

The copper-catalyzed azide-alkyne cycloaddition reaction[1] (CuAAC) (Scheme 1, a) is known for its high fidelity in the presence of many functional groups and under demanding reaction conditions.[2] Experimental simplicity and high selectivity of this process have been exploited in many applications in synthetic and medicinal chemistry,[3] bioconjugations,[4] materials science,[5] and polymer chemistry.[6] The efficiency and selectivity of this transformation are a direct consequence of the reactivity of in situ-generated copper(I) acetylides. Coordination of the organic azide to the copper center of the acetylide increases the nucleophilicity of the triple bond and initiates a sequence of steps which ultimately results in the formation of the new C—N bond between the nucleophilic β-carbon atom of the acetylide and the terminal, electrophilic nitrogen atom of the azide (Scheme 2). Naturally, internal alkynes are devoid of such reactivity, and therefore CuAAC is limited to terminal acetylenes, producing only 1,4-disubstituted triazoles. Although the ruthenium-catalyzed azide-alkyne cycloaddition reaction (Scheme 1, b)[7] and methods of functionalization of the triazole heterocycle itself[8] partially address these deficiencies, a general method for the regiocontrolled synthesis of differently substituted 1,2,3-triazoles would be a valuable addition to the family of catalytic cycloaddition reactions.
Scheme 1.
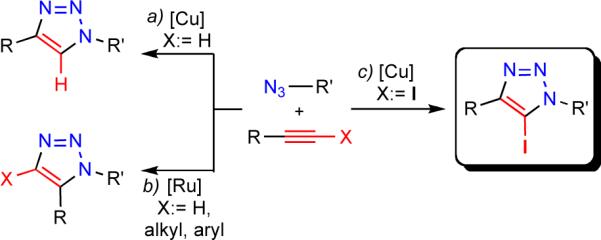
Various routes to substituted 1,2,3-triazoles.
Scheme 2.
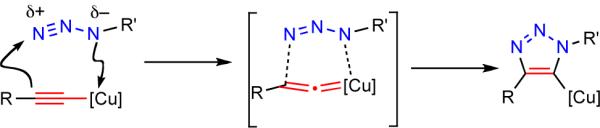
Reactivity of organic azides with copper(I) acetyides.
In this communication we report that 1-iodoalkynes, stable and readily accessible (vide infra) internal acetylenes, exhibit exceptional reactivity in the copper-catalyzed annulation reaction with organic azides (Scheme 1, c). Indeed, their reactivity appears to surpass that of terminal alkynes. As an added benefit, the products of the reaction, 5-iodo-1,2,3-triazoles, are versatile synthetic intermediates amenable for further functionalization. Although several syntheses of iodotriazoles are known, the reactions require stoichiometric amounts of copper catalysts and employ reactive electrophilic halogenating reagents (e.g., iodine monochloride, N-iodosuccinimide).[9] In addition, some procedures require extended reaction times and generate mixtures of 5-H and 5-iodo-triazoles.[10]
Disclosed here is a general, rapid and operationally simple method for the chemo- and regioselective synthesis of 5-iodo-1,4,5-trisubstituted-1,2,3-triazoles from organic azides and iodoalkynes. The catalysis is effected by copper(I) iodide in the presence of an amine ligand.
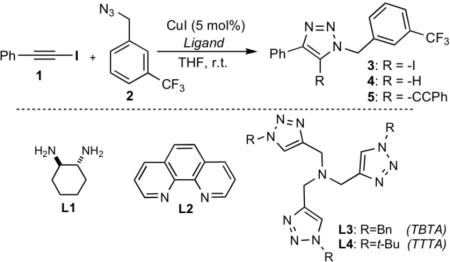
The initial survey of experimental conditions, which included a broad array of copper(I) and copper(II) salts, solvents and ligands, revealed that the reaction of iodoalkyne 1 and azide 2 was catalyzed by copper(I) iodide/triethylamine (TEA) in THF, giving 5-iodo-1,2,3-triazole 3 as the major product,[11] along with 5-proto- and 5-alkynyl triazoles 4 and 5, respectively (Table 1, entry 2).
Table 1.
Optimization of the reaction conditions.[a]
| Entry | Additive | equiv | 3 : 4 : 5[b] | Yield[%][c] |
|---|---|---|---|---|
| 1 | – | – | – | n.r. |
| 2 | TEA | 0.5 | 10 : 3: 1 | 55 |
| 3 | TEA | 1 | 25 : 2 : 1 | 75 |
| 4 | TEA | 2 | 1 : 0 : 0 | 90 |
| 5 | DIPEA | 0.5 | 15 : 1 : 2 | 47 |
| 6 | DIPEA | 2 | 1 : 0 : 0 | 73 |
| 7 | 2,6-lutidine | 0.5 | 30 : 1 : 0 | 12 |
| 8 | TMEDA | 0.5 | 20 : 1 : 0 | 26 |
| 9 | L1 | 0.5 | 1 : 1 : 15 | 25 |
| 10 | L2 | 0.5 | – | n.r. |
| 11 | L3 | 0.05 | 1 : 0 : 0 | 60[d] |
| 12 | L4 | 0.05 | 1 : 0 : 0 | 93 [d] |
General reaction conditions: CuI (0.02 mmol) and ligand in THF (2 mL), 1 (0.40 mmol) 2 (0.40 mmol), room temperature, 6h.
Product ratio determined by HPLC-MS.
Isolated yield of 3.
Reaction time was 45 min.
Inclusion of an amine ligand was crucial, as no reaction was observed when TEA was omitted (Table 1, entry 1). Furthermore, the reaction displayed a sharp dependence on the quantity of TEA (Table 1, entries 1 – 4). Thus, 5-iodo-triazole 3 was generated as the sole product in excellent yield by simply using an excess (2 equiv.) of TEA. This trend extended to other tertiary amine ligands, although the desired 5-iodotriazole was obtained in lower yield (Table 1, cf. entries 4 and 6, 8).
The observed rate and chemoselectivity of the reaction were strongly dependent on the nature of the amine ligand. For example, the efficiency of the reaction was markedly reduced, and 5-alkynyl-triazole 5 was formed as the major product,[12] when TEA was replaced with 1,2-diamines (Table 1, entries 8 and 9). Pyridines, such as 2,6-lutidine, and 1,10-phenanthroline, were also ineffective (Table 1, entries 7 and 10). By contrast, tris((1,2,3-triazolyl)methyl)amine ligands[13] were found to be highly efficient in promoting this cycloaddition. Both tris((1-benzyl-1H-1,2,3-triazolyl)methyl)amine (TBTA) and its tert-butyl analog, tris((1-tert-butyl-1H-1,2,3-triazolyl)methyl)amine (TTTA) (Table 1, entries 11 and 12) gave 5-iodotriazole 3 as the exclusive product in excellent yield. In addition, these ligands markedly accelerated the reaction, reducing the time to completion from 6 hrs to 45 min.
The increased chemoselectivity of the reaction in the presence of these ligands is a consequence of the rate acceleration of the triazoles-forming pathway. Both iodoalkyne 1 and 5-iodotriazole 3 slowly undergo reductive dehalogenation in the presence of various copper salts, generating the corresponding terminal alkyne and 5-prototriazole 4. These pathways likely account for the generation of the observed byproducts, but are far too slow in the presence of accelerating ligands.
Based on these observations, TTTA emerged as the optimal ligand for the rapid and chemoselective construction of 5-iodo-1,2,3 triazoles. It is noteworthy that both CuI-TTTA and CuI-TEA systems were found to be compatible with wide variety of solvents (Table 2). While some did have a large effect on the reaction rate, the selectivity was not affected, even when the reaction was performed in protic solvents.
Table 2.
Solvent compatibility study.[a]
| TTTA (5 mol%) | TEA (2equiv) | |||
|---|---|---|---|---|
| Solvent | Time [h] | Yield[%] | Time [h] | Yield[%] |
| THF | 1 | 93 | 6 | 90 |
| MeCN | 1 | 94 | 6 | 85 |
| DMF | 2 | 91 | 18 | 86 |
| Water | 2 | 85 | 6 | 76 |
| EtOH | 4 | 78 | 24 | 69 |
| DCM | 4 | 79 | 24 | 62 |
| toluene | 5 | 62 | 24 | 73 |
General conditions: CuI (0.05 mmol) and ligand in solvent (5 mL), 1 (1.00 mmol) 2 (1.00 mmol)
[b] Isolated yield of 3.
The CuI-TTTA catalyst system was applied to a series of structurally and functionally diverse azides and 1-iodoalkynes (Figure 1). In all cases, the 5-iodo-1,2,3-triazoles were obtained as the exclusive products. Due to the mild reaction conditions, high chemoselectivity, and low copper catalyst loading, reaction workup was usually as simple as trituration followed by filtration. As a result, this method is highly amenable to scale-up, and representative 5-iodotriazoles 6 and 15 were prepared in multigram quantities. In addition, the diverse array of functional groups tolerated by this annulation stands out as a particularly exceptional feature. Both sterically demanding (e.g., 10, 22) and functionally dense (e.g., 7, 17) substrates could be utilized. As such, the azideiodoalkyne cycloaddition provides a highly orthogonal means of chemical ligation, similar to the more conventional CuAAC reaction.
Figure 1.

Substrate scope for the Cu(I)-catalyzed azide-iodoalkyne cycloaddition.[a] [b]
[a] General reaction conditions: azide (1.00 mmol), 1-iodoalkyne (1.00 mmol), CuI (0.05 mmol), TTTA (0.05 mmol), THF (5mL), room temperature, 2h, [b] Values in parentheses represent isolated yield, [c] Reaction time was 6 h, [d] Reaction was performed at 10 mmol scale.
The utility of this cycloaddition was enhanced through the development of a simple and highly efficient synthesis of 1-iodoalkynes from terminal acetylenes (Scheme 3). Terminal alkynes were treated with N-iodomorpholine[14] 23, in the presence of CuI, giving the corresponding 1-iodoakynes within 30 to 60 minutes. The products could be isolated by simply passing the reaction mixture through a pad of silica gel or alumina, yielding the desired 1-iodoalkynes in good to excellent yield.
Scheme 3.
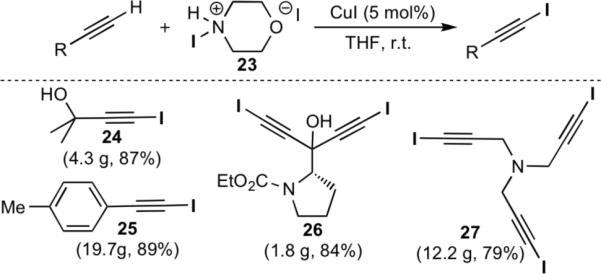
Synthesis of 1-iodoalkynes using N-iodomorpholine•HI.
Given the speed and fidelity with which the 1-iodoalkynes could be synthesized, a one-pot, two-stage protocol was developed (Scheme 4). The 1-iodoalkyne was partially purified via filtration through neutral alumina prior to the introduction of the azide component.[15] This method gave 5-iodotriazoles 28–30 with an efficiency comparable to that observed with the isolated 1-iodoalkynes.
Scheme 4.
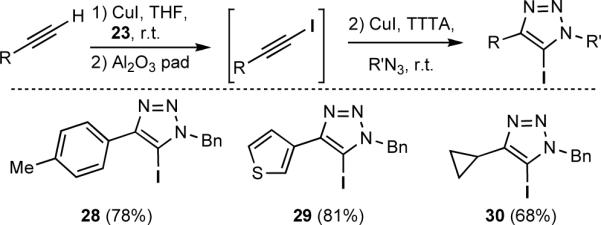
One-pot, two-step synthesis of 5-iodo-1,2,3-triazoles.
This sequence could be further extended to the synthesis of 1,4,5-triaryl-1,2,3-triazoles 31–33 (Scheme 5) by assembling the 5-iodotriazole and immediately employing Pd(0)-catalyzed cross-coupling with an appropriate arylboronic acid.[16] This simple stepwise construction obviates purification of any intermediates and simultaneously provides complete control over the placement of substituents around the 1,2,3-triazole core, allowing facile access to all regioisomeric permutations of triaryltriazoles 31–33. This achievement is notable, as a similar regiocontrolled synthesis would not be possible via thermal or ruthenium-catalyzed 1,3-dipolar cycloaddition due to the high degree of similarity between the aryl groups (phenyl, tolyl, and p-methoxyphenyl).
Scheme 5.
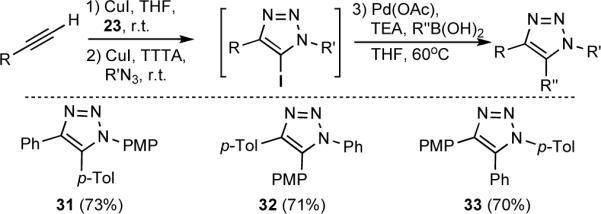
One-pot, three-step synthesis of 1,4,5-triaryltriazoles. PMP = p-methoxyphenyl, p-Tol = p-methylphenyl
Although this newly discovered Cu(I)-catalyzed cycloaddition clearly shares some similarities with the CuAAC process, modes of activation of iodo- and terminal alkynes by copper are likely distinctly different. Our mechanistic proposals are outlined in Scheme 6. One possible pathway is similar to that proposed for the CuAAC[1a,17] and involves the formation of the σ-acetylide complex 35 as the first key intermediate (Scheme 6, a).[18] Coordination of the azide via the proximal nitrogen is followed by the cyclization, yielding the cuprated triazoles 38. Cu-I exchange via σ-bond metathesis with iodoalkyne 34 completes the cycle, liberating iodotriazole 39 and regenerating acetylide 35.
Scheme 6.
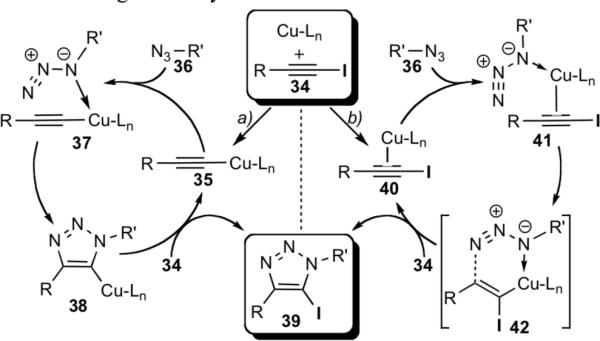
Proposed mechanisms for the Cu(I)-catalyzed azide-iodoalkyne cycloaddition.
Alternatively, copper may activate the iodoalkyne via the formation of a π-complex intermediate (Scheme 6, b), which then engages the azide, producing complex 41. Cyclization then proceeds via a vinylidene-like transition state, 42, to give iodotriazole 39. A similar transition state has been proposed to expliain the involvement of di-copper intermediates in the CuAAC reaction.[19] The distinctive feature of this pathway is that the C—I bond is never severed during the catalysis.
Although a detailed examination of the mechanism has not been completed, we currently favor pathway b based on our preliminary studies and the results from the reaction optimization experiments. The main argument in support of this hypothesis is the exclusive formation of the 5-iodotriazole even when the reaction is performed in protic solvents (Table 2) or with the substrates containing acidic protons (Figure 1, compounds 11, 15, 22). If pathway a were operational, the cuprated triazole intermediate 38 could be trapped with other electrophiles, including a proton, thereby producing a mixture of the 5-iodo and 5-prototriazoles. The absence of the latter products supports our proposal that pathway a is not dominant.
The new catalytic cycloaddition reaction enables rapid, controlled, and practical synthesis of 1,4,5-trisubstituted-1,2,3-triazoles. This reaction displays broad substrate scope, excellent functional group and solvent compatibility, and remarkably high rates which may exceed those of the more familiar CuAAC. In addition to these immediate practical benefits, the unprecedented and exquisite reactivity, as well as facile synthesis, of 1-iodoactylenes disclosed here will serve as a powerful tool to probe the mechanism of other copper-catalyzed transformations of alkynes, including the CuAAC reaction.
Experimental Section
Typical procedure for the synthesis of 1-iodoalkynes - synthesis of 1-iodo-phenylacetylene (1)
Phenylacetylene (8.17 g, 80.0 mmol) was dissolved in THF (200 mL) and treated with CuI (0.762 g, 4.00 mmol) and N-iodomorpholine (30.0 g, 88.0 mmol). The reaction mixture was stirred at room temperature for 45 minutes, after which a fine white precipitate had formed. The suspension was poured onto a pad of activated neutral alumina (400 mL) and the filtrate was collected under vacuum. The solid phase was washed with DCM (4×100mL) and the combined organic fractions were pooled and concentrated by evaporation, giving 1 (16.6 g, 72.8 mmol, 91%) as a yellow oil. This material was used without further purification.
Typical procedure for the synthesis of 5-iodotriazoles - synthesis of 5-iodo-4-phenyl-1-(3-(trifluoromethyl)benzyl)-1H-1,2,3-triazole (3)
CuI (9.52 mg, 0.050 mmol) and TTTA (0.021 g, 0.050 mmol) were stirred in THF (4.5 mL) at room temperature for 20 min, after which a homogeneous solution was obtained. 1 (0.228 g, 1.00 mmol) and 2 (0.201 g, 1.00 mmol) were dissolved in THF (0.5 mL) and added in a single portion to the catalyst solution. The reaction mixture was allowed to stir for 45 min, and then quenched by adding 1 mL of 10% NH4OH solution. The volatile components were removed by evaporation, and the resulting residue was suspended in water and diethyl ether. A precipitate formed upon vigorous stirring and was isolated by filtration, giving 3 (0.399 g, 0.930 mmol, 93%) as a fine white powder.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health, National Institutes of General Medical Sciences (GM28384, K.B.S.; GM087620 and GM083658, V.V.F.) and the Skaggs Institute for Chemical Biology. Postdoctoral fellowships were provided by the NIH (J.C.T) and NSERC (J.E.H).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- [1].a Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem., Int. Ed. Engl. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; b Tornøe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- [2].a Wu P, Fokin VV. Aldrichimica Acta. 2007;40:7. [Google Scholar]; b Meldal M, Tornøe CW. Chem. Rev. 2008;108:2952. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- [3].a Kolb HC, Sharpless KB. Drug Discovery Today. 2003;8:1128. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]; b Whiting M, Tripp JC, Lin YC, Lindstrom W, Olson AJ, Elder JH, Sharpless KB, Fokin VV. J. Med. Chem. 2006;49:7697. doi: 10.1021/jm060754+. [DOI] [PubMed] [Google Scholar]; c Wilkinson BL, Bornaghi LF, Houston TA, Poulsen S-A. In: Drug Design Research Perspectives. Kaplan SP, editor. Nova, Hauppauge; 2007. p. 57. [Google Scholar]
- [4].a Link AJ, Tirrell DA. J. Am. Chem. Soc. 2003;125:11164. doi: 10.1021/ja036765z. [DOI] [PubMed] [Google Scholar]; b Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. J. Am. Chem. Soc. 2003;125:3192. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]; c Lutz J-F, Zarafshani Z. Adv. Drug Delivery Rev. 2008;60:958. doi: 10.1016/j.addr.2008.02.004. [DOI] [PubMed] [Google Scholar]
- [5].Hawker CJ, Fokin VV, Finn MG, Sharpless KB. Aust. J. Chem. 2007;60:381. [Google Scholar]
- [6].a Evans RA. Aust. J. Chem. 2007;60:384. [Google Scholar]; b Johnson JA, Koberstein JT, Finn MG, Turro NJ. Macromol. Rapid Comm. 2008;29:1052. [Google Scholar]
- [7].a Zhang L, Chen X, Xue P, Sun HHY, Williams ID, Sharpless KB, Fokin VV, Jia G. J. Am. Chem. Soc. 2005;127:15998. doi: 10.1021/ja054114s. [DOI] [PubMed] [Google Scholar]; b Boren BC, Narayan S, Rasmussen LK, Zhang L, Zhao H, Lin Z, Jia G, Fokin VV. J. Am. Chem. Soc. 2008;130:8923. doi: 10.1021/ja0749993. [DOI] [PubMed] [Google Scholar]
- [8].a Chuprakov S, Chernyak N, Dudnik AS, Gevorgyan V. Org. Lett. 2007;9:2333. doi: 10.1021/ol070697u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fukuzawa S-I, Shimizu E, Ogata K. Heterocycles. 2009;78:645. [Google Scholar]
- [9].a Wu YM, Deng J, Li Y, Chen QY. Synthesis. 2005:1314. [Google Scholar]; b Li L, Zhang G, Zhu A, Zhang L. J. Org. Chem. 2008;73:3630. doi: 10.1021/jo800035v. [DOI] [PubMed] [Google Scholar]
- [10].a Perez-Castro I, Caamano O, Fernandez F, Garcia MD, Lopez C, De Clercq E. Org. Biomol. Chem. 2007;5:3805. doi: 10.1039/b710348d. [DOI] [PubMed] [Google Scholar]; b Kuijpers BHM, Dijkmans GCT, Groothuys S, Quaedflieg PJLM, Blaauw RH, van Delft FL, Rutjes FPJT. Synlett. 2005:3059. Kuijpers et al. recently reported an elegant synthesis of 5-bromo-1,2,3-triazoles from 1-bromoalkynes, however reactions required 40 mol% Cu(I)/Cu(II), elevated temperature or 16–50 h to reach completion; [Google Scholar]
- [11].The regiochemistry of 3 was assigned by reducing the 5-iodo center, giving 5-H-triazole, 4. See Supporting Information for details.
- [12].Gerard B, Ryan J, Beeler AB, Porco JA., Jr. Tetrahedron. 2006;62:6405. [Google Scholar]
- [13].Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org. Lett. 2004;6:2853. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- [14].Rice RV, Beal GD. 2,290,710. U.S. Patent. 1943
- [15].Addition of electrophilic iodinating reagents (N-iodomorpholine, ICl, NIS, etc.) to a solution containing CuI-TTTA, the target azide and terminal alkyne rapidly gave the corresponding 1-iodoalkyne, but failed to promote the subsequent cycloaddition. This failure is likely due to the disruption of the catalytically active complex, either via oxidation of the metal or displacement/destruction of the ligand.
- [16].Deng J, Wu Y-M, Chen Q-Y. Synthesis. 2005:2730. [Google Scholar]
- [17].Himo F, Lovell T, Hilgraf R, Rostovtsev VV, Noodleman L, Sharpless KB, Fokin VV. J. Am. Chem. Soc. 2005;127:210. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- [18].Siemsen P, Livingston RC, Diederich F. Angew. Chem. Int. Ed. 2000;39:2632. [PubMed] [Google Scholar]
- [19].a Ahlquist M, Fokin VV. Organometallics. 2007;26:4389. [Google Scholar]; b Straub BF. Chem. Comm. 2007:3868. doi: 10.1039/b706926j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.