saperstein DS, Briemberg HR, Ericsson M, Park P, Amato AA. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005;57:664–78. doi: 10.1002/ana.20464. [DOI] [PubMed] [Google Scholar]
- 42.Baechler EC, Bauer JW, Slattery CA, Ortmann WA, Espe KJ, Novitzke J, Ytterberg SR, Gregersen PK, Behrens TW, Reed AM. An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol Med. 2007;13:59–68. doi: 10.2119/2006-00085.Baechler. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan FK, Zhou X, Mayes MD, Gourh P, Guo X, Marcum C, Jin L, Arnett FC., Jr Signatures of differentially regulated interferon gene expression and vasculotrophism in the peripheral blood cells of systemic sclerosis patients. Rheumatology (Oxford) 2006;45:694–702. doi: 10.1093/rheumatology/kei244. [DOI] [PubMed] [Google Scholar]
- 44.York MR, Nagai T, Mangini AJ, Lemaire R, van Seventer JM, Lafyatis R. A macrophage marker, Siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and toll-like receptor agonists. Arthritis Rheum. 2007;56:1010–20. doi: 10.1002/art.22382. [DOI] [PubMed] [Google Scholar]
- 45.Alcorta DA, Barnes DA, Dooley MA, Sullivan P, Jonas B, Liu Y, Lionaki S, Reddy CB, Chin H, Dempsey AA, Jennette JC, Falk RJ. Leukocyte gene expression signatures in antineutrophil cytoplasmic autoantibody and lupus glomerulonephritis. Kidney Int. 2007;72:853–64. doi: 10.1038/sj.ki.5002371. [DOI] [PubMed] [Google Scholar]
- 46.Potti A, Bild A, Dressman HK, Lewis DA, Nevins JR, Ortel TL. Gene-expression patterns predict phenotypes of immune-mediated thrombosis. Blood. 2006;107:1391–6. doi: 10.1182/blood-2005-07-2669. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Lempicki RA, Polis MA, Yang J, McLaughlin M, Koratich C, Huang DW, Fullmer B, Wu L, Rehm CA, Masur H, Lane HC, Sherman KE, Fauci AS, Kottilil S. Gene expression profiles in hepatitis C virus (HCV) and HIV coinfection: class prediction analyses before treatment predict the outcome of anti-HCV therapy among HIV-coinfected persons. J Infect Dis. 2006;193:1172–7. doi: 10.1086/501365. [DOI] [PubMed] [Google Scholar]
- 48.Thach DC, Agan BK, Olsen C, Diao J, Lin B, Gomez J, Jesse M, Jenkins M, Rowley R, Hanson E, Tibbetts C, Stenger DA, Walter E. Surveillance of transcriptomes in basic military trainees with normal, febrile respiratory illness, and convalescent phenotypes. Genes Immun. 2005;6:588–95. doi: 10.1038/sj.gene.6364244. [DOI] [PubMed] [Google Scholar]
- 49.Popper SJ, Watson VE, Shimizu C, Kanegaye JT, Burns JC, Relman DA. Gene transcript abundance profiles distinguish Kawasaki disease from adenovirus infection. J Infect Dis. 2009;200:657–66. doi: 10.1086/603538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramilo O, Allman W, Chung W, Mejias A, Ardura M, Glaser C, Wittkowski KM, Piqueras B, Banchereau J, Palucka AK, Chaussabel D. Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood. 2007;109:2066–77. doi: 10.1182/blood-2006-02-002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simmons CP, Popper S, Dolocek C, Chau TN, Griffiths M, Dung NT, Long TH, Hoang DM, Chau NV, Thao le TT, Hien TT, Relman DA, Farrar J. Patterns of host genome-wide gene transcript abundance in the peripheral blood of patients with acute dengue hemorrhagic fever. J Infect Dis. 2007;195:1097–107. doi: 10.1086/512162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ardura MI, Banchereau R, Mejias A, Di Pucchio T, Glaser C, Allantaz F, Pascual V, Banchereau J, Chaussabel D, Ramilo O. Enhanced monocyte response and decreased central memory T cells in children with invasive Staphylococcus aureus infections. PLoS One. 2009;4:e5446. doi: 10.1371/journal.pone.0005446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jacobsen M, Repsilber D, Gutschmidt A, Neher A, Feldmann K, Mollenkopf HJ, Ziegler A, Kaufmann SH. Candidate biomarkers for discrimination between infection and disease caused by Mycobacterium tuberculosis. J Mol Med. 2007;85:613–21. doi: 10.1007/s00109-007-0157-6. [DOI] [PubMed] [Google Scholar]
- 54.Ockenhouse CF, Hu WC, Kester KE, Cummings JF, Stewart A, Heppner DG, Jedlicka AE, Scott AL, Wolfe ND, Vahey M, Burke DS. Common and divergent immune response signaling pathways discovered in peripheral blood mononuclear cell gene expression patterns in presymptomatic and clinically apparent malaria. Infect Immun. 2006;74:5561–73. doi: 10.1128/IAI.00408-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang BM, McLean AS, Dawes IW, Huang SJ, Lin RC. The Use of Gene-Expression Profiling to Identify Candidate Genes in Human Sepsis. Am J Respir Crit Care Med. 2007 doi: 10.1164/rccm.200612-1819OC. [DOI] [PubMed] [Google Scholar]
- 56.Zhang HQ, Lu H, Enosawa S, Takahara S, Sakamoto S, Suzuki S. Identification of novel genes associated with immunosuppression in renal transplant patients. Transplant Proc. 2002;34:2733–5. doi: 10.1016/s0041-1345(02)03391-2. [DOI] [PubMed] [Google Scholar]
- 57.Martinez-Llordella M, Puig-Pey I, Orlando G, Ramoni M, Tisone G, Rimola A, Lerut J, Latinne D, Margarit C, Bilbao I, Brouard S, Hernandez-Fuentes M, Soulillou JP, Sanchez-Fueyo A. Multiparameter immune profiling of operational tolerance in liver transplantation. Am J Transplant. 2007;7:309–19. doi: 10.1111/j.1600-6143.2006.01621.x. [DOI] [PubMed] [Google Scholar]
- 58.Deng MC, Eisen HJ, Mehra MR, Billingham M, Marboe CC, Berry G, Kobashigawa J, Johnson FL, Starling RC, Murali S, Pauly DF, Baron H, Wohlgemuth JG, Woodward RN, Klingler TM, Walther D, Lal PG, Rosenberg S, Hunt S. Noninvasive discrimination of rejection in cardiac allograft recipients using gene expression profiling. Am J Transplant. 2006;6:150–60. doi: 10.1111/j.1600-6143.2005.01175.x. [DOI] [PubMed] [Google Scholar]
- 59.Baron C, Somogyi R, Greller LD, Rineau V, Wilkinson P, Cho CR, Cameron MJ, Kelvin DJ, Chagnon P, Roy DC, Busque L, Sekaly RP, Perreault C. Prediction of graft-versus-host disease in humans by donor gene-expression profiling. PLoS Med. 2007;4:e23. doi: 10.1371/journal.pmed.0040023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang Y, Xu H, Du X, Lit L, Walker W, Lu A, Ran R, Gregg JP, Reilly M, Pancioli A, Khoury JC, Sauerbeck LR, Carrozzella JA, Spilker J, Clark J, Wagner KR, Jauch EC, Chang DJ, Verro P, Broderick JP, Sharp FR. Gene expression in blood changes rapidly in neutrophils and monocytes after ischemic stroke in humans: a microarray study. J Cereb Blood Flow Metab. 2006;26:1089–102. doi: 10.1038/sj.jcbfm.9600264. [DOI] [PubMed] [Google Scholar]
- 61.Solmi R, Ugolini G, Rosati G, Zanotti S, Lauriola M, Montroni I, del Governatore M, Caira A, Taffurelli M, Santini D, Coppola D, Guidotti L, Carinci P, Strippoli P. Microarray-based identification and RT-PCR test screening for epithelial-specific mRNAs in peripheral blood of patients with colon cancer. BMC Cancer. 2006;6:250. doi: 10.1186/1471-2407-6-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Osman I, Bajorin DF, Sun TT, Zhong H, Douglas D, Scattergood J, Zheng R, Han M, Marshall KW, Liew CC. Novel blood biomarkers of human urinary bladder cancer. Clin Cancer Res. 2006;12:3374–80. doi: 10.1158/1078-0432.CCR-05-2081. [DOI] [PubMed] [Google Scholar]
- 63.Llorente L, Zou W, Levy Y, Richaud-Patin Y, Wijdenes J, Alcocer-Varela J, Morel-Fourrier B, Brouet JC, Alarcon-Segovia D, Galanaud P, Emilie D. Role of interleukin 10 in the B lymphocyte hyperactivity and autoantibody production of human systemic lupus erythematosus. J Exp Med. 1995;181:839–44. doi: 10.1084/jem.181.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Llorente L, Richaud-Patin Y, Garcia-Padilla C, Claret E, Jakez-Ocampo J, Cardiel MH, Alcocer-Varela J, Grangeot-Keros L, Alarcon-Segovia D, Wijdenes J, Galanaud P, Emilie D. Clinical and biologic effects of anti-interleukin-10 monoclonal antibody administration in systemic lupus erythematosus. Arthritis Rheum. 2000;43:1790–800. doi: 10.1002/1529-0131(200008)43:8<1790::AID-ANR15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 65.Yokoyama H, Takabatake T, Takaeda M, Wada T, Naito T, Ikeda K, Goshima S, Takasawa K, Tomosugi N, Kobayashi K, et al. Up-regulated MHC-class II expression and gamma-IFN and soluble IL-2R in lupus nephritis. Kidney Int. 1992;42:755–63. doi: 10.1038/ki.1992.344. [DOI] [PubMed] [Google Scholar]
- 66.Haas C, Ryffel B, Le Hir M. IFN-gamma is essential for the development of autoimmune glomerulonephritis in MRL/Ipr mice. J Immunol. 1997;158:5484–91. [PubMed] [Google Scholar]
- 67.Horwitz DA, Gray JD, Behrendsen SC, Kubin M, Rengaraju M, Ohtsuka K, Trinchieri G. Decreased production of interleukin-12 and other Th1-type cytokines in patients with recent-onset systemic lupus erythematosus. Arthritis Rheum. 1998;41:838–44. doi: 10.1002/1529-0131(199805)41:5<838::AID-ART10>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 68.Steinberg AD, Baron S, Talal N. The pathogenesis of autoimmunity in New Zealand mice, I. Induction of antinucleic acid antibodies by polyinosinic-polycytidylic acid. Proc Natl Acad Sci U S A. 1969;63:1102–7. doi: 10.1073/pnas.63.4.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. 1979;301:5–8. doi: 10.1056/NEJM197907053010102. [DOI] [PubMed] [Google Scholar]
- 70.Stewart TA. Neutralizing interferon alpha as a therapeutic approach to autoimmune diseases. Cytokine Growth Factor Rev. 2003;14:139–54. doi: 10.1016/s1359-6101(02)00088-6. [DOI] [PubMed] [Google Scholar]
- 71.Cederblad B, Blomberg S, Vallin H, Perers A, Alm GV, Ronnblom L. Patients with systemic lupus erythematosus have reduced numbers of circulating natural interferon-alpha- producing cells. J Autoimmun. 1998;11:465–70. doi: 10.1006/jaut.1998.0215. [DOI] [PubMed] [Google Scholar]
- 72.Blomberg S, Eloranta ML, Cederblad B, Nordlin K, Alm GV, Ronnblom L. Presence of cutaneous interferon-alpha producing cells in patients with systemic lupus erythematosus. Lupus. 2001;10:484–90. doi: 10.1191/096120301678416042. [DOI] [PubMed] [Google Scholar]
- 73.Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL. Plasmacytoid Dendritic Cells (Natural Interferon- alpha/beta-Producing Cells) Accumulate in Cutaneous Lupus Erythematosus Lesions. Am J Pathol. 2001;159:237–43. doi: 10.1016/s0002-9440(10)61689-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vallin H, Blomberg S, Alm GV, Cederblad B, Ronnblom L. Patients with systemic lupus erythematosus (SLE) have a circulating inducer of interferon-alpha (IFN-alpha) production acting on leucocytes resembling immature dendritic cells. Clin Exp Immunol. 1999;115:196–202. doi: 10.1046/j.1365-2249.1999.00772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bave U, Vallin H, Alm GV, Ronnblom L. Activation of natural interferon-alpha producing cells by apoptotic U937 cells combined with lupus IgG and its regulation by cytokines. J Autoimmun. 2001;17:71–80. doi: 10.1006/jaut.2001.0519. [DOI] [PubMed] [Google Scholar]
- 76.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–3. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 77.Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19:225–34. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 78.Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med. 1999;189:521–30. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–50. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Le Bon A, Schiavoni G, D'Agostino G, Gresser I, Belardelli F, Tough DF. Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity. 2001;14:461–70. doi: 10.1016/s1074-7613(01)00126-1. [DOI] [PubMed] [Google Scholar]
- 81.Preble OT, Black RJ, Friedman RM, Klippel JH, Vilcek J. Systemic lupus erythematosus: presence in human serum of an unusual acid-labile leukocyte interferon. Science. 1982;216:429–31. doi: 10.1126/science.6176024. [DOI] [PubMed] [Google Scholar]
- 82.Tsao BP. Update on human systemic lupus erythematosus genetics. Curr Opin Rheumatol. 2004;16:513–21. doi: 10.1097/01.bor.0000132648.62680.81. [DOI] [PubMed] [Google Scholar]
- 83.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 84.Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, Dummer R, Simon HU, Yousefi S. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem. 2004;279:44123–32. doi: 10.1074/jbc.M405883200. [DOI] [PubMed] [Google Scholar]
- 85.de Weerd NA, Samarajiwa SA, Hertzog PJ. Type I interferon receptors: biochemistry and biological functions. J Biol Chem. 2007;282:20053–7. doi: 10.1074/jbc.R700006200. [DOI] [PubMed] [Google Scholar]
- 86.de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, Silverman RH, Williams BR. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69:912–20. [PubMed] [Google Scholar]
- 87.Ding C. Belimumab, an anti-BLyS human monoclonal antibody for potential treatment of inflammatory autoimmune diseases. Expert Opin Biol Ther. 2008;8:1805–14. doi: 10.1517/14712598.8.11.1805. [DOI] [PubMed] [Google Scholar]
- 88.Shodell M, Shah K, Siegal FP. Circulating human plasmacytoid dendritic cells are highly sensitive to corticosteroid administration. Lupus. 2003;12:222–30. doi: 10.1191/0961203303lu362xx. [DOI] [PubMed] [Google Scholar]
- 89.Peterson KS, Huang JF, Zhu J, D'Agati V, Liu X, Miller N, Erlander MG, Jackson MR, Winchester RJ. Characterization of heterogeneity in the molecular pathogenesis of lupus nephritis from transcriptional profiles of laser-captured glomeruli. J Clin Invest. 2004;113:1722–33. doi: 10.1172/JCI19139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nzeusseu Toukap A, Galant C, Theate I, Maudoux AL, Lories RJ, Houssiau FA, Lauwerys BR. Identification of distinct gene expression profiles in the synovium of patients with systemic lupus erythematosus. Arthritis Rheum. 2007;56:1579–88. doi: 10.1002/art.22578. [DOI] [PubMed] [Google Scholar]
- 91.Tucci M, Quatraro C, Lombardi L, Pellegrino C, Dammacco F, Silvestris F. Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: role of interleukin-18. Arthritis Rheum. 2008;58:251–62. doi: 10.1002/art.23186. [DOI] [PubMed] [Google Scholar]
- 92.Gill MA, Blanco P, Arce E, Pascual V, Banchereau J, Palucka AK. Blood dendritic cells and DC-poietins in systemic lupus erythematosus. Hum Immunol. 2002;63:1172–80. doi: 10.1016/s0198-8859(02)00756-5. [DOI] [PubMed] [Google Scholar]
- 93.Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, Theofilopoulos AN. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med. 2003;197:777–88. doi: 10.1084/jem.20021996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Braun D, Geraldes P, Demengeot J. Type I Interferon controls the onset and severity of autoimmune manifestations in lpr mice. J Autoimmun. 2003;20:15–25. doi: 10.1016/s0896-8411(02)00109-9. [DOI] [PubMed] [Google Scholar]
- 95.Mathian A, Weinberg A, Gallegos M, Banchereau J, Koutouzov S. IFN-{alpha} Induces Early Lethal Lupus in Preautoimmune (New Zealand Black × New Zealand White)F1 but Not in BALB/c Mice. J Immunol. 2005;174:2499–506. doi: 10.4049/jimmunol.174.5.2499. [DOI] [PubMed] [Google Scholar]
- 96.Zagury D, Le Buanec H, Mathian A, Larcier P, Burnett R, Amoura Z, Emilie D, Peltre G, Bensussan A, Bizzini B, Gallo RC, Koutouzov S. IFNalpha kinoid vaccine-induced neutralizing antibodies prevent clinical manifestations in a lupus flare murine model. Proc Natl Acad Sci U S A. 2009;106:5294–9. doi: 10.1073/pnas.0900615106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Izui S, Ibnou-Zekri N, Fossati-Jimack L, Iwamoto M. Lessons from BXSB and related mouse models. Int Rev Immunol. 2000;19:447–72. doi: 10.3109/08830180009055507. [DOI] [PubMed] [Google Scholar]
- 98.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–72. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 99.Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, Liang C, Bartov G, McDaniel LD, Zhou XJ, Schultz RA, Wakeland EK. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A. 2006;103:9970–5. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bave U, Alm GV, Ronnblom L. The combination of apoptotic U937 cells and lupus IgG is a potent IFN-alpha inducer. J Immunol. 2000;165:3519–26. doi: 10.4049/jimmunol.165.6.3519. [DOI] [PubMed] [Google Scholar]
- 101.Vollmer J, Tluk S, Schmitz C, Hamm S, Jurk M, Forsbach A, Akira S, Kelly KM, Reeves WH, Bauer S, Krieg AM. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J Exp Med. 2005;202:1575–85. doi: 10.1084/jem.20051696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, Coffman RL. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202:1131–9. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–7. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27:801–10. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, Arnold CN, Baccala R, Silverman GJ, Beutler BA, Theofilopoulos AN. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci U S A. 2009;106:12061–6. doi: 10.1073/pnas.0905441106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Armstrong DL, Reiff A, Myones BL, Quismorio FP, Jr, Klein-Gitelman M, McCurdy D, Wagner-Weiner L, Silverman E, Ojwang JO, Kaufman KM, Kelly JA, Merrill JT, Harley JB, Bae SC, Vyse TJ, Gilkeson GS, Gaffney PM, Moser KL, Putterman C, Edberg JC, Brown EE, Ziegler J, Langefeld CD, Zidovetzki R, Jacob CO. Identification of new SLE-associated genes with a two-step Bayesian study design. Genes Immun. 2009;10:446–56. doi: 10.1038/gene.2009.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sigurdsson S, Nordmark G, Goring HH, Lindroos K, Wiman AC, Sturfelt G, Jonsen A, Rantapaa-Dahlqvist S, Moller B, Kere J, Koskenmies S, Widen E, Eloranta ML, Julkunen H, Kristjansdottir H, Steinsson K, Alm G, Ronnblom L, Syvanen AC. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76:528–37. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Graham RR, Kozyrev SV, Baechler EC, Reddy MV, Plenge RM, Bauer JW, Ortmann WA, Koeuth T, Gonzalez Escribano MF, Pons-Estel B, Petri M, Daly M, Gregersen PK, Martin J, Altshuler D, Behrens TW, Alarcon-Riquelme ME. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38:550–5. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- 109.Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD, Nath SK, Guthridge JM, Cobb BL, Mirel DB, Marion MC, Williams AH, Divers J, Wang W, Frank SG, Namjou B, Gabriel SB, Lee AT, Gregersen PK, Behrens TW, Taylor KE, Fernando M, Zidovetzki R, Gaffney PM, Edberg JC, Rioux JD, Ojwang JO, James JA, Merrill JT, Gilkeson GS, Seldin MF, Yin H, Baechler EC, Li QZ, Wakeland EK, Bruner GR, Kaufman KM, Kelly JA. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–10. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum. 2008;58:2481–7. doi: 10.1002/art.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8:492–502. doi: 10.1038/sj.gene.6364408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F, Li W, Masters SL, Booty MG, Carulli JP, Padyukov L, Alfredsson L, Klareskog L, Chen WV, Amos CI, Criswell LA, Seldin MF, Kastner DL, Gregersen PK. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357:977–86. doi: 10.1056/NEJMoa073003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jacob CO, Reiff A, Armstrong DL, Myones BL, Silverman E, Klein-Gitelman M, McCurdy D, Wagner-Weiner L, Nocton JJ, Solomon A, Zidovetzki R. Identification of novel susceptibility genes in childhood-onset systemic lupus erythematosus using a uniquely designed candidate gene pathway platform. Arthritis Rheum. 2007;56:4164–73. doi: 10.1002/art.23060. [DOI] [PubMed] [Google Scholar]
- 114.Jacob CO, Zhu J, Armstrong DL, Yan M, Han J, Zhou XJ, Thomas JA, Reiff A, Myones BL, Ojwang JO, Kaufman KM, Klein-Gitelman M, McCurdy D, Wagner-Weiner L, Silverman E, Ziegler J, Kelly JA, Merrill JT, Harley JB, Ramsey-Goldman R, Vila LM, Bae SC, Vyse TJ, Gilkeson GS, Gaffney PM, Moser KL, Langefeld CD, Zidovetzki R, Mohan C. Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2009;106:6256–61. doi: 10.1073/pnas.0901181106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O'Hara A, Herrick AL, Bowden AP, Perrino FW, Lindahl T, Barnes DE, Crow YJ. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;80:811–5. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hubner N. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065–7. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 117.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–98. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lehtinen DA, Harvey S, Mulcahy MJ, Hollis T, Perrino FW. The TREX1 double-stranded DNA degradation activity is defective in dominant mutations associated with autoimmune disease. J Biol Chem. 2008;283:31649–56. doi: 10.1074/jbc.M806155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A. 2009;106:2770–5. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Moser KL, Kelly JA, Lessard CJ, Harley JB. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun. 2009;10:373–9. doi: 10.1038/gene.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Palucka AK, Blanck JP, Bennett L, Pascual V, Banchereau J. Cross-regulation of TNF and IFN-{alpha} in autoimmune diseases. Proc Natl Acad Sci U S A. 2005;102:3372–7. doi: 10.1073/pnas.0408506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Arce E, Jackson DG, Gill MA, Bennett LB, Banchereau J, Pascual V. Increased frequency of pre-germinal center B cells and plasma cell precursors in the blood of children with systemic lupus erythematosus. J Immunol. 2001;167:2361–9. doi: 10.4049/jimmunol.167.4.2361. [DOI] [PubMed] [Google Scholar]
- 123.Odendahl M, Jacobi A, Hansen A, Feist E, Hiepe F, Burmester GR, Lipsky PE, Radbruch A, Dorner T. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol. 2000;165:5970–9. doi: 10.4049/jimmunol.165.10.5970. [DOI] [PubMed] [Google Scholar]
- 124.Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng NY, Mays I, Garman L, Helms C, James J, Air GM, Capra JD, Ahmed R, Wilson PC. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453:667–71. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yao Y, Richman L, Higgs BW, Morehouse CA, de los Reyes M, Brohawn P, Zhang J, White B, Coyle AJ, Kiener PA, Jallal B. Neutralization of interferon-alpha/beta-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum. 2009;60:1785–96. doi: 10.1002/art.24557. [DOI] [PubMed] [Google Scholar]
- 126.Prieur AM, Roux-Lombard P, Dayer JM. Dynamics of fever and the cytokine network in systemic juvenile arthritis. Rev Rhum Engl Ed. 1996;63:163–70. [PubMed] [Google Scholar]
- 127.Muller K, Herner EB, Stagg A, Bendtzen K, Woo P. Inflammatory cytokines and cytokine antagonists in whole blood cultures of patients with systemic juvenile chronic arthritis. Br J Rheumatol. 1998;37:562–9. doi: 10.1093/rheumatology/37.5.562. [DOI] [PubMed] [Google Scholar]
- 128.Fitzgerald AA, Leclercq SA, Yan A, Homik JE, Dinarello CA. Rapid responses to anakinra in patients with refractory adult-onset Still's disease. Arthritis Rheum. 2005;52:1794–803. doi: 10.1002/art.21061. [DOI] [PubMed] [Google Scholar]
- 129.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–147. [PubMed] [Google Scholar]
- 130.Wittkowski H, Frosch M, Wulffraat N, Goldbach-Mansky R, Kallinich T, Kuemmerle-Deschner J, Fruhwald MC, Dassmann S, Pham TH, Roth J, Foell D. S100A12 is a novel molecular marker differentiating systemic-onset juvenile idiopathic arthritis from other causes of fever of unknown origin. Arthritis Rheum. 2008;58:3924–31. doi: 10.1002/art.24137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Frosch M, Ahlmann M, Vogl T, Wittkowski H, Wulffraat N, Foell D, Roth J. The myeloid-related proteins 8 and 14 complex, a novel ligand of toll-like receptor 4, and interleukin-1beta form a positive feedback mechanism in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2009;60:883–91. doi: 10.1002/art.24349. [DOI] [PubMed] [Google Scholar]
- 132.Korman BD, Alba MI, Le JM, Alevizos I, Smith JA, Nikolov NP, Kastner DL, Remmers EF, Illei GG. Variant form of STAT4 is associated with primary Sjogren's syndrome. Genes Immun. 2008;9:267–70. doi: 10.1038/gene.2008.1. [DOI] [PubMed] [Google Scholar]
- 133.Nordmark G, Kristjansdottir G, Theander E, Eriksson P, Brun JG, Wang C, Padyukov L, Truedsson L, Alm G, Eloranta ML, Jonsson R, Ronnblom L, Syvanen AC. Additive effects of the major risk alleles of IRF5 and STAT4 in primary Sjogren's syndrome. Genes Immun. 2009;10:68–76. doi: 10.1038/gene.2008.94. [DOI] [PubMed] [Google Scholar]
- 134.Hjelmervik TO, Petersen K, Jonassen I, Jonsson R, Bolstad AI. Gene expression profiling of minor salivary glands clearly distinguishes primary Sjogren's syndrome patients from healthy control subjects. Arthritis Rheum. 2005;52:1534–44. doi: 10.1002/art.21006. [DOI] [PubMed] [Google Scholar]
- 135.Hu S, Wang J, Meijer J, Ieong S, Xie Y, Yu T, Zhou H, Henry S, Vissink A, Pijpe J, Kallenberg C, Elashoff D, Loo JA, Wong DT. Salivary proteomic and genomic biomarkers for primary Sjogren's syndrome. Arthritis Rheum. 2007;56:3588–600. doi: 10.1002/art.22954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Greenberg SA. A gene expression approach to study perturbed pathways in myositis. Curr Opin Rheumatol. 2007;19:536–41. doi: 10.1097/BOR.0b013e3282efe261. [DOI] [PubMed] [Google Scholar]
- 137.Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies. I: Quantitation of subsets according to diagnosis and sites of accumulation and demonstration and counts of muscle fibers invaded by T cells. Ann Neurol. 1984;16:193–208. doi: 10.1002/ana.410160206. [DOI] [PubMed] [Google Scholar]
- 138.Wenzel J, Schmidt R, Proelss J, Zahn S, Bieber T, Tuting T. Type I interferon-associated skin recruitment of CXCR3+ lymphocytes in dermatomyositis. Clin Exp Dermatol. 2006;31:576–82. doi: 10.1111/j.1365-2230.2006.02150.x. [DOI] [PubMed] [Google Scholar]
- 139.Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008;371:2201–12. doi: 10.1016/S0140-6736(08)60955-1. [DOI] [PubMed] [Google Scholar]
- 140.Tezak Z, Hoffman EP, Lutz JL, Fedczyna TO, Stephan D, Bremer EG, Krasnoselska-Riz I, Kumar A, Pachman LM. Gene expression profiling in DQA1*0501+ children with untreated dermatomyositis: a novel model of pathogenesis. J Immunol. 2002;168:4154–63. doi: 10.4049/jimmunol.168.8.4154. [DOI] [PubMed] [Google Scholar]
- 141.Walsh RJ, Kong SW, Yao Y, Jallal B, Kiener PA, Pinkus JL, Beggs AH, Amato AA, Greenberg SA. Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum. 2007;56:3784–92. doi: 10.1002/art.22928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866–73. doi: 10.1038/nature05663. [DOI] [PubMed] [Google Scholar]
- 143.Oestreicher JL, Walters IB, Kikuchi T, Gilleaudeau P, Surette J, Schwertschlag U, Dorner AJ, Krueger JG, Trepicchio WL. Molecular classification of psoriasis disease-associated genes through pharmacogenomic expression profiling. Pharmacogenomics J. 2001;1:272–87. doi: 10.1038/sj.tpj.6500067. [DOI] [PubMed] [Google Scholar]
- 144.Nomura I, Gao B, Boguniewicz M, Darst MA, Travers JB, Leung DY. Distinct patterns of gene expression in the skin lesions of atopic dermatitis and psoriasis: a gene microarray analysis. J Allergy Clin Immunol. 2003;112:1195–202. doi: 10.1016/j.jaci.2003.08.049. [DOI] [PubMed] [Google Scholar]
- 145.Bowcock AM, Shannon W, Du F, Duncan J, Cao K, Aftergut K, Catier J, Fernandez-Vina MA, Menter A. Insights into psoriasis and other inflammatory diseases from large-scale gene expression studies. Hum Mol Genet. 2001;10:1793–805. doi: 10.1093/hmg/10.17.1793. [DOI] [PubMed] [Google Scholar]
- 146.Zhou X, Krueger JG, Kao MC, Lee E, Du F, Menter A, Wong WH, Bowcock AM. Novel mechanisms of T-cell and dendritic cell activation revealed by profiling of psoriasis on the 63,100-element oligonucleotide array. Physiol Genomics. 2003;13:69–78. doi: 10.1152/physiolgenomics.00157.2002. [DOI] [PubMed] [Google Scholar]
- 147.Imboden JB. The immunopathogenesis of rheumatoid arthritis. Annu Rev Pathol. 2009;4:417–34. doi: 10.1146/annurev.pathol.4.110807.092254. [DOI] [PubMed] [Google Scholar]
- 148.Devauchelle V, Marion S, Cagnard N, Mistou S, Falgarone G, Breban M, Letourneur F, Pitaval A, Alibert O, Lucchesi C, Anract P, Hamadouche M, Ayral X, Dougados M, Gidrol X, Fournier C, Chiocchia G. DNA microarray allows molecular profiling of rheumatoid arthritis and identification of pathophysiological targets. Genes Immun. 2004;5:597–608. doi: 10.1038/sj.gene.6364132. [DOI] [PubMed] [Google Scholar]
- 149.van der Pouw Kraan TC, van Gaalen FA, Kasperkovitz PV, Verbeet NL, Smeets TJ, Kraan MC, Fero M, Tak PP, Huizinga TW, Pieterman E, Breedveld FC, Alizadeh AA, Verweij CL. Rheumatoid arthritis is a heterogeneous disease: evidence for differences in the activation of the STAT-1 pathway between rheumatoid tissues. Arthritis Rheum. 2003;48:2132–45. doi: 10.1002/art.11096. [DOI] [PubMed] [Google Scholar]
- 150.van der Pouw Kraan TC, Wijbrandts CA, van Baarsen LG, Rustenburg F, Baggen JM, Verweij CL, Tak PP. Responsiveness to anti-tumour necrosis factor alpha therapy is related to pre-treatment tissue inflammation levels in rheumatoid arthritis patients. Ann Rheum Dis. 2008;67:563–6. doi: 10.1136/ard.2007.081950. [DOI] [PubMed] [Google Scholar]
- 151.Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360:1989–2003. doi: 10.1056/NEJMra0806188. [DOI] [PubMed] [Google Scholar]
- 152.Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, Whitfield ML. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS One. 2008;3:e2696. doi: 10.1371/journal.pone.0002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bos CL, van Baarsen LG, Timmer TC, Overbeek MJ, Basoski NM, Rustenburg F, Baggen JM, Thiesen HJ, Dijkmans BA, van der Pouw Kraan TC, Voskuyl AE, Verweij CL. Molecular subtypes of systemic sclerosis in association with anti-centromere antibodies and digital ulcers. Genes Immun. 2009;10:210–8. doi: 10.1038/gene.2008.98. [DOI] [PubMed] [Google Scholar]
- 154.Rueda B, Broen J, Simeon C, Hesselstrand R, Diaz B, Suarez H, Ortego-Centeno N, Riemekasten G, Fonollosa V, Vonk MC, van den Hoogen FH, Sanchez-Roman J, Aguirre-Zamorano MA, Garcia-Portales R, Pros A, Camps MT, Gonzalez-Gay MA, Coenen MJ, Airo P, Beretta L, Scorza R, van Laar J, Gonzalez-Escribano MF, Nelson JL, Radstake TR, Martin J. The STAT4 gene influences the genetic predisposition to systemic sclerosis phenotype. Hum Mol Genet. 2009;18:2071–7. doi: 10.1093/hmg/ddp119. [DOI] [PubMed] [Google Scholar]
- 155.Frohman EM, Racke MK, Raine CS. Multiple sclerosis--the plaque and its pathogenesis. N Engl J Med. 2006;354:942–55. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 156.Mycko MP, Papoian R, Boschert U, Raine CS, Selmaj KW. cDNA microarray analysis in multiple sclerosis lesions: detection of genes associated with disease activity. Brain. 2003;126:1048–57. doi: 10.1093/brain/awg107. [DOI] [PubMed] [Google Scholar]
- 157.Mycko MP, Papoian R, Boschert U, Raine CS, Selmaj KW. Microarray gene expression profiling of chronic active and inactive lesions in multiple sclerosis. Clin Neurol Neurosurg. 2004;106:223–9. doi: 10.1016/j.clineuro.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 158.Iglesias AH, Camelo S, Hwang D, Villanueva R, Stephanopoulos G, Dangond F. Microarray detection of E2F pathway activation and other targets in multiple sclerosis peripheral blood mononuclear cells. J Neuroimmunol. 2004;150:163–77. doi: 10.1016/j.jneuroim.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 159.Satoh J, Nakanishi M, Koike F, Miyake S, Yamamoto T, Kawai M, Kikuchi S, Nomura K, Yokoyama K, Ota K, Kanda T, Fukazawa T, Yamamura T. Microarray analysis identifies an aberrant expression of apoptosis and DNA damage-regulatory genes in multiple sclerosis. Neurobiol Dis. 2005;18:537–50. doi: 10.1016/j.nbd.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 160.Koike F, Satoh J, Miyake S, Yamamoto T, Kawai M, Kikuchi S, Nomura K, Yokoyama K, Ota K, Kanda T, Fukazawa T, Yamamura T. Microarray analysis identifies interferon beta-regulated genes in multiple sclerosis. J Neuroimmunol. 2003;139:109–18. doi: 10.1016/s0165-5728(03)00155-3. [DOI] [PubMed] [Google Scholar]
- 161.Sturzebecher S, Wandinger KP, Rosenwald A, Sathyamoorthy M, Tzou A, Mattar P, Frank JA, Staudt L, Martin R, McFarland HF. Expression profiling identifies responder and non-responder phenotypes to interferon-beta in multiple sclerosis. Brain. 2003;126:1419–29. doi: 10.1093/brain/awg147. [DOI] [PubMed] [Google Scholar]
- 162.Pappas DJ, Coppola G, Gabatto PA, Gao F, Geschwind DH, Oksenberg JR, Baranzini SE. Longitudinal system-based analysis of transcriptional responses to type I interferons. Physiol Genomics. 2009;38:362–71. doi: 10.1152/physiolgenomics.00058.2009. [DOI] [PubMed] [Google Scholar]
- 163.Huang X, Yuang J, Goddard A, Foulis A, James RF, Lernmark A, Pujol-Borrell R, Rabinovitch A, Somoza N, Stewart TA. Interferon expression in the pancreases of patients with type I diabetes. Diabetes. 1995;44:658–64. doi: 10.2337/diab.44.6.658. [DOI] [PubMed] [Google Scholar]
- 164.Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–26. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 165.Larsen CM, Faulenbach M, Vaag A, Ehses JA, Donath MY, Mandrup-Poulsen T. Sustained Effects of Interleukin-1-Receptor Antagonist Treatment in Type 2 Diabetes Mellitus. Diabetes Care. 2009 doi: 10.2337/dc09-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cassidy JT, Ross E. Juvenile Rheumatoid Arthritis. Textbook of Pediatric Rheumatology. 2001:218–321. [Google Scholar]
- 167.Chaussabel D, Allman W, Mejias A, Chung W, Bennett L, Ramilo O, Pascual V, Palucka AK, Banchereau J. Analysis of significance patterns identifies ubiquitous and disease-specific gene-expression signatures in patient peripheral blood leukocytes. Ann N Y Acad Sci. 2005;1062:146–54. doi: 10.1196/annals.1358.017. [DOI] [PubMed] [Google Scholar]
- 168.Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, Bolouri MS, Ray HN, Sihag S, Kamal M, Patterson N, Lander ES, Mann M. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–40. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- 169.Segal E, Yelensky R, Koller D. Genome-wide discovery of transcriptional modules from DNA sequence and gene expression. Bioinformatics. 2003;19 1:i273–82. doi: 10.1093/bioinformatics/btg1038. [DOI] [PubMed] [Google Scholar]
- 170.Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet. 2006;7:55–65. doi: 10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- 171.Horvath S, Dong J. Geometric interpretation of gene coexpression network analysis. PLoS Comput Biol. 2008;4:e1000117. doi: 10.1371/journal.pcbi.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chaussabel D, Quinn C, Shen J, Patel P, Glaser C, Baldwin N, Stichweh D, Blankenship D, Li L, Munagala I, Bennett L, Allantaz F, Mejias A, Ardura M, Kaizer E, Monnet L, Allman W, Randall H, Johnson D, Lanier A, Punaro M, Wittkowski KM, White P, Fay J, Klintmalm G, Ramilo O, Palucka AK, Banchereau J, Pascual V. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity. 2008;29:150–64. doi: 10.1016/j.immuni.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liang MH, Socher SA, Larson MG, Schur PH. Reliability and validity of six systems for the clinical assessment of disease activity in systemic lupus erythematosus. Arthritis Rheum. 1989;32:1107–18. doi: 10.1002/anr.1780320909. [DOI] [PubMed] [Google Scholar]
- 174.Gladman DD, Ibanez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol. 2002;29:288–91. [PubMed] [Google Scholar]
- 175.Snyder M, Weissman S, Gerstein M. Personal phenotypes to go with personal genomes. Mol Syst Biol. 2009;5:273. doi: 10.1038/msb.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Whitney AR, Diehn M, Popper SJ, Alizadeh AA, Boldrick JC, Relman DA, Brown PO. Individuality and variation in gene expression patterns in human blood. Proc Natl Acad Sci U S A. 2003;100:1896–901. doi: 10.1073/pnas.252784499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Debey S, Zander T, Brors B, Popov A, Eils R, Schultze JL. A highly standardized, robust, and cost-effective method for genome-wide transcriptome analysis of peripheral blood applicable to large-scale clinical trials. Genomics. 2006;87:653–64. doi: 10.1016/j.ygeno.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 178.Debey-Pascher S, Eggle D, Schultze JL. RNA stabilization of peripheral blood and profiling by bead chip analysis. Methods Mol Biol. 2009;496:175–210. doi: 10.1007/978-1-59745-553-4_13. [DOI] [PubMed] [Google Scholar]
- 179.Tian Z, Palmer N, Schmid P, Yao H, Galdzicki M, Berger B, Wu E, Kohane IS. A practical platform for blood biomarker study by using global gene expression profiling of peripheral whole blood. PLoS One. 2009;4:e5157. doi: 10.1371/journal.pone.0005157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Vartanian K, Slottke R, Johnstone T, Casale A, Planck SR, Choi D, Smith JR, Rosenbaum JT, Harrington CA. Gene expression profiling of whole blood: comparison of target preparation methods for accurate and reproducible microarray analysis. BMC Genomics. 2009;10:2. doi: 10.1186/1471-2164-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Li L, Ying L, Naesens M, Xiao W, Sigdel T, Hsieh S, Martin J, Chen R, Liu K, Mindrinos M, Davis R, Sarwal M. Interference of globin genes with biomarker discovery for allograft rejection in peripheral blood samples. Physiol Genomics. 2008;32:190–7. doi: 10.1152/physiolgenomics.00216.2007. [DOI] [PubMed] [Google Scholar]
- 182.Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006;24:1151–61. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Shi L, Perkins RG, Fang H, Tong W. Reproducible and reliable microarray results through quality control: good laboratory proficiency and appropriate data analysis practices are essential. Curr Opin Biotechnol. 2008;19:10–8. doi: 10.1016/j.copbio.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 184.Tan PK, Downey TJ, Spitznagel EL, Jr, Xu P, Fu D, Dimitrov DS, Lempicki RA, Raaka BM, Cam MC. Evaluation of gene expression measurements from commercial microarray platforms. Nucleic Acids Res. 2003;31:5676–84. doi: 10.1093/nar/gkg763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Butte AJ, Kohane IS. Creation and implications of a phenome-genome network. Nat Biotechnol. 2006;24:55–62. doi: 10.1038/nbt1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci U S A. 2004;101:9309–14. doi: 10.1073/pnas.0401994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fraser ID, Germain RN. Navigating the network: signaling cross-talk in hematopoietic cells. Nat Immunol. 2009;10:327–31. doi: 10.1038/ni.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zak DE, Aderem A. Systems biology of innate immunity. Immunol Rev. 2009;227:264–82. doi: 10.1111/j.1600-065X.2008.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, James JJ, Maysuria M, Mitton JD, Oliveri P, Osborn JL, Peng T, Ratcliffe AL, Webster PJ, Davidson EH, Hood L, Dimitrov K. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–25. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 190.Wold B, Myers RM. Sequence census methods for functional genomics. Nat Methods. 2008;5:19–21. doi: 10.1038/nmeth1157. [DOI] [PubMed] [Google Scholar]
- 191.Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, Schmidt D, O'Keeffe S, Haas S, Vingron M, Lehrach H, Yaspo ML. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–60. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 192.Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–69. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 193.Martin DA, Elkon KB. Autoantibodies make a U-turn: the toll hypothesis for autoantibody specificity. J Exp Med. 2005;202:1465–9. doi: 10.1084/jem.20052228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–96. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 195.Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, Cao W, Wang YH, Su B, Nestle FO, Zal T, Mellman I, Schroder JM, Liu YJ, Gilliet M. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–9. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 196.Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, Homey B, Barrat FJ, Zal T, Gilliet M. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009;206:1983–94. doi: 10.1084/jem.20090480. [DOI] [PMC free article] [PubMed] [Google Scholar]