

SUMMARY
Blood platelets provide the initial response to vascular endothelial injury, becoming activated as they adhere to the injured site. Activated platelets recruit leukocytes, and initiate proliferation and migration of vascular smooth muscle cells (SMC) within the injured vessel wall, leading to development of neointimal hyperplasia. Endothelial CD39/NTPDase1 and recombinant solCD39 rapidly metabolize nucleotides, including stimulatory ADP released from activated platelets, thereby suppressing additional platelet reactivity. Using a murine model of vascular endothelial injury, we investigated whether circulating human solCD39 could reduce platelet activation and accumulation, thus abating leukocyte infiltration and neointimal formation following vascular damage. Intraperitoneally-administered solCD39 ADPase activity in plasma peaked 1 hr post-injection, with an elimination half-life of 43 hr. Accordingly, mice were administered solCD39 or saline 1 hr prior to vessel injury, then either sacrificed 24 hr post-injury or treated with solCD39 or saline (3X weekly) for an additional 18 days. 24 hr post-injury, solCD39-treated mice displayed a reduction in platelet activation and recruitment, P-selectin expression, and leukocyte accumulation in the arterial lumen. Furthermore, repeated administration of solCD39 modulated the late stage of vascular injury by suppressing leukocyte deposition, macrophage infiltration and SMC proliferation/migration, resulting in abrogation of neointimal thickening. In contrast, injured femoral arteries of saline-injected mice exhibited massive platelet thrombus formation, marked P-selectin expression, and leukocyte infiltration. Pronounced neointimal growth with macrophage and SMC accretion was also observed (intimal-to-medial area ratio 1.56±0.34 at 19 days). Thus, systemic administration of solCD39 profoundly affects injury-induced cellular responses, minimizing platelet deposition and leukocyte recruitment, and suppressing neointimal hyperplasia.
Keywords: CD39, endothelial E-NTPDase1, arterial injury, platelet activation, vascular stenosis
What is known about this topic?
-
-
Endothelial damage promotes platelet activation and deposition. Activated platelets recruit leukocytes, and initiate proliferation and migration of vascular smooth muscle cells to sites of injury, leading to development of neointimal hyperplasia.
-
-
Controlling platelet activation and recruitment is important for limiting downstream occlusive vascular sequelae.
-
-
Endothelial CD39/NTPDase1 and recombinant solCD39 rapidly metabolize nucleotides, including stimulatory ADP released from activated platelets, thereby suppressing platelet reactivity.
What does this paper add?
-
-
Human solCD39 remains enzymatically active in murine plasma for a sustained period of time following intraperitoneal administration, with an elimination phase half-life of 43 hr.
-
-
Circulating solCD39 was found to mitigate thrombotic and post-thrombotic cellular responses following vascular endothelial injury.
-
-
24 hr post-injury, platelet activation and recruitment, P-selectin expression, and leukocyte accumulation in the arterial lumen was markedly reduced by human solCD39.
-
-
SolCD39 administration modulated the late stage of vascular injury by suppressing leukocyte deposition, macrophage infiltration and smooth muscle cell proliferation/migration, resulting in protection from neointimal thickening.
-
-
In addition to its potential as an antithrombotic agent, human solCD39 may be of therapeutic importance for treatment of platelet-driven vascular stenosis.
INTRODUCTION
Vascular endothelial cells (EC) maintain the integrity and function of the vessel wall. Endothelial dysfunction or disruption promotes platelet activation, recruitment and deposition as well as leukocyte accumulation at the site of injury. There is also increased local expression of adhesion molecules (P-selectin (CD62P), intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1)) which participate in “homing” and infiltration of monocytes (1-6). Activated platelets release biologically active compounds, such as growth factors, cytokines, and chemokines, which recruit leukocytes and initiate proliferation and migration of vascular smooth muscle cells (SMC) from the media to the intima. This culminates in development of neointimal hyperplasia and resultant luminal narrowing (1-3). Thus, platelet-endothelial and platelet-leukocyte interactions play major roles in the initiation and acceleration of pathophysiological processes that lead to vascular thrombosis and stenosis (3;4;6).
Important mediators of these cell-cell interactions are extracellular nucleotides (e.g. adenosine diphosphate (ADP), adenosine triphosphate (ATP), and uridine triphosphate (UTP)) that are released from activated platelets, leukocytes, EC, and SMC. Released nucleotides bind to purinergic receptors of the P2X and P2Y families on platelets, leukocytes, endothelium, and vascular SMC, leading to modulation of cellular homeostatic responses (7;8). As platelets adhere to sites of vascular damage they become activated and release ADP and ATP (and the autacoids thromboxane A2 and serotonin) (6;9). ADP in the platelet releasate is the major activating and recruiting agent responsible for further stimulation and recruitment of additional platelets in the microenvironment, via engagement of platelet P2Y1 and P2Y12 ADP-responsive purinoreceptors (6;8). Metabolism of ADP results in diminution of platelet reactivity, recruitment and aggregation, returning platelets to their baseline state.
Cell surface ecto-nucleoside triphosphate diphosphohydrolases (E-NTPDases) are the primary enzymes involved in controlling availability of extracellular nucleotides to P2 receptors. The action of NTPDase1/CD39 (ecto-ATPDase, EC 3.6.1.5), expressed on vascular endothelium (and leukocytes) (10-16), plays a critical role in maintenance of blood fluidity. CD39 on the EC surface rapidly metabolizes ATP and ADP (to AMP) in the releasate from activated platelets, thereby inhibiting further platelet accumulation (9-11;14). This limits thrombus growth and subsequent vessel occlusion. Thromboregulation by CD39 occurs in the absence of other regulators of platelet reactivity (nitric oxide and prostacyclin). Significantly, only platelet responsiveness is blocked via extracellular metabolism of ADP (from the platelet releasate) by EC ecto-ADPase/CD39, thus platelet function is not compromised (9).
Structurally, CD39 consists of a large extracellular domain which is anchored to the cell membrane via flanking transmembrane domains (12;17). The nucleotide-hydrolyzing activity of CD39 resides within the extracellular domain (17-19). Although membrane anchoring is important for optimal enzymatic activity, it is not essential (18;19). We developed a 66 kDa recombinant, soluble form of human CD39 (‘solCD39’), consisting of the extracellular domain of CD39, with enzymatic and biological properties similar to native CD39 (20). This recombinant form has proven invaluable for elucidating structure-function relationships within the enzyme, including the active site (21;22), as well as exploring potential antithrombotic therapeutic applications of CD39 (23-25).
The important role of activated and adherent platelets, the ‘initial responders’ following vascular EC damage, in triggering events that subsequently lead to development of neointimal hyperplasia is well established. We and others have demonstrated the effectiveness of the enzymatic action of CD39 in suppressing ADP-induced platelet activation and recruitment. In the present study we used a murine model of vascular injury to examine the effect of solCD39 treatment on platelet-driven vascular neointimal thickening. We show for the first time that systemic administration of human solCD39 minimized injury-induced platelet deposition and leukocyte recruitment, and abrogated neointimal hyperplasia. Thus, human solCD39 can play an active role in suppressing the thrombotic, inflammatory, and proliferative cellular responses following vascular injury.
MATERIALS AND METHODS
Pharmacokinetic analyses of solCD39 in murine plasma
Animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at Weill Cornell Medical Center. Human solCD39 (4 mg/kg) was administered intraperitoneally (IP) to male C57Bl/6 mice (Jackson Laboratories, Bar Harbor, ME), 8 weeks of age, and blood samples subsequently collected at 20 min, 1, 2, 4, 24, 48 hr, 8 and 17 days after injection (n = 2-4 mice per time point). For the 8 and 17 day bleeds, mice were administered solCD39 IP every other day (three times per week, as appropriate). Blood (500 μl) was collected by cardiac puncture into a syringe containing 50 μl 3.8% trisodium citrate (anticoagulant), then mixed gently by inversion, and centrifuged (8,000 RPM for 5 min, 4°C) to separate cells from plasma. Plasma samples were frozen until day of assay. The activity of solCD39 in murine plasma was determined on two dilutions of each sample, in duplicate, using our radio-TLC assay for ADPase activity under standard conditions (50 μM ADP, 3 mM calcium in a total volume of 50 μl for 5 min at 37°C). Products of enzymatic metabolism were quantified by radio-TLC scanning (InstantImager, Perkin-Elmer), and values calculated as averages of duplicate measurements (10;21;22;26). The enzymatic activity in each sample was expressed as picomoles of ADP metabolized per minute per microliter plasma. Plasma from mice who did not receive solCD39 was used as control. This control plasma exhibited no endogenous ADPase activity. SolCD39 in vivo half-life (distribution and elimination phases) was determined using two phase exponential decay nonlinear regression analyses (GraphPad Prism3).
Transluminal wire-induced endothelial injury of the femoral artery
Male C57Bl/6 mice (Jackson Laboratories), aged 8-12 weeks, were utilized. Wire-induced injury of the femoral artery was performed as previously described (1;27). Mice were anesthetized (IP injection of ketamine and xylazine), and the femoral artery between the epigastric and saphenous arteries dissected free from the vein (visualized by surgical microscopy). The femoral vessels were clamped below the inguinal ligament, and an arteriotomy was performed on the femoral artery approximately 1 mm distal to the epigastric branch. A 0.01 inch (0.25 mm) diameter angioplasty guidewire was introduced into the arterial lumen, the clamp was removed, and the wire advanced to the level of the aortic bifurcation and pulled back a total of three times. The guidewire was then removed and the arteriotomy site ligated with an 8-0 nylon suture. After ligation, the skin incisions were closed with interrupted 5-0 absorbable sutures using a subcuticular closure method for wound apposition (IACUC guidelines).
SolCD39 or saline administration
Human solCD39 (4 mg/kg) or saline (control) was administered IP to mice 1 hr prior to arterial injury. Mice were sacrificed 24 hr following injury to analyze early responses, including platelet deposition and leukocyte recruitment. Later responses, including vascular lesion development, were assessed 19 days post-injury (4 mice per group). For solCD39-administered mice sacrificed at 19 days, solCD39 was injected three times per week in order to maintain circulatory levels. SolCD39 displays an elimination phase t½ of 43 hr in murine plasma following IP injection.
Histological and morphological analyses of injured femoral arteries
24 hr and 19 days after injury, mice were sacrificed, and femoral arteries harvested from the iliac bifurcation down to the arteriotomy site and rinsed in PBS. Specimens were placed in 30% sucrose/OCT (1:1) and snap-frozen for cryopreservation. The femoral artery was then sectioned, and cross-sections (10 μm thick) prepared for staining with hematoxylin-eosin or immunohistochemical analyses. The entire vessel was examined for lesion development.
Deposition of activated platelets and leukocytes was assessed by histological and immunohistochemical staining. Activated EC, SMC, and macrophages were also detected immunohistochemically. Arterial specimens were analyzed by computerized morphometry, and the different vessel regions quantified (Image-Pro Plus-5.1 software). On each section, measurements of the luminal area, the area inside the internal elastic lamina (IEL), and the area beneath the external elastic lamina (EEL) were made. Intimal area was calculated by subtracting the luminal area from the area inside the IEL; medial area was calculated by subtracting the area inside the IEL from the area inside the EEL; and the ratio of intimal area to medial area (I/M ratio) was also calculated (1;27). For morphometric analyses, vessel region quantification was assessed in 6 representative sections per vessel, covering a distance of 500 μm. Results from the segments of each artery were then averaged.
Immunohistochemical analyses
Representative sections were immunohistochemically stained for platelets [rat anti-mouse CD41 antibody, clone MWReg30], leukocytes [rat anti-mouse CD45 antibody, clone 30-F11], EC [rat anti-mouse CD31 (PECAM-1, clone MEC13.3) and rat anti-mouse panendothelial cell antigen, clone MECA-32, antibodies], SMC [mouse anti-actin, α-SM antibody, clone 1A4], macrophages [rat anti-mouse Mac-3 antibody, clone M3/84], P-selectin [rat anti-mouse CD62P antibody, clone RB40.34], ICAM-1 [hamster anti-mouse CD54 antibody, clone 3E2], and VCAM-1 [rat anti-mouse CD106 antibody, clone 429]. Antibodies were obtained from BD Biosciences, except for anti-α-SM actin which was from Sigma.
Frozen artery specimen slides were air dried (15 min, room temperature), then rinsed with PBS and fixed in acetone:methanol (1:1) (−20°C, 5 min). Fixed sections were rinsed with PBS for 5 min (3X), then 0.1% Triton X-100 in 1% BSA (5 min, room temperature), followed by incubation with non-immune serum (30 min). To identify specific cell types and expression of specific proteins (e.g. adhesion molecules), sections were incubated with the individual 1° antibody or corresponding IgG isotype control overnight (4°C), followed by the biotinylated IgG 2° antibody (1 hr, room temperature). Staining was carried out by reaction with horseradish peroxidase-conjugated streptavidin and developed with the AEC chromogen (Vector Laboratories); sections were counterstained with hematoxylin-eosin. Photomicroscopy of stained specimens was performed under bright-field illumination. For immunofluorescent imaging, sections were incubated with the 1° antibody or isotype control, washed with 1% BSA for 5 min (4X), and then incubated with the Alexa Fluor 488- or 594-conjugated IgG 2° antibody (Molecular Probes) (1 hr, room temperature). Slides were then rinsed with 1% BSA for 5 min (4X) and coverslipped in diazabicyclo (2,2,2)-octane 70% glycerol mounting medium (Prolong Antifade Kit, Molecular Probes). Immunofluorescently-labeled sections were examined by fluorescence microscopy. Platelet-, leukocyte-, macrophage-, SMC (actin)-, and P-selectin-positive antibody staining was quantified using Image-Pro Plus-5.1 software. The percentage expression of a given marker in a specific area (thrombus, lumen, media, or neointima) was determined by dividing the positive-stained area by its total area.
Statistical analyses
Data are presented as mean ± SD. Comparisons were made between treatment groups (solCD39 versus saline) at 24 hr and 19 days using Student’s unpaired, two-tailed t test. Values of P<0.05 were considered statistically significant.
RESULTS
In this study, we established that IP-delivered solCD39 remains enzymatically active in the murine circulation for an extended period of time, similar to that of IV administration. Moreover, using a murine model of transluminal endothelial injury of the femoral artery, we determined the efficacy of human solCD39 to minimize the early sequelae of vascular injury by controlling excessive platelet and leukocyte reactivity. In addition, we demonstrated the ability of solCD39 to ameliorate neointimal hyperplasia, a downstream consequence of the inflammatory milieu evoked by activated platelet and leukocyte accumulation.
In vivo half-life of human solCD39 in murine plasma following IP injection
Pharmacokinetic analyses revealed that solCD39 ADPase activity in murine plasma peaked at 1 hr following IP injection, and gradually declined over a 48 hr time course (Fig. 1). The ADPase activity of 17.1 μg/ml solCD39 in murine plasma is shown in the figure for comparison (Fig. 1). The data best fit a biphasic exponential decay curve with a calculated distribution phase half-life (t½α) of 195 min and an elimination phase half-life (t½β) of 43 hr. This indicated that IP-administered solCD39 displays a similarly long persistence in the murine circulation as previously observed for IV-delivered solCD39 (20). The effect of repeated solCD39 administration was also tested on a second group of mice, where additional injections were performed (three times per week) after the initial injection. The ADPase activity observed in murine plasma at day 8 (24 hr following an injection) was 2.1 times the activity seen at 24 hr (day 1), indicating plasma accumulation of solCD39. However, solCD39 ADPase activity was minimal by day 17, suggesting induction of a neutralizing immune response against CD39.
Fig. 1. Pharmacokinetics of IP-administered human solCD39 in mice.

The activity of solCD39 in murine plasma was measured using our ADPase radio-TLC assay, as described in “Materials and Methods”. The enzymatic activity of each sample was expressed as picomoles ADP metabolized per min per μl plasma. Each data point represents the mean ADPase activity (± SD) in plasma samples from 2-4 mice; values for each plasma sample were obtained from 2-3 separate assays. The dashed line, which signifies the ADPase activity of 17.1 μg/ml solCD39 in murine plasma, is shown as a comparison. SolCD39 in vivo half-life was determined using two phase exponential decay nonlinear regression analyses.
Human solCD39 modulation of the early (thrombotic) stage following vascular injury
24 hr post-injury, solCD39-treated mice displayed a marked reduction in platelet deposition (17.6±1.8% CD41-positive expression) in the lumen of the injured artery (Fig. 2B, F), compared to the pronounced accumulation of platelets (34.0±3.4% CD41-positive expression) observed in the saline control mice (P<0.002) (Fig. 2A, E). In the control mice platelets localized primarily in a large thrombus within the lumen, while in the solCD39-administered animals they only lined the luminal wall. Importantly, the sizable platelet-containing thrombus (81.1±3.5% CD41-positive expression) in the saline-administered mice (Fig. 2A, E) strongly expressed P-selectin (75.6±5.7% CD62P-positive expression) (Fig. 2G), indicating the presence of a high percentage of activated platelets. In contrast, minimal expression of platelet-associated P-selectin (5.0±1.9% CD62P-positive expression) (Fig. 2H) was observed in solCD39-infused animals. Additionally, 24 hr post-injury, leukocyte accumulation was significantly reduced (1.0±0.04% CD45-positive expression) in the lumen of the injured artery of solCD39-treated mice (Fig. 2J) relative to saline control mice (10.2±1.0% CD45-positive expression) (P<0.001) (Fig. 2I). Moreover, few macrophages were found in the lumen (2.2±0.8% Mac-3-positive expression) of the injured artery of solCD39-infused animals (Fig. 2L), whereas macrophages had accumulated in the thrombus (55.8±8.5% Mac-3-positive expression) within the lumen of vessels of control mice (Fig. 2K). In addition, expression of adhesion molecules ICAM-1 and VCAM-1 was not observed on the luminal wall in solCD39-infused animals (Fig. 3D, F). Slight luminal ICAM-1 and VCAM-1 expression was detected in the saline-administered mice (Fig. 3C, E), possibly on undenuded activated EC or from circulating ICAM-1 and VCAM-1 molecules (1;5;28;29). To determine whether residual EC lined the injured vessel lumen in the saline-injected control mice, arterial sections were stained for PECAM-1. PECAM-1-positive expression was detected on the vessel lumen not occupied by the thrombus as well as on the luminal surface of the thrombus itself (Fig. 3G). However, since PECAM-1 is found on platelets and leukocytes in addition to EC (30), sections were also probed with a more EC-specific antibody against the panendothelial cell antigen. Using this antibody, limited positive staining for EC was observed on the luminal surface of the control vessel, but not on the inner-most surface of the thrombus (Fig. 3I). This suggested that some of the PECAM-1 staining may be due to macrophages adherent to the lumen and the inner surface of the thrombus in the saline-injected mice (Fig. 2K). Minimal PECAM-1 and panendothelial cell antigen expression was detected on the injured arterial lumen in sections examined from solCD39-administered mice (Fig. 3H, J), indicating few residual EC were present. As expected, in both groups the medial layer of the artery was composed of SMC (Fig. 3K, L). Significantly, solCD39 did not prevent adhesion of a platelet monolayer on the denuded endothelium, but did prevent recruitment of additional platelets and thrombus formation (Fig. 2F).
Fig. 2. Immunohistochemical analysis of mouse femoral arteries 24 hr after wire-induced endothelial injury.
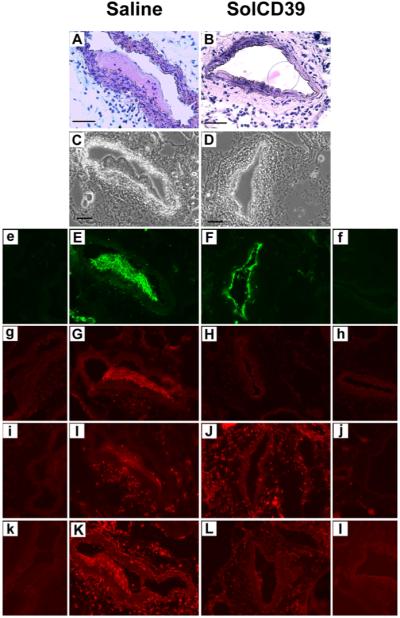
Immunohistochemical analysis was performed on representative cross-sections of femoral arteries of mice injected with human solCD39 or saline to identify specific cell types or adhesion molecule expression. Sections were stained with hematoxylin-eosin (A, B; 40x) as well as visualized under bright-field illumination (C, D; 20x). Sections were also immunofluorescently stained for platelets (E, F; anti-CD41 antibody, 20x), adhesion molecule P-selectin (G, H; anti-CD62P antibody, 20x), leukocytes (I, J; anti-CD45 antibody, 20x), and macrophages (K, L; anti-Mac-3 antibody, 20x). Images of the corresponding IgG isotype control to each specific antibody are labeled with lower case letters (e–l). Bar = 50 μm.
Fig. 3. Immunohistochemical analysis of mouse femoral arteries 24 hr after wire-induced endothelial injury.
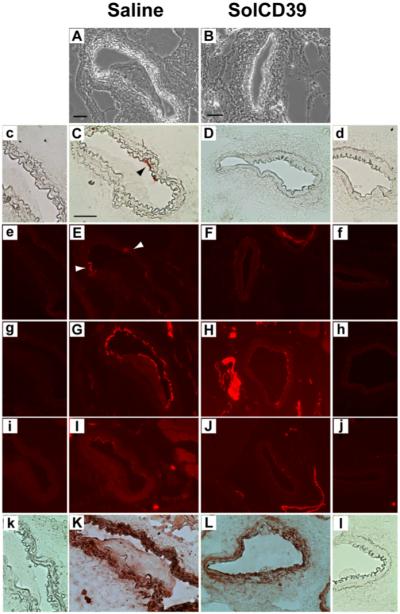
Representative cross-sections of femoral arteries of mice injected with human solCD39 or saline were visualized under bright-field illumination (A, B; 20x) as well as immunohistochemically probed for adhesion molecule ICAM-1 (C, D; anti-CD54 antibody, 40x) and for SMC (K, L; anti-actin, α-SM antibody, 40x), followed by colorimetric staining. In addition, sections were immunofluorescently stained for adhesion molecule VCAM-1 (E, F; anti-CD106 antibody, 20x) and for EC (G, H; anti-CD31 (PECAM-1) and I, J; anti-panendothelial cell antigen antibodies, 20x). Arrowheads indicate ICAM-1 (C) and VCAM-1 (E) expression on the luminal surface of the injured vessel in saline-treated control mice. Images of the corresponding IgG isotype control to each specific antibody are labeled with lower case letters (c-l). Bar = 50 μm.
Human solCD39 modulation of the late stage following vascular injury
Distinct differences were also seen between treatment groups at 19 days after injury. In saline-injected mice, excessive leukocyte accumulation (59.2±5.6% CD45-positive expression) (Fig. 4E) and pronounced neointimal hyperplasia (Fig. 4A) was observed. However, 19 days after injury and repeated (3X weekly) injections of solCD39, leukocyte deposition was absent within the arterial lumen (1.0±0.9% CD45-positive expression) (Fig. 4F), and neointimal formation was not seen (Fig. 4B). Importantly, 19 days post-injury, both solCD39- and saline-injected mice showed expression of PECAM-1 and panendothelial cell antigen (EC markers) on the luminal surface, indicating re-endothelialization of the artery (Fig. 4G, H, I, J). In addition, there was a lack of expression of adhesion molecules ICAM-1 and VCAM-1 on the luminal surface (Fig. 5C, D, E, F). However, in the saline-administered mice, ICAM-1 and VCAM-1 expression was detected on the medial cells (Fig. 5C, E), likely on SMC and possibly macrophages (Fig. 5G, I) (31-33). Significantly, in the injured artery of solCD39-treated mice, macrophages did not infiltrate from the lumen (0.5±0.2% Mac-3-positive expression) (Fig. 5H) and SMC were present only in the media (81.2±5.0% SM actin-positive expression) (Fig. 5J), indicating a quiescent uninjured state. Most importantly, solCD39 administration ameliorated neointimal hyperplasia (Fig. 4B). This was in sharp contrast to the injured vessels of saline-injected mice, where macrophage (28.9±9.0% Mac-3-positive expression) (Fig. 5G) and SMC accumulation (68.3±1.0% SM actin-positive expression) (Fig. 5I) was observed in the hyperplastic neointima (Fig. 4A). Thus, treatment with solCD39 minimized injury-induced platelet deposition and leukocyte recruitment, and suppressed neointimal hyperplasia.
Fig. 4. Immunohistochemical analysis of mouse femoral arteries 19 days after wire-induced endothelial injury.
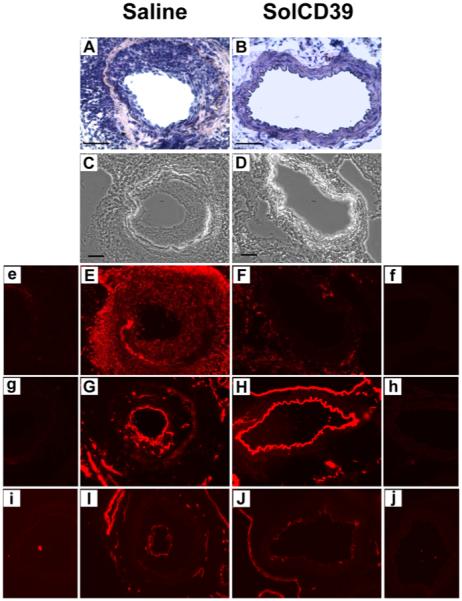
Immunohistochemical analysis was performed on representative cross-sections of femoral arteries of mice injected with human solCD39 or saline to identify specific cell types. Sections were stained with hematoxylin-eosin (A, B; 40x) as well as visualized under bright-field illumination (C, D; 20x). Sections were also immunofluorescently stained for leukocytes (E, F; anti-CD45 antibody, 20x), and EC (G, H; anti-CD31 (PECAM-1) and I, J; anti-panendothelial cell antigen antibodies, 20x). Images of the corresponding IgG isotype control to each specific antibody are labeled with lower case letters (e-j). Bar = 50 μm.
Fig. 5. Immunohistochemical analysis of mouse femoral arteries 19 days after wire-induced endothelial injury.
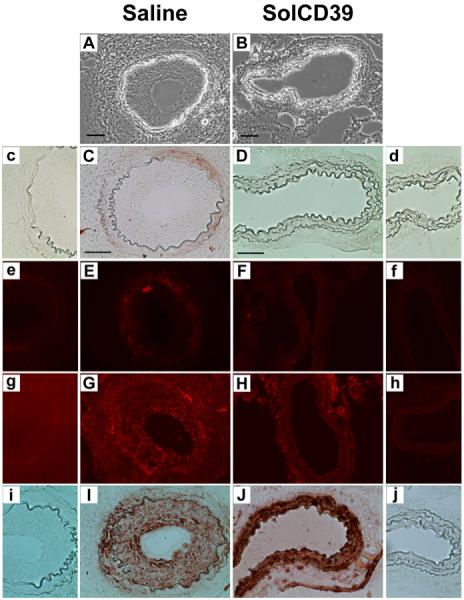
Representative cross-sections of femoral arteries of mice injected with human solCD39 or saline were visualized under bright-field illumination (A, B; 20x). Sections were also immunohistochemically probed for adhesion molecule ICAM-1 (C, D; anti-CD54 antibody, 40x) and for SMC (I, J; anti-actin, α-SM antibody, 40x), followed by colorimetric staining. In addition, sections were immunofluorescently stained for adhesion molecule VCAM-1 (E, F; anti-CD106 antibody, 20x) and for macrophages (G, H; anti-Mac-3 antibody, 20x). Images of the corresponding IgG isotype control to each specific antibody are labeled with lower case letters (c–j). Bar = 50 μm.
Morphological analysis of injured femoral arteries following administration of human solCD39
In addition to histological examination of mouse femoral arteries following infusion of solCD39 or saline (24 hr and 19 days post-injury), vessels were analyzed by morphometry (Table 1). At 24 hr post-injury, neointimal hyperplasia was not observed in the injured vessels of solCD39- and saline-injected mice; the intimal area as well as intimal-to-medial area (I/M) ratio was zero (Table 1). Notably, 19 days following vessel injury, the injured arteries of solCD39-administered mice lacked neointimal thickening as compared to saline-infused mice (P<0.001). The mean intimal area in the saline-treated mice was 26311±4490 at 19 days as compared to 0±0 in the solCD39-treated animals (P<0.001) (Table 1). This measurement translated into an I/M ratio of 1.56±0.34 in the saline-injected mice as compared to 0±0 in the solCD39-mice (P<0.001) (Table 1). We conclude that human solCD39 provides protection against injury-induced development of neointimal hyperplasia.
Table 1.
Morphometric Analysis of Injured Mouse Femoral Arteries following Injection of Human SolCD39 or Saline
| 24 hr | 19 days | |||
|---|---|---|---|---|
| Saline | SolCD39 | Saline | SolCD39 | |
| Intimal area, μm2 | 0±0 | 0±0 | 26311±4490 | 0±0†§ |
| Medial area, μm2 | 20996±2390 | 13672±2896 | 17157±2436 | 18298±1438 |
| I/M ratio | 0±0 | 0±0 | 1.56±0.34 | 0±0†§ |
I/M ratio indicates ratio of intimal area-to-medial area.
Data represent mean ± SD from 4 animals.
P<0.001 versus saline value.
The endothelial cell monolayer appeared as a line, therefore its area was considered to be 0, and mean I/M ratio was also 0.
DISCUSSION
Platelet deposition and leukocyte recruitment following endothelial denudation characterize the early phase of vascular injury. Studies have shown the presence of platelets covering the denuded injured artery, and adherent leukocytes (predominantly neutrophils), as early as 1 hr (to 24 hr) post injury (1;27;34). This is associated with increased expression of the adhesion molecules P-selectin, ICAM-1, and VCAM-1 (1;27). Neointimal growth, resulting from migration and proliferation of SMC into the intima, occurs in the late phase of vascular injury, promoted by the inflammatory milieu elicited by activated platelet and leukocyte accumulation. In particular, platelet-derived growth factor, TGF-β, cytokines IL-1, IL-6, IL-8, and thrombin, released from activated platelets, provide a major proliferative stimulus to SMC (1-3). Significant neointimal hyperplasia (primarily consisting of SMC, as well as macrophages and T-lymphocytes) is observed at 2 and 4 weeks post-injury, and EC regrowth within 3-4 weeks (1;27;35;36). In this study we demonstrate that systemic administration of human solCD39 can modulate these early and late events occurring in the lumen of the femoral artery following vascular injury.
Since platelets play a pivotal role in vascular occlusive processes, including stenosis, control of pronounced platelet activation and recruitment following adherence of the initial platelet monolayer to sites of vascular damage is of critical importance. Significantly, we observed that human solCD39 minimized the early sequelae of induced vascular injury (24 hr) by restricting platelet recruitment. Local diminution of P-selectin expression indicated that inhibition of platelet recruitment by circulating solCD39 was associated with suppression of platelet activation. Consequently, only a platelet monolayer developed on the denuded endothelium in the presence of systemic solCD39, which effectively limited leukocyte accretion within the lumen and macrophage infiltration into the luminal wall of the injured femoral artery.
In mouse models of arterial injury using wild-type and P-selectin-null mice, P-selectin has been shown to play an important role in thrombus formation and the inflammatory response (leukocyte and SMC recruitment), leading to neointimal lesion development (27;34;37). In the absence of P-selectin-mediated accumulation of adherent platelets, leukocyte recruitment is abrogated and hyperplasia diminished (27;37). In addition, systemic platelet activation and platelet P-selectin expression has been linked with development of intima-media wall thickening of the carotid artery in humans with atherosclerosis and type 2 diabetes (38;39). In our control animals, we observed the characteristic formation of an activated platelet thrombus expressing high levels of P-selectin, and subsequent leukocyte infiltration, SMC proliferation and infiltration, and neointimal growth (I/M ratio of 1.56±0.34 at 19 days) following vascular injury. The extent of intimal thickening seen here is typical, as others have reported similar I/M ratios at 4 weeks following transluminal endothelial injury to the mouse femoral artery (1;27). In contrast, in the PECAM-1 positive re-endothelialized artery of solCD39-infused mice, a quiescent uninjured state was achieved, free of neointimal expansion. Leukocyte deposition was absent within the arterial lumen, macrophages had not infiltrated the vessel wall, and SMC were present only in the media of the artery. Thus, the combined reduction of platelet activation, P-selectin expression and leukocyte recruitment, engendered by circulating human solCD39, led to significant modulation of the late stage (19 days) of vascular injury, effectively suppressing neointimal hyperplasia.
Based on our findings, it is likely that the primary underlying basis for the suppressive effect of solCD39 on injury-induced neointimal thickening is the inhibition of activated platelet-driven events, including SMC proliferation. Some of the protective effect of solCD39 may also stem from the conversion of pro-inflammatory AMP to anti-inflammatory adenosine via the action of ecto-5′-nucleotidase (CD73). Studies with CD73 knockout mice have demonstrated a role for CD73-mediated generation of adenosine in suppression of neointimal hyperplasia (40). Another potential mechanism contributing to the suppression of hyperplastic thickening is direct inhibition of extracellular nucleotide-induced recruitment and SMC growth. It is now recognized that ATP, ADP, and UTP play major signaling roles in vascular SMC proliferation and migration via P2X and P2Y receptors (7;8). Therefore, extracellular ATP, ADP and UTP, released from activated and damaged cells of the vasculature (platelets, leukocytes, and EC) (41), could provide significant signals for intimal infiltration and expansion of SMC. Studies have shown that in addition to ADP and ATP, CD39 can also efficiently hydrolyze UTP (42). Significantly, it was recently shown that CD39 overexpression in mice decreased SMC proliferation following angioplasty (43). Thus, metabolic deletion of ATP and ADP (and UTP) by solCD39 could be removing the mitogenic and migrational signals derived from these nucleotides, preventing SMC proliferation and resultant neointimal thickening.
Given the direct role of endothelial cell CD39 and recombinant solCD39 in modulating platelet reactivity, antithrombotic therapeusis based on the action of CD39/solCD39 has been investigated. Recent studies have shown the protective effects of human CD39 overexpression (via transgenic expression or targeted viral gene delivery) as well as liposomally-delivered CD39 in animal models of thrombosis, vascular injury, and transplantation (43-47). Furthermore, the antithrombotic potential of human solCD39 has been demonstrated in vitro, ex vivo and in vivo in our laboratory and by others (20-25). SolCD39 inhibited ADP-induced human, porcine, and murine platelet aggregation in vitro and ex vivo, and strongly inhibited collagen- and thrombin receptor agonist peptide (TRAP6)-induced human platelet reactivity as well as ex vivo collagen- and arachidonate-induced murine platelet aggregation (9;23;24). The platelet-inhibitory effects of solCD39 were also shown in vivo in a murine model of stroke driven by excessive platelet activation and recruitment (23). When administered prior to transient intraluminal right middle cerebral artery occlusion, solCD39 reduced ipsilateral fibrin deposition, decreased platelet deposition, and increased post-ischemic laser Doppler blood flow 2-fold at 24 hr without an increase in intracerebral hemorrhage. Even if administered 3 hr following stroke induction, solCD39 reduced cerebral infarction volume (23). Our current study suggests that in addition to the direct antithrombotic effects of CD39 supplementation, systemic solCD39 administration also minimizes the vasculopathy mediated by activated platelets.
In summary, using a murine model of vascular endothelial injury, we demonstrated the efficacy of circulating human solCD39 to modulate both the early and late sequelae of vascular injury. SolCD39 markedly reduced platelet deposition and leukocyte recruitment as well as macrophage infiltration into the arterial lumen. Moreover, solCD39 protected against injury-induced development of neointimal hyperplasia. Thus, our data suggest that in addition to its potential as an antithrombotic agent, human solCD39 could be of therapeutic importance for the treatment of platelet-driven vascular stenosis.
ACKNOWLEDGEMENTS
We thank Mr. Nikolay Novikov (Weill Cornell Medical College) for his technical assistance with the animal experiments, and Dr. M. Johan Broekman (Weill Cornell Medical College) for his helpful suggestions and critical reading of the manuscript.
GRANT SUPPORT: This work was supported by a Department of Veterans Affairs Merit Review grant and NIH grants HL046403 and HL047073.
REFERENCES
- (1).Roque M, Fallon JT, Badimon JJ, et al. Mouse model of femoral artery denudation injury associated with the rapid accumulation of adhesion molecules on the luminal surface and recruitment of neutrophils. Arterioscler Thromb Vasc Biol. 2000;20:335–42. doi: 10.1161/01.atv.20.2.335. [DOI] [PubMed] [Google Scholar]
- (2).Mitra AK, Gangahar DM, Agrawal DK. Cellular, molecular and immunological mechanisms in the pathophysiology of vein graft intimal hyperplasia. Immunol Cell Biol. 2006;84:115–24. doi: 10.1111/j.1440-1711.2005.01407.x. [DOI] [PubMed] [Google Scholar]
- (3).May AE, Langer H, Seizer P, et al. Platelet-leukocyte interactions in inflammation and atherothrombosis. Semin Thromb Hemost. 2007;33:123–7. doi: 10.1055/s-2007-969023. [DOI] [PubMed] [Google Scholar]
- (4).Siegel-Axel DI, Gawaz M. Platelets and endothelial cells. Semin Thromb Hemost. 2007;33:128–35. doi: 10.1055/s-2007-969025. [DOI] [PubMed] [Google Scholar]
- (5).van Gils JM, Zwaginga JJ, Hordijk PL. Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J Leukoc Biol. 2009;85:195–204. doi: 10.1189/jlb.0708400. [DOI] [PubMed] [Google Scholar]
- (6).Jennings LK. Mechanisms of platelet activation: Need for new strategies to protect against platelet-mediated atherothrombosis. Thromb Haemost. 2009;102:248–57. doi: 10.1160/TH09-03-0192. [DOI] [PubMed] [Google Scholar]
- (7).Seye CI, Kong Q, Yu N, et al. P2 receptors in atherosclerosis and postangioplasty restenosis. Purinergic Signal. 2007;3:153–62. doi: 10.1007/s11302-006-9047-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Erlinge D, Burnstock G. P2 receptors in cardiovascular regulation and disease. Purinergic Signal. 2008;4:1–20. doi: 10.1007/s11302-007-9078-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Marcus AJ, Broekman MJ, Drosopoulos JH, et al. Heterologous cell-cell interactions: thromboregulation, cerebroprotection and cardioprotection by CD39 (NTPDase-1) J Thromb Haemost. 2003;1:2497–509. doi: 10.1111/j.1538-7836.2003.00479.x. [DOI] [PubMed] [Google Scholar]
- (10).Marcus AJ, Broekman MJ, Drosopoulos JHF, et al. The endothelial cell ecto-ADPase responsible for inhibition of platelet function is CD39. J Clin Invest. 1997;99:1351–60. doi: 10.1172/JCI119294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kaczmarek E, Koziak K, Sévigny J, et al. Identification and characterization of CD39 vascular ATP diphosphohydrolase. J Biol Chem. 1996;271:33116–22. doi: 10.1074/jbc.271.51.33116. [DOI] [PubMed] [Google Scholar]
- (12).Maliszewski CR, Delespesse GJ, Schoenborn MA, et al. The CD39 lymphoid cell activation antigen. Molecular cloning and structural characterization. J Immunol. 1994;153:3574–83. [PubMed] [Google Scholar]
- (13).Koziak K, Sévigny J, Robson SC, et al. Analysis of CD39/ATP diphosphohydrolase (ATPDase) expression in endothelial cells, platelets and leukocytes. Thromb Haemost. 1999;82:1538–44. [PubMed] [Google Scholar]
- (14).Sevigny J, Sundberg C, Braun N, et al. Differential catalytic properties and vascular topography of murine nucleoside triphosphate diphosphohydrolase 1 (NTPDase1) and NTPDase2 have implications for thromboregulation. Blood. 2002;99:2801–9. doi: 10.1182/blood.v99.8.2801. [DOI] [PubMed] [Google Scholar]
- (15).Glenn JR, White AE, Johnson A, et al. Leukocyte count and leukocyte ecto-nucleotidase are major determinants of the effects of adenosine triphosphate and adenosine diphosphate on platelet aggregation in human blood. Platelets. 2005;16:159–70. doi: 10.1080/09537100500063889. [DOI] [PubMed] [Google Scholar]
- (16).Pulte ED, Broekman MJ, Olson KE, et al. CD39/NTPDase-1 activity and expression in normal leukocytes. Thromb Res. 2007;121:309–17. doi: 10.1016/j.thromres.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zimmermann H. Ectonucleotidases: Some recent developments and a note on nomenclature. Drug Dev Res. 2001;52:44–56. [Google Scholar]
- (18).Wang TF, Ou Y, Guidotti G. The transmembrane domains of ectoapyrase (CD39) affect its enzymatic activity and quaternary structure. J Biol Chem. 1998;273:24814–21. doi: 10.1074/jbc.273.38.24814. [DOI] [PubMed] [Google Scholar]
- (19).Schulte am Esch J, Sévigny J, Kaczmarek E, et al. Structural elements and limited proteolysis of CD39 influence ATP diphosphohydrolase activity. Biochemistry. 1999;38:2248–58. doi: 10.1021/bi982426k. [DOI] [PubMed] [Google Scholar]
- (20).Gayle RB, Maliszewski CR, Gimpel SD, et al. Inhibition of platelet function by recombinant soluble ecto-ADPase/CD39. J Clin Invest. 1998;101:1851–9. doi: 10.1172/JCI1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Drosopoulos JHF, Broekman MJ, Islam N, et al. Site-directed mutagenesis of human endothelial cell ecto-ADPase/soluble CD39. Requirement of glutamate174 and serine218 for enzyme activity and inhibition of platelet recruitment. Biochemistry. 2000;39:6936–43. doi: 10.1021/bi992581e. [DOI] [PubMed] [Google Scholar]
- (22).Drosopoulos JHF. Roles of Asp54 and Asp213 in Ca2+ utilization by soluble human CD39/ecto-nucleotidase. Arch Biochem Biophys. 2002;406:85–95. doi: 10.1016/s0003-9861(02)00414-9. [DOI] [PubMed] [Google Scholar]
- (23).Pinsky DJ, Broekman MJ, Peschon JJ, et al. Elucidation of the thromboregulatory role of CD39/ectoapyrase in the ischemic brain. J Clin Invest. 2002;109:1031–40. doi: 10.1172/JCI10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Buergler JM, Maliszewski CR, Broekman MJ, et al. Effects of SolCD39, a novel inhibitor of platelet aggregation, on platelet deposition and aggregation after PTCA in a porcine model. J Thromb Thrombolysis. 2005;19:115–22. doi: 10.1007/s11239-005-1381-y. [DOI] [PubMed] [Google Scholar]
- (25).Belayev L, Khoutorova L, Deisher TA, et al. Neuroprotective effect of SolCD39, a novel platelet aggregation inhibitor, on transient middle cerebral artery occlusion in rats. Stroke. 2003;34:758–63. doi: 10.1161/01.STR.0000056169.45365.15. [DOI] [PubMed] [Google Scholar]
- (26).Musi E, Islam N, Drosopoulos JHF. Constraints imposed by transmembrane domains affect enzymatic activity of membrane-associated human CD39/NTPDase1 mutants. Arch Biochem Biophys. 2007;461:30–9. doi: 10.1016/j.abb.2007.02.009. [DOI] [PubMed] [Google Scholar]
- (27).Smyth SS, Reis ED, Zhang W, et al. β3-Integrin-deficient mice but not P-selectin-deficient mice develop intimal hyperplasia after vascular injury: Correlation with leukocyte recruitment to adherent platelets 1 hour after injury. Circulation. 2001;103:2501–7. doi: 10.1161/01.cir.103.20.2501. [DOI] [PubMed] [Google Scholar]
- (28).Kanda K, Hayman GT, Silverman MD, et al. Comparison of ICAM-1 and VCAM-1 expression in various human endothelial cell types and smooth muscle cells. Endothelium. 1998;6:33–44. doi: 10.3109/10623329809053403. [DOI] [PubMed] [Google Scholar]
- (29).Muller WA. Leukocyte-endothelial cell interactions in the inflammatory response. Lab Invest. 2002;82:521–33. doi: 10.1038/labinvest.3780446. [DOI] [PubMed] [Google Scholar]
- (30).Woodfin A, Voisin MB, Nourshargh S. PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler Thromb Vasc Biol. 2007;27:2514–23. doi: 10.1161/ATVBAHA.107.151456. [DOI] [PubMed] [Google Scholar]
- (31).Voisard R, Osswald M, Baur R, et al. Expression of intercellular adhesion molecule-1 in human coronary endothelial and smooth muscle cells after stimulation with tumor necrosis factor-alpha. Coron Artery Dis. 1998;9:737–45. doi: 10.1097/00019501-199809110-00006. [DOI] [PubMed] [Google Scholar]
- (32).Manka DR, Wiegman P, Din S, et al. Arterial injury increases expression of inflammatory adhesion molecules in the carotid arteries of apolipoprotein-E-deficient mice. J Vasc Res. 1999;36:372–8. doi: 10.1159/000025676. [DOI] [PubMed] [Google Scholar]
- (33).O’Brien KD, Allen MD, McDonald TO, et al. Vascular cell adhesion molecule-1 is expressed in human coronary atherosclerotic plaques. Implications for the mode of progression of advanced coronary atherosclerosis. J Clin Invest. 1993;92:945–51. doi: 10.1172/JCI116670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Osaka M, Hagita S, Haraguchi M, et al. Real-time imaging of mechanically injured femoral artery in mice reveals a biphasic pattern of leukocyte accumulation. Am J Physiol. 2007;292:H1876–H1882. doi: 10.1152/ajpheart.00708.2006. [DOI] [PubMed] [Google Scholar]
- (35).Kikuchi S, Umemura K, Kondo K, et al. Photochemically induced endothelial injury in the mouse as a screening model for inhibitors of vascular intimal thickening. Arterioscler Thromb Vasc Biol. 1998;18:1069–78. doi: 10.1161/01.atv.18.7.1069. [DOI] [PubMed] [Google Scholar]
- (36).Tanaka H, Sukhova GK, Swanson SJ, et al. Sustained activation of vascular cells and leukocytes in the rabbit aorta after balloon injury. Circulation. 1993;88:1788–803. doi: 10.1161/01.cir.88.4.1788. [DOI] [PubMed] [Google Scholar]
- (37).Wang K, Zhou X, Zhou Z, et al. Platelet, not endothelial, P-selectin is required for neointimal formation after vascular injury. Arterioscler Thromb Vasc Biol. 2005;25:1584–9. doi: 10.1161/01.ATV.0000172687.01179.d4. [DOI] [PubMed] [Google Scholar]
- (38).Koyama H, Maeno T, Fukumoto S, et al. Platelet P-selectin expression is associated with atherosclerotic wall thickness in carotid artery in humans. Circulation. 2003;108:524–9. doi: 10.1161/01.CIR.0000081765.88440.51. [DOI] [PubMed] [Google Scholar]
- (39).Fateh-Moghadam S, Li Z, Ersel S, et al. Platelet degranulation is associated with progression of intima-media thickness of the common carotid artery in patients with diabetes mellitus type 2. Arterioscler Thromb Vasc Biol. 2005;25:1299–303. doi: 10.1161/01.ATV.0000165699.41301.c5. [DOI] [PubMed] [Google Scholar]
- (40).Zernecke A, Bidzhekov K, Ozuyaman B, et al. CD73/ecto-5′-nucleotidase protects against vascular inflammation and neointima formation. Circulation. 2006;113:2120–7. doi: 10.1161/CIRCULATIONAHA.105.595249. [DOI] [PubMed] [Google Scholar]
- (41).Elliott MR, Chekeni FB, Trampont PC, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–6. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kukulski F, Levesque SA, Lavoie EG, et al. Comparative hydrolysis of P2 receptor agonists by NTPDases 1, 2, 3 and 8. Purinergic Signal. 2005;1:193–204. doi: 10.1007/s11302-005-6217-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Koziak K, Bojakowska M, Robson SC, et al. Overexpression of CD39/nucleoside triphosphate diphosphohydrolase-1 decreases smooth muscle cell proliferation and prevents neointima formation after angioplasty1. J Thromb Haemost. 2008;6:1191–7. doi: 10.1111/j.1538-7836.2008.03019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Imai M, Takigami K, Guckelberger O, et al. Recombinant adenoviral mediated CD39 gene transfer prolongs cardiac xenograft survival. Transplantation. 2000;70:864–70. doi: 10.1097/00007890-200009270-00003. [DOI] [PubMed] [Google Scholar]
- (45).Gangadharan SP, Imai M, Rhynhart KK, et al. Targeting platelet aggregation: CD39 gene transfer augments nucleoside triphosphate diphosphohydrolase activity in injured rabbit arteries. Surgery. 2001;130:296–303. doi: 10.1067/msy.2001.116032. [DOI] [PubMed] [Google Scholar]
- (46).Dwyer KM, Robson SC, Nandurkar HH, et al. Thromboregulatory manifestations in human CD39 transgenic mice and the implications for thrombotic disease and transplantation. J Clin Invest. 2004;113:1440–6. doi: 10.1172/JCI19560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Haller CA, Cui W, Wen J, et al. Reconstitution of CD39 in liposomes amplifies nucleoside triphosphate diphosphohydrolase activity and restores thromboregulatory properties. J Vasc Surg. 2006;43:816–23. doi: 10.1016/j.jvs.2005.11.057. [DOI] [PubMed] [Google Scholar]