

Abstract
We present a program, named Promega, to predict the Xaa-Pro peptide bond conformation on the basis of backbone chemical shifts and the amino acid sequence. Using a chemical shift database of proteins of known structure together with the PDB-extracted amino acid preference of cis Xaa-Pro peptide bonds, a cis/trans probability score is calculated from the backbone and 13Cβ chemical shifts of the proline and its neighboring residues. For an arbitrary number of input chemical shifts, which may include Pro-13Cγ, Promega calculates the statistical probability that a Xaa-Pro peptide bond is cis. Besides its potential as a validation tool, Promega is particularly useful for studies of larger proteins where Pro-13Cγ assignments can be challenging, and for on-going efforts to determine protein structures exclusively on the basis of backbone and 13Cβ chemical shifts.
Keywords: backbone chemical shifts, CS-Rosetta, NMR, omega angle, proline, structure prediction
In proteins, the majority (~99.7%) of peptide bonds is found to be in the trans conformation (Weiss et al. 1998), which is energetically favorable due to (1) stronger favorable electrostatic interactions between Oi and C’i+1 and (2) the steric repulsion between the Cα/Hα atoms of the two bonded amino acids in a cis conformation (Wedemeyer et al. 2002). However, Pro residues exhibit a higher preference to form a cis Xaa-Pro peptide bond, attributed to the steric similarity between the cis configuration and the corresponding trans form. In proteins, the population of Pro residues engaged in a cis Xaa-Pro peptide bond is about 5%, making the correct identification of the Xaa-Pro peptide bond conformation an important task during NMR protein structure determination. Proline isomerization plays an important role in protein folding and can modulate protein function, and therefore remains the subject of active on-going research (Jahn et al. 2006)(Baldwin 2008; Lindfors et al. 2008; Andrews et al. 2009; Day et al. 2009; Pascal et al. 2009; Severin et al. 2009; Weininger et al. 2009).
When following standard NMR procedures, identification of the cis or trans form of a Xaa-Pro peptide bond generally relies on the observation of a strong Hα-Hα or Hα-Hδ sequential NOE (Wüthrich 1986). However, considering that the isomer-specific 1Hα-1Hα or 1Hα-1Hδ NOE intensities can be difficult to quantify, due to the proximity of such resonances to the intense water signal, the often crowded nature of the 1Hα-1Hδ spectral region when considering larger proteins, and the absence of such signals when studying fully perdeuterated proteins, identification of the correct Xaa-Pro peptide bond conformation can remain a non-trivial task (Torchia et al. 1989). An alternate method for distinguishing cis and trans Xaa-Pro peptide bonds relies on the empirical finding that the 13C chemical shifts for Pro in cis and trans configurations differ significantly, both in small peptides and in proteins (Dorman and Bovey 1973; Siemion et al. 1975; Howarth and Lilley 1978; Schubert et al. 2002). In particular, the chemical shift difference between 13Cβ and 13Cγ has proven to be a reliable indicator for identifying the conformation of Xaa-Pro peptide bonds (Siemion et al. 1975; Schubert et al. 2002). In view of recent work aimed at determining protein structures solely from backbone NMR chemical shifts (Cavalli et al. 2007; Robustelli et al. 2008; Shen et al. 2008; Wishart et al. 2008; Shen et al. 2009b), it is particularly important to develop a robust procedure for identifying cis-Pro peptide bonds without recourse to NOE data or the often unavailable 13Cγ chemical shifts. The current study aims to present such a procedure.
Nowadays, a large number of proteins with both NMR chemical shifts and high-quality structures are available for studying the relations between NMR chemical shifts and local protein structure (Cornilescu et al. 1999; Neal et al. 2003; Shen and Bax 2007; London et al. 2008; Kohlhoff et al. 2009; Shen et al. 2009a), providing a good basis to further explore the relation between NMR chemical shifts and the cis/trans state of Xaa-Pro peptide bonds. Here, we first evaluate the relation between both the local amino acid sequence as well as the chemical shift patterns of Pro and its neighboring residues and the cis/trans state of the Xaa-Pro peptide bond, using a database newly constructed from proteins for which high resolution X-ray coordinates are available in the PDB, and chemical shifts have been deposited in the BMRB (Markley et al. 2008). Then, we present a scoring function to calculate the probability of any given Pro-centered tripeptide to contain a cis Xaa-Pro peptide bond.
We explore the relation between residue sequence and the probability of finding any given Xaa-Pro peptide bond in the cis or trans configuration by using the protein database originally developed for the CS-Rosetta program (Shen et al. 2009b). This database contains 109,396 Pro-centered tri-peptides of which 4,919 with a cis Xaa-Pro peptide bond, spread over 9446 proteins for which high-resolution (≤2.5Å) X-ray structures are available. This protein sequence database is about three times larger than the database previously used for the same purpose, which also had a less stringent X-ray resolution cutoff (Pahlke et al. 2005), but shows quite similar amino acid preferences for the residues neighboring cis and trans Pro (Table S1; Figure S1). Analysis of these structures indicates that Gly and the aromatic amino acids Phe, Trp, and Tyr are more prevalent in the Xaa position preceding a cis-Pro, and Phe, His, Trp, and Tyr are favored in the position following a cis Xaa-Pro peptide bond. Asp, Ile, Leu, Val, and Met are disfavored to precede a cis Pro, and Asp, Glu, and Ile are disfavored to follow a cis Pro.
A second database, containing 1,746 Pro-centered tripeptides (114 with a cis and 1,632 with a trans Xaa-Pro peptide bond), was constructed from 580 proteins for which both a high-resolution X-ray structure and (nearly) complete BMRB chemical shifts (δ15N, δ13C’, δ13Cα, δ13Cβ, δ1Hα and δ1HN) are available. The preparation of this chemical shift database, including the calculation of secondary chemical shifts, chemical shift re-referencing, exclusion of residues with large B-factors in the X-ray reference structure, and exclusion of chemical shift outliers follows the same procedure as was used for generating TALOS (Cornilescu et al. 1999) and SPARTA (Shen and Bax 2007) protein databases. Moreover, 89 proteins in the database were found to be studied using perdeuterated sample, with corresponding substantial 2H isotope effects on 13Cα/β chemical shifts. Uniform corrections for these isotope effects were applied for those proteins following the same procedure described previously (Shen et al. 2009b).
Using this pruned database, the average secondary chemical shift , as well as the standard deviation , are calculated for each backbone atom x [x=(15N, 13C’, 13Cα, 13Cβ, 1Hα and 1HN)] at position j [j = i-1,i,i+1] of the Pro-centered tripeptides with Xaa-Pro peptide bond conformation Y (Y=cis or trans), with results listed in Table 1 and Supplementary Table S2, and shown in Figures 1A and S2. For slightly more than half of the tri-peptides in the data base (940, of which 45 have a cis Pro), Pro 13Cγ chemical shifts were also available (Fig. 1D).
Table 1.
Average secondary chemical shift and standard deviation for backbone and 13Cβ and 13Cγ nuclei of Pro residues preceded by a cis or trans peptide bond.a
| X | ||||
|---|---|---|---|---|
| 1Hα | 0.17 | 0.53 | −0.03 | 0.39 |
| 13C’ | −1.78 | 1.45 | −0.26 | 1.58 |
| 13Cα | −0.19 | 1.07 | 0.55 | 1.47 |
| 13Cβ | 2.42 | 1.27 | 0.21 | 0.97 |
| 13Cγ b | 24.41 | 0.74 | 27.45 | 0.86 |
Using the random coil chemical shifts used by TALOS+ (Shen et al. 2009a).
The regular chemical shift instead of the secondary chemical shift is reported
Figure 1.
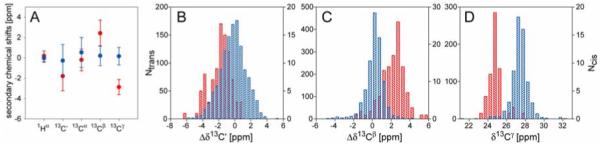
Chemical shift distribution for Pro residues in folded proteins. (A) Plot of the average 1Hα, 13C’, 13Cα, 13Cβ and 13Cγ secondary chemical shifts and their standard deviations for Pro with cis (red) or trans (blue) Xaa-Pro peptide bond. Δδ13Cγ are calculated using an average chemical shift of 27.28 ppm provided by BMRB, all other secondary chemical shifts are calculated relative to the random coil values taken from TALOS+ program (Shen et al. 2009a). (B-D) Histograms of 13C’ (B), 13Cβ (C) secondary chemical shifts and 13Cγ (D) chemical shifts of Pro residues preceded by a cis (red, right y-axis) or trans (blue, left y-axis) peptide bond.
As previously found, our data show that the 13Cβ and 13Cγ chemical shifts of Pro engaged in cis and trans Xaa-Pro peptide bonds differ considerably (Fig. 1, Table 1). We find δ13Cβ values for Pro with trans and cis conformations at 31.8±1.0 and 33.8±1.2 ppm, respectively, while δ13Cγ values fall at 27.4±0.9 and 24.4±0.7 ppm, respectively, and therefore are much more discriminating. When considering the difference of δ13Cβ and δ13Cγ, or Δβγ, our values of 4.5±1.2 and 9.4±1.3 ppm, respectively, agree closely with those of previous studies (Siemion et al. 1975; Schubert et al. 2002), confirming their utility as an indicator for cis and trans Pro peptide bonds. Furthermore, the 13C’ chemical shifts are found to be different for cis and trans Pro (Fig. 1B; Table 1), albeit to a lesser extent than 13Cβ and 13Cγ. With the exception of δ1Hα of the preceding residue, which is found to be more up-field when this residue precedes a cis peptide bond, the chemical shifts of the two Pro-neighboring residues correlate only very weakly with the cis/trans form of the peptide bond (Table S2; Fig. S2).
The above residue type and chemical shift analysis for Pro and its immediate neighbors can be used to estimate the probability for the Xaa-Pro peptide bond to be cis or trans. Below, we describe the computational procedure used by the program Promega to predict the Pro omega angle. Promega utilizes a probability scoring function which includes both the amino acid type and the chemical shifts of a Pro-centered tripeptide to predict its Xaa-Pro peptide bond conformation.
At first, a normalized relative occurrence H (Pahlke et al. 2005) is defined to account for the occurrence of each amino acid type (X) in the neighboring positions (j) of a Pro residue at position i:
| [1] |
where stands for the observed occurrence of amino acid X at position j of a given Pro-centered tripeptide, NX is the natural occurrence of amino acid X in the database.
Next, assuming a Gaussian distribution for the chemical shifts (Fig. 1 B-D), a chemical shift score function, , is generated to account for the compatibility of a given Pro-centered tri-peptide with either a cis or trans Xaa-Pro peptide bond:
| [2] |
where x=[15N, 13C’, 13Cα, 13Cβ, 1Hα and 1HN] for the two neighboring residues, x=[13C’, 13Cα, 13Cβ, 1Hα, and 13Cγ (if available)] for the center Pro residue, Y=[cis, trans], and
| [3] |
where is the experimental secondary chemical shift (corrected for 2H isotope shifts when applicable).
An overall score function, taking into account both the amino acid type and the chemical shift patterns, is then used to estimate the compatibility of the chemical shifts and residue types of a given Pro-centered tripeptide Xaa-Pro-Xbb with those of a cis or trans Xaa-Pro peptide bond:
| [4a] |
where
| [4b] |
The propensity of a given Pro-centered tri-peptide to have a cis Xaa-Pro peptide bond is given by:
| [5] |
Note that Pcis does not represent a normalized probability, as it does not yet account for the fact that the total likelihood of any given Pro to be in the cis form, in the absence of chemical shift or residue type information, is only ca 5%. The Pcis value of 0.5, corresponding to such a case, simply indicates that the Pro residue has the database average likelihood of 5% to be cis. Therefore, Pcis simply signifies the above or below average likelihood for a Pro residue to be in the cis form. Pcis can be converted to a normalized, true probability by:
| [6] |
The effectiveness of Pcis to identify cis-Pro peptide bonds is illustrated in Figure 2. As expected on the basis of previous results (Schubert et al. 2002), when 13Cγ chemical shifts are present along with the 13Cβ chemical shifts (Fig. 2A), the Pcis score is an excellent indicator for the Xaa-Pro peptide bond. Figure 2A shows that 42 out of 45 tripeptides with cis Xaa-Pro peptide bond having a Pcis score close to 1.0, and NOE data indicates that for at least two of the three cis-Pro residues with low Pcis scores the peptide bond differs in conformation from the crystal structure (Table S3). For 846 out of 895 tripeptides with a trans Xaa-Pro peptide bond, Pcis ≤ 0.1, corresponding to (Fig.2A).
Figure 2.

Prediction of Xaa-Pro peptide bond conformation from NMR chemical shifts by the program Promega. (A,B) Plot of the crystallographically observed ω angles, versus their probability score, Pcis, calculated by eq 5, (A) for residues that included 13Cγ chemical shift assignments, and (B) for all database residues with at least three out of four (13C’, 13Cα, 13Cβ, 1Hα) Pro assignments, and ignoring 13Cγ chemical shifts. Note that Pcis (eq 5) corresponds to a relative probability, i.e., Pcis = 0.5 refers to the case where the cis probability equals the average cis probability in the database (ca 5%). (C,D) Histograms of the normalized, true probability score , calculated by using eq 6 for Pro residues in the database in (C) the presence and (D) the absence of 13Cγ chemical shifts. The solid line in (D) corresponds to eq 6. Note that in the presence of 13Cγ chemical shifts the Pcis result is essentially binary, and the below unity value observed in the histogram for the highest Pcis bin largely reflects cases where solution and crystal structures differ.
Manual inspection of several of the prediction outliers, for which additional NMR data were available, including an NMR reference structures and/or NOE restraints (Table S3), suggests differences between the crystal and solution structures for at least a significant fraction of the outliers. For example, the two tripeptide segments with smallest Pcis score but with a cis Pro in the X-ray reference structure (Fig. 2A; Table S3), exhibit strong sequential Hαi-1 to Pro-Hδi NOEs, characteristic of a trans peptide bond in the NMR structure. Similarly, for a number of residues with a high Pcis value (≥0.99) but a trans peptide bond in the X-ray structure, NOE data strongly point to cis peptide bonds (Table S3).
13Cγ chemical shifts are often difficult to assign, in particular for larger proteins and when using perdeuterated samples. Therefore, the ability of Pcis scores to identify cis peptide bonds without recourse to δ13Cγ is particularly important. Such Pcis scores were calculated for all Xaa-Pro-Xbb tripeptides contained in the chemical shift database (Figure 2B), and show that Pcis remains a strong indicator for the Xaa-Pro peptide bond. For example, 66 out of 82 tripeptides with cis Xaa-Pro peptide bond have a Pcis score >0.8, and 969 out of 1436 tripeptides with trans Xaa-Pro peptide bond have a Pcis score <0.2. When viewed in histogram format, the fraction of Pro residues with a cis Pro peptide bond as a function of its Pcis score closely follows the expected probability of eq 6, i.e., the actual probability for a cis Pro peptide bond (Fig. 2D).
When no chemical shifts are available, i.e., only the residue type information is included in eq 4, the ability of Promega to identify cis Xaa-Pro peptide bonds is much decreased, and with Pcis values then falling in the 0.2-0.8 range (Figure S3), these values are only suggestive of whether a given Pro residue has above (Pcis > 0.5) or below (Pcis < 0.5) average likelihood to be in the cis configuration.
Promega output serves as an alert to exercise caution when building molecular models in the absence of complete sets of NOE data, where the possibility of a cis peptide bond is often ignored. In this respect, an important application concerns the chemical shift based protein structure generation process (Cavalli et al. 2007; Shen et al. 2008; Wishart et al. 2008). These protocols rely on empirically optimized search procedures to select protein fragments from a protein database, followed by an assembly and relaxation procedure. The character of the Xaa-Pro peptide bond is generally not considered during the fragment selection procedure and its chemical shifts provide insufficient weight when selecting larger fragments. Therefore, such fragment pools usually do not differ significantly in the fraction that contains a cis Xaa-Pro peptide bond from the database average of ~5%. This makes it likely that true cis peptide bonds are not identified during such procedures. Therefore, the MFR fragment search procedure (Kontaxis et al. 2005) has been adapted such that Pcis functions as an additional term defining the ω torsion angle, improving the quality of input fragments for the CS-Rosetta protocol. As an example, we consider the protein XcR50, for which models were calculated in our original study of the effectiveness of CS-Rosetta.(Shen et al. 2008) This 76-residue protein contains five Pro residues, of which three have cis peptide bonds in the X-ray reference structure (Table S5). Nine out of ten lowest energy models of our previous CS-Rosetta study correctly identified cis-Pro62, but failed to identify cis-Pro49. NOE and chemical shift data indicate that the C-terminal residue Pro76 is trans in solution. Including a Promega term in MFR fragment selection results in CS-Rosetta models that have the correct cis conformations for both Pro49 and Pro62, and clearly identify the other three as trans. Even though the local backbone rmsd with the Promega-term included in MFR fragment selection removes the discrepancy at Pro49 (Fig. 3B), the impact on the overall structure is minimal and the backbone rmsd relative to the X-ray structure remains unchanged at 1.7±0.3Å.
Figure 3.
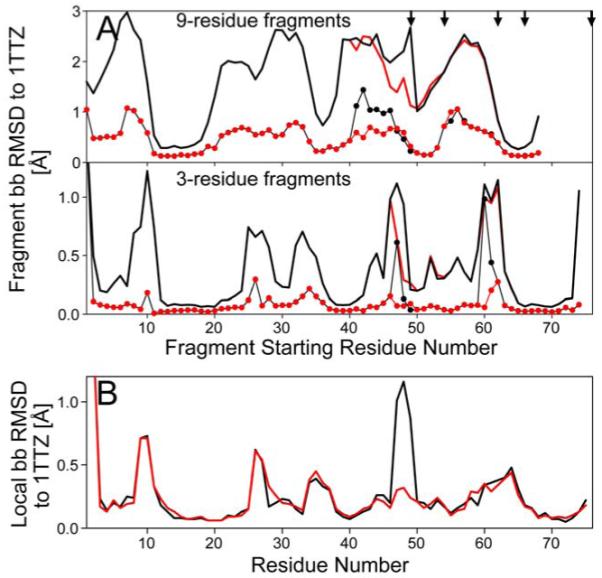
Impact of adding Promega output to CS-Rosetta. (A) Backbone RMSD of 200 MFR-selected CS-Rosetta fragments for protein XcR50. Plots of the lowest (thin lines with dots) and average (bold lines) backbone coordinate rmsd’s (N, Cα and C’) between any given segment in the experimental structure (PDB entry 1TTZ) and 200 fragments, as a function of starting position of the query segment (note that the 68 is the last starting residue for a 9-residue fragment in this 76-residue protein). Results from the standard MFR method with chemical shifts are plotted in black, while those selected using the MFR method that includes a Promega term (with no δ13Cγ shifts considered), are in red. The locations of five Pro residues are marked by arrows. (B) Local accuracy of the XcR50 CS-Rosetta structures, relative to the reference Xray structure (1TTZ). For the 10 lowest energy CS-Rosetta models obtained using standard (black line) and Promega-included MFR (red line), the average local backbone RMSD (N, Cα, C’) values are plotted against the center residue number for fragments of 3 residues.
Promega is written in the C++ language, and reports both the Pcis and normalized scores, calculated by Eqs 4-6. The backbone and 13Cβ chemical shifts of each Pro-centered tripeptide, the 13Cγ chemical shift of the center Proline residue if available, as well as the amino acid types of the two neighboring residues, serve as input for the prediction. A script is included to correct the 13Cα/β chemical shifts for 2H isotope effects, if required.
In conclusion, the cis/trans configuration of Xaa-Pro peptide bonds in proteins can be predicted with reasonable accuracy on the basis of amino acid type and backbone and 13Cβ chemical shifts. When both Pro 13Cγ and 13 Cβ chemical shifts are available, the Xaa-Pro cis or trans state will be identified uniquely for the vast majority of cases. In the absence of Pro 13Cγ chemical shifts, Promega merely provides a statistical probability for the peptide bond in question to be cis or trans.
Supplementary Material
Acknowledgments
This work was funded by the Intramural Research Program of the NIDDK, NIH, and by the Intramural AIDS-Targeted Antiviral Program of the Office of the Director, NIH. CS-Rosetta calculations were carried out on the NIH CIT Biowulf cluster.
Footnotes
Software availability
Promega and the CS-Rosetta package with its updated MFR protocol can be downloaded from http://spin.niddk.nih.gov/bax/.
References
- Andrews BT, Roy M, Jennings PA. Chromophore packing leads to hysteresis in GFP. J. Mol. Biol. 2009;392:218–227. doi: 10.1016/j.jmb.2009.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin RL. The search for folding intermediates and the mechanism of protein folding. Annual Review of Biophysics. 2008;37:1–21. doi: 10.1146/annurev.biophys.37.032807.125948. [DOI] [PubMed] [Google Scholar]
- Cavalli A, Salvatella X, Dobson CM, Vendruscolo M. Protein structure determination from NMR chemical shifts. Proc. Natl. Acad. Sci. U. S. A. 2007;104:9615–9620. doi: 10.1073/pnas.0610313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- Day IJ, Maeda K, Paisley HJ, Mok KH, Hore PJ. Refolding of ribonuclease A monitored by real-time photo-CIDNP NMR spectroscopy. J. Biomol. NMR. 2009;44:77–86. doi: 10.1007/s10858-009-9322-2. [DOI] [PubMed] [Google Scholar]
- Dorman DE, Bovey FA. C-13 magnetic resonance spectroscopy - spectrum of proline in oligopeptides. J. Org. Chem. 1973;38:2379–2383. [Google Scholar]
- Howarth OW, Lilley DMJ. Carbon-13 NMR of peptides and proteins. Prog. Nucl. Magn. Reson. Spectrosc. 1978;12:1–40. [Google Scholar]
- Jahn TR, Parker MJ, Homans SW, Radford SE. Amyloid formation under physiological conditions proceeds via a native-like folding intermediate. Nature Structural & Molecular Biology. 2006;13:195–201. doi: 10.1038/nsmb1058. [DOI] [PubMed] [Google Scholar]
- Kohlhoff KJ, Robustelli P, Cavalli A, Salvatella X, Vendruscolo M. Fast and Accurate Predictions of Protein NMR Chemical Shifts from Interatomic Distances. J. Am. Chem. Soc. 2009;131:13894–13895. doi: 10.1021/ja903772t. [DOI] [PubMed] [Google Scholar]
- Kontaxis G, Delaglio F, Bax A. Molecular fragment replacement approach to protein structure determination by chemical shift and dipolar homology database mining. Meth. Enzymol. 2005;394:42–78. doi: 10.1016/S0076-6879(05)94003-2. [DOI] [PubMed] [Google Scholar]
- Lindfors HE, de Koning PE, Drijfhout JW, Venezia B, Ubbink M. Mobility of TOAC spin-labelled peptides binding to the Src SH3 domain studied by paramagnetic NMR. J. Biomol. NMR. 2008;41:157–167. doi: 10.1007/s10858-008-9248-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London RE, Wingad BD, Mueller GA. Dependence of amino acid side chain C-13 shifts on dihedral angle: Application to conformational analysis. J. Am. Chem. Soc. 2008;130:11097–11105. doi: 10.1021/ja802729t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markley JL, Ulrich EL, Berman HM, Henrick K, Nakamura H, Akutsu H. BioMagResBank (BMRB) as a partner in the Worldwide Protein Data Bank (wwPDB): new policies affecting biomolecular NMR depositions. J. Biomol. NMR. 2008;40:153–155. doi: 10.1007/s10858-008-9221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S, Nip AM, Zhang HY, Wishart DS. Rapid and accurate calculation of protein H-1, C-13 and N-15 chemical shifts. J. Biomol. NMR. 2003;26:215–240. doi: 10.1023/a:1023812930288. [DOI] [PubMed] [Google Scholar]
- Pahlke D, Freund C, Leitner D, Labudde D. Statistically significant dependence of the Xaa-Pro peptide bond conformation on secondary structure and amino acid sequence. Bmc Structural Biology. 2005:5. doi: 10.1186/1472-6807-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal C, Pate F, Cheynier V, Delsuc MA. Study of the Interactions Between a Proline-Rich Protein and a Flavan-3-ol by NMR: Residual Structures in the Natively Unfolded Protein Provides Anchorage Points for the Ligands. Biopolymers. 2009;91:745–756. doi: 10.1002/bip.21221. [DOI] [PubMed] [Google Scholar]
- Robustelli P, Cavalli A, Vendruscolo M. Determination of Protein Structures in the Solid State from NMR Chemical Shifts. Structure. 2008;16:1764–1769. doi: 10.1016/j.str.2008.10.016. [DOI] [PubMed] [Google Scholar]
- Schubert M, Labudde D, Oschkinat H, Schmieder P. A software tool for the prediction of Xaa-Pro peptide bond conformations in proteins based on C-13 chemical shift statistics. J. Biomol. NMR. 2002;24:149–154. doi: 10.1023/a:1020997118364. [DOI] [PubMed] [Google Scholar]
- Severin A, Joseph RE, Boyken S, Fulton DB, Andreotti AH. Proline Isomerization Preorganizes the Itk SH2 Domain for Binding to the Itk SH3 Domain. J. Mol. Biol. 2009;387:726–743. doi: 10.1016/j.jmb.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Bax A. Protein backbone chemical shifts predicted from searching a database for torsion angle and sequence homology. J. Biomol. NMR. 2007;38:289–302. doi: 10.1007/s10858-007-9166-6. [DOI] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+ : a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 2009a;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu GH, Eletsky A, Wu YB, Singarapu KK, Lemak A, Ignatchenko A, Arrowsmith CH, Szyperski T, Montelione GT, Baker D, Bax A. Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. U. S. A. 2008;105:4685–4690. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Vernon R, Baker D, Bax A. De novo protein structure generation from incomplete chemical shift assignments. J. Biomol. NMR. 2009b;43:63–78. doi: 10.1007/s10858-008-9288-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemion IZ, Wieland T, Pook KH. Infuence of distance of proline carbonyl from beta and gamma carbon on C-13 chemical shifts. Angew. Chem.-Int. Edit. Engl. 1975;14:702–703. doi: 10.1002/anie.197507021. [DOI] [PubMed] [Google Scholar]
- Torchia DA, Sparks SW, Young PE, Bax A. Proline Assignments and Identification of the Cis K116/P117 Peptide-Bond in Liganded Staphylococcal Nuclease Using Isotope Edited 2d Nmr-Spectroscopy. J. Am. Chem. Soc. 1989;111:8315–8317. [Google Scholar]
- Wedemeyer WJ, Welker E, Scheraga HA. Proline cis-trans isomerization and protein folding. Biochemistry. 2002;41:14637–14644. doi: 10.1021/bi020574b. [DOI] [PubMed] [Google Scholar]
- Weininger U, Jakob RP, Eckert B, Schweimer K, Schmid FX, Balbach J. A remote prolyl isomerization controls domain assembly via a hydrogen bonding network. Proc. Natl. Acad. Sci. U. S. A. 2009;106:12335–12340. doi: 10.1073/pnas.0902102106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MS, Jabs A, Hilgenfeld R. Peptide bonds revisited. Nature Structural Biology. 1998;5:676–676. doi: 10.1038/1368. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Arndt D, Berjanskii M, Tang P, Zhou J, Lin G. CS23D: a web server for rapid protein structure generation using NMR chemical shifts and sequence data. Nucleic Acids Res. 2008;36:496–502. doi: 10.1093/nar/gkn305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüthrich K. NMR of Proteins and Nucleic Acids. John Wiley & Sons; New York: 1986. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.