

Abstract
Upon recognition of influenza virus (Flu) via TLR7, plasmacytoid dendritic cells (pDCs) produce type I IFN in significant amounts. Synthetic TLR7 ligands induce the maturation of pDCs, as evidenced by the expression of costimulatory molecules and the production of pro-inflammatory cytokines; however, they only induce low-level production of IFN-α. In order to dissect the TLR7 signaling in pDCs and how these different profiles are induced, we studied the effects of two TLR7 ligands (Flu and CL097) on the activation of blood-isolated pDCs and the human GEN2.2 pDC cell line.
Type I IFN production by pDCs correlates with differential Interferon Regulatory Factor (IRF) 7 translocation into the nucleus induced by the two TLR7 ligands. Surprisingly, with both activators we nevertheless observed the rapid expression of the IFN-inducible genes MxA, CXCL10 and TRAIL within 4 h of stimulation. This expression, controlled by STAT1 phosphorylation, was independent of type I IFN. STAT1 activation was found to be strictly dependent on the PI3K-p38MAPK pathway, demonstrating a new signaling pathway leading to rapid expression of IFN-inducible genes after TLR7 triggering. Thus pDCs, through this unusual TLR7 signaling, have the capacity to promptly respond to viral infection during the early phases of the innate immune response.
Keywords: Cell Differentiation; Cell Line; Cell Nucleus; metabolism; Dendritic Cells; cytology; Humans; Inflammation; Interferon Type I; metabolism; Interferons; metabolism; Leukocytes, Mononuclear; cytology; Ligands; Models, Biological; Phosphorylation; Signal Transduction; Toll-Like Receptor 7; metabolism
Introduction
Toll-like receptors (TLRs) contain a leucine-rich repeat ectodomain which enables the recognition of pathogen-associated molecular patterns1. Among the 10 TLRs identified in humans, TLR7 is the least studied. This TLR binds single-stranded viral RNA, it localises to endosomal compartments and is particularly expressed by plasmacytoid dendritic cells (pDCs)2. Ligand binding to TLR7 activates human pDCs which respond by producing either pro-inflammatory cytokines or substantial levels of type I IFN and activating specific T cells3–5. In addition, we have recently demonstrated that Flu- and TLR7 agonist- activated pDCs express TNF-related apoptosis-inducing ligand (TRAIL). This renders them capable of direct cytotoxic activity towards infected and tumor cells6. This has been observed in vivo: Stary et al. describe the presence of TRAIL-expressing and IFN-α producing pDCs in tumors after topical use of the TLR7 ligand imiquimod in basal cell carcinoma (BCC) treatment7. Taken together, these data suggest that pDCs represent key target cells in the striking anti-tumor effects observed with synthetic TLR7 agonists, such as imidazoquinolines, in the treatment of cutaneous virus-induced neoplasia (e.g., condyloma-HPV)8 and other skin tumors, such as BCC9.
The signaling pathways triggered by TLR7 activation have been partially described. Following ligand recognition, endosomal TLR7 interacts with the key adaptor molecule MyD88 (myeloid differentiation primary-response gene 88) which recruits a signal complex comprised of IRAK1 (interleukin- 1- receptor- associated kinase 1), IRAK4 and TRAF6 (tumor- necrosis factor- receptor- associated factor 6). TLR7 triggering leads to the activation of NF-κB (nuclear factor-κB) and MAPKs (mitogen-activated protein kinases) inducing the expression of pro-inflammatory cytokines and co-stimulatory molecules and the translocation of IRF7 (interferon-regulatory factor 7) into the nucleus where it can induce the transcription of type I IFN genes10. Agonistic engagement of TLR7 also leads to the expression of the IFN-inducible genes MxA, CXCL10 and TRAIL11. A type I IFN autocrine loop is thought to explain expression of these genes, as has been described in mouse bone-marrow derived DCs12. However, the precise pathway downstream of TLR7 leading to human pDC activation remains to be determined.
In this article we analyze the activation profile and signaling events triggered by two TLR7 ligands: Influenza virus (Flu, natural ligand) and CL097 (synthetic agonist). We studied the effects of these two ligands on both blood-isolated pDCs and the pDC model cell line GEN2.2. Results of these studies demonstrate the existence of a novel pathway downstream of TLR7 involving early STAT1 phosphorylation and expression of IFN-inducible genes in the absence of type I IFN.
Materials and methods
Antibodies, Flow cytometry
Surface or intracellular phenotype was determined by flow cytometry on a FACS Canto II (Becton-Dickinson, Mountain View, CA, USA), using specific antibodies, by direct or indirect labelling. The following antibodies were from Immunotech (Beckman Coulter, Marseille, France): PE-conjugated anti-CD40 (mAb89), PE-conjugated anti-CD80 (mAb104) and PE-conjugated goat anti-mouse IgG (H + L). PE-conjugated anti-CD123 (9F5) was purchased from Pharmingen (San Diego, CA, USA). PE-conjugated anti-phospho-STAT1 (4a/pY701-stat1) was from BD (Becton-Dickinson). FITC-conjugated anti-BDCA-2 (AC144) and FITC-conjugated anti-IFN-α (LT27:295) were from Miltenyi Biotec (Paris, France) and purified anti-TRAIL (2E5) from Alexis (Lausen, Switzerland).
Cells
Normal pDCs were isolated from PBMC of healthy volunteers with a BDCA-4 cell isolation kit (Miltenyi Biotec). Their purity, as determined with anti-BDCA-2 and anti-CD123 mAbs, was about 80% (Figure 1.A).
Figure 1. Influenza virus and CL097 induce human pDC activation by triggering TLR7.
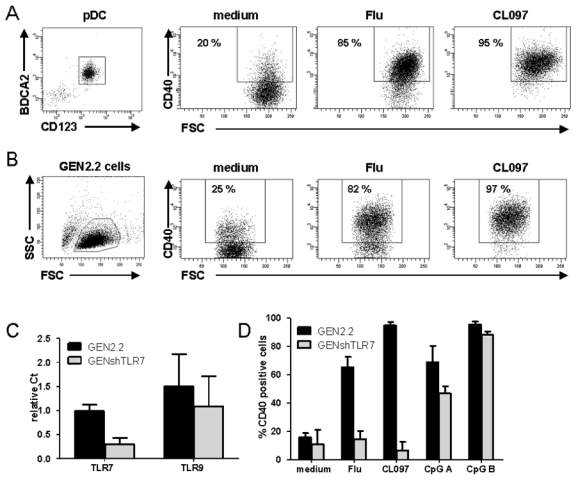
(A) Left panel: Flow cytometry evaluation of purity of unstimulated pDCs enriched from healthy donor blood labeled with anti-BDCA2 and anti-CD123 antibodies. Cells were untreated (medium) or stimulated with UV-formol-inactivated Influenza virus (Flu) or synthetic TLR7 ligand (CL097). Expression of CD40 was measured by flow cytometry on CD123+ cells. Dot plots are shown, the percentage of CD40 positive cells is indicated on each plot. Results shown are representative of four independent experiments. (B) GEN2.2 cells were untreated or stimulated with Flu or CL097 for 24 h. CD40 expression was evaluated on forward scatter (FSC)/side scatter (SSC) gated live cells by flow cytometry. Percentages indicated on dot correspond to the proportion of CD40 positive cells. Results shown are representative of at least five independent experiments. (C) Expression levels of TLR7 and TLR9 in the GEN2.2 cell line and in lentiviral shRNA TLR7 transfected GEN2.2 cells (GENshTLR7) measured by real-time PCR. Expression levels are normalized to G6PDH. Data are shown as the mean and standard deviation from duplicate values of two independent experiments. (D) GEN2.2 and GENshTLR7 cells were untreated or stimulated with Flu, CL097 or two different synthetic TLR9 ligands (CpG A and CpG B) for 24 h. Expression of CD40 was measured by flow cytometry. The mean percentages and standard deviation from duplicate values of three independent experiments are shown.
The pDC cell line GEN2.2 was grown in complete medium (RPMI 1640 Glutamax (GibcoBRL, Cergy-Pontoise, France) supplemented with 1 mM sodium pyruvate, 20 μg/ml gentamicin, nonessential amino acids) to which 10 % heat inactivated Fetal Calf Serum was added (FCS, Gibco).
Generation of lentiviral shRNA TLR7 transfected GEN2.2 cells
GEN2.2 cells were transfected with MISSION Lentiviral transduction particles targeting TLR7 (NM_016562) Clone ID: TRCN0000056975 (Sigma Aldrich, St Quentin Fallavier, France) at MOI 2. Transfected cells were maintained in the presence of 10 μg/ml puromycin for 2 weeks to allow the selection of resistant clones which we called GENshTLR7 cells. The level of TLR7 mRNA expression in GENshTLR7 cells was assessed by real-time PCR. Silencing was found to diminish TLR7 mRNA expression by 80 %.
Activation of GEN2.2 cells
Cells were cultured at 106 cells/ml in complete medium with 10 % FCS. Cells were stimulated with either 640 UHA/ml UV-formol-inactivated influenza virus strain A/H3N2/Wisconsin/67/05 (Sanofi Pasteur), or 1 μg/ml CL097 (TLR7/8 ligand, Invivogen, Toulouse, France), or 10 μg/ml CpG-A ODN 2336 (TLR9 ligand, Coley Pharmaceuticals, Ottawa, Canada), or 10 μg/ml CpG-B ODN 2216 (TLR9 ligand, Invivogen) or 50,000 U/ml human recombinant IFN-α (PeproTech, Neuilly sur Seine, France). For some experiments, blocking anti-IFN-α (50,000 U/ml, PBL medical laboratories), anti-IFN-β (25,000 U/ml, PBL) and anti-IFN-α/βR2 (5 μg/ml, PBL) or inhibitors from Calbiochem (Nottingham, UK) were added: 5 μM BAY11-7082, 5μM BMS-345541, 10 μM LY-294002 or 50 μM SB203580. After stimulation, phenotypic analyses were performed by flow cytometry. Culture supernatants were cryopreserved for cytokine measurements. These supernatants were tested for IFN-α content by ELISA (PBL) and for IL-6, IL-8, TNF-α and CXCL10 by Cytometric Bead Array multiplex (CBA, BD Biosciences).
Protein extraction and signaling factor analysis
After stimulation of GEN2.2 cells, cytosolic and nuclear fractions were extracted using the protein extraction kit from Active Motif (Rixensart, Belgium). Nuclear extracts were probed for NFκB subunits c-Rel, p50, p52, p65 and RelB content using the TransAM NFκB family kit (Active Motif). Whole cell extracts were used for quantification of phospho-STAT1 and phospho-p38MAPK by CBA multiplex (BD Biosciences).
Western-blot analysis
Following activation, GEN2.2 cells were washed in phosphate-buffered saline (PBS), lysed in 100 ml of sample buffer, and heated at 100 °C for 5 minutes. Whole-cell extract (20 μg) was loaded onto a 12 % SDS-polyacrylamide gel. After electrophoresis, proteins were transferred to a PVDF membrane (BIORAD, Marne la Coquette, France). Non-specific binding sites were blocked with 5 % nonfat milk in PBS Tween20 0.1 %. Membranes were then incubated with primary antibodies: anti-phospho-STAT1 (4a/pY701), anti-phospho-STAT2 (7a/pY) (Pharmingen) and anti-actin (Sigma). Antibody labeling was revealed using horseradish peroxidase (HRP)-conjugated secondary antibodies (Dako, Glostrup, Denmark) and was visualized using enhanced chemiluminescence (ECL, Amersham Life Science, Les Ulis, France).
Immunofluorescence
Twenty one well Teflon printed slides (Immuno-Cell International, Mechelen, Belgium) were coated with 1 μg/ml poly-L-lysine (Sigma). 104 GEN2.2 cells were added to each well and allowed to adhere for 1 h. Cells were then stimulated for 2 and 3 h with 640 UHA/ml UV-formol-inactivated influenza virus strain A/H3N2/Wisconsin/67/05, 1 μg/ml CL097 or 5,000 U/ml human recombinant IFN-α. After activation, medium was removed, slides were fixed in −20 °C methanol for 10 min and dried at room temperature for 1 h. Slides were rehydrated in PBS for 10 min and then incubated with primary antibody anti-IRF7 (H-246, Santa Cruz Biotechnology, San Diego, CA, USA). Antibody labeling was revealed using FITC-conjugated goat anti-rabbit secondary antibody (Pharmingen). Cells were also colored with 1 mg/ml Evans blue (Sigma) before visualization by fluorescence microscopy.
Quantitative RT-PCR
Total RNA was isolated from stimulated GEN2.2 cells using RNeasy kit (Qiagen, Courtaboeuf, France). Reverse transcription to cDNA was carried out by standard methods using reverse transcriptase (Roche Diagnostics, Meylan, France) and dNTP (Roche). These cDNA were amplified using naked primers, LightCycler TaqMan Master mix and Universal ProbeLibrary probe (Roche). Polymerase Chain Reactions (PCR) were conducted in a LightCycler instrument (Roche). Primers were synthetised by Roche, their sequences were as follows (listed 5′-3′): G6PDH (F: AACAGAGTGAGCCCTTCTTCA, R: GGAGGCTGCATCATCGTACT); human TLR7 (F: GCTAGACTGTCTCAAAAGAACAAAAA, R: GCCCACACTCAATCTGCAC); human TLR9 (F: ATAGCCGTGAGCCGGAAT, R: GCAGGCAGAGGTGAGGTG); IFN-α1 (F: CCCTCTCTTTATCAACAAACTTGC, R: TTGTTTTCATGTTGGACCAGA); IFN-α2 (F: TCCTGCTTGAAGGACAGACA, R: TTTCAGCCTTTTGGAACTGG); IFN-β1 (F: CTTTGCTATTTTCAGACAAGATTCA, R: GCCAGGAGGTTCTCAACAAT); IFN-ω1 (F: ACCAGCTATAGCCCTGTTGG, R: AAGTAGGCCATGGTTCTGAGG); MxA (F: TCCAGCCACCATTCCAAG, R: CAACAAGTTAAATGGTATCACAGAGC). Relative threshold cycle (Ct) values for each gene were normalized to the housekeeping gene G6PDH using the equation 2(−dCp) where Cp is the mean cross point of duplicate runs calculated by Lightcycler software and dCp = gene Cp − G6PDH Cp.
Results
Influenza virus and CL097 induce human pDC activation via TLR7
This study focuses on TLR7 signaling in pDCs. The recently developed human pDC cell line, GEN2.2, was used as a model of pDC13,14. We evaluated 2 kinds of TLR ligands: CL097, an imidazoquinoline15,16 and influenza virus (Flu)17. We first verified their activity and their specificity for TLR7. After Flu or CL097 stimulation, both enriched primary pDCs (Figure 1.A) and the GEN2.2 cells (Figure 1.B) displayed the same activation profile with a high increase of CD40 expression, thus both ligands were capable of inducing pDC activation. To verify the TLR7-specificity of the effect, TLR7 shRNA lentiviral transfection of GEN2.2 cells was performed, stably-transfected cells were selected leading to the establishment of the GENshTLR7 cell-line. These cells showed an 80 % decrease in TLR7 mRNA expression, while levels of TLR9 mRNA were unchanged (Figure 1.C). Upon Flu or CL097 stimulation, GENshTLR7 cells were unable to up-regulate CD40 but maintained their capacity to respond to TLR9 ligands (CpG A and B) (Figure 1.D). Moreover, GENshTLR7 cells produced neither IFN-α nor inflammatory cytokines following stimulation (data not shown). Control-shRNA transfected cells displayed the same activation profile as untransfected GEN2.2 cells, confirming that the previous results were not due to an alteration caused by lentiviral transfection (data not shown). Thus, both Flu and CL097 act specifically on TLR7 to induce CD40 upregulation and cytokine production. Altogether, these data show that Flu and CL097 activate human pDC by triggering TLR7.
Influenza virus and CL097 induce differential activation of pDCs
PDCs are known to secrete pro-inflammatory cytokines or substantial amounts of type I IFN upon activation3,4. The nature of the cytokines produced by GEN2.2 cells following activation by the two TLR7 ligands was evaluated. CL097-stimulated GEN2.2 cells produced the pro-inflammatory cytokines IL-6, IL-8 and TNF-α whereas Flu-activated cells mainly secreted IFN-α (Figure 2.A). To further characterize this secretion, the transcription of 4 type I IFN genes was assessed. Only Flu activation induced the transcription of IFN-α1, IFN-α2, IFN-β1 and IFN-ω1 genes in GEN2.2 cells, while CL097 activation did not result in transcription of any of the IFN genes tested (Figure 2.B). These results were confirmed with human blood pDCs. A higher number of IFN-α-secreting cells were observed after Flu-activation (56 %) than after CL097 stimulation (7 %) (Figure 2.C). Thus, the two TLR7 ligands chosen induced different activation profiles for pDCs.
Figure 2. Differential pDC maturation is triggered by different TLR7 ligands.
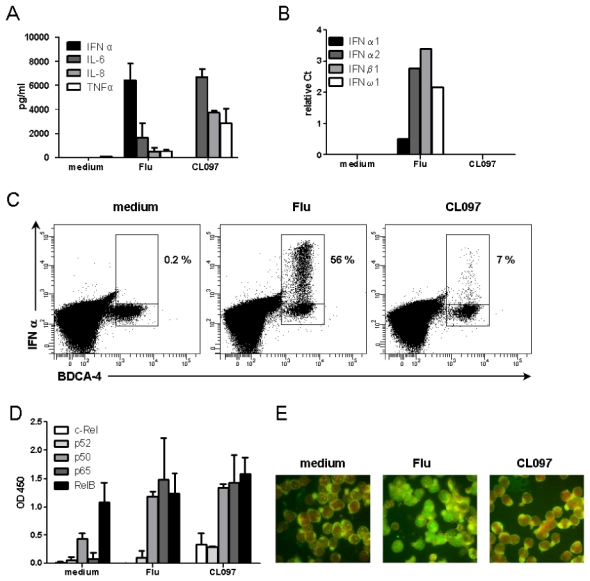
GEN2.2 cells were untreated or stimulated with Flu or CL097 for 24 h. (A) Production of pro-inflammatory cytokines was measured in culture supernatants by ELISA and CBA. The mean and standard deviation from duplicate values of three independent experiments are shown. (B) After 24 h of culture, RNA was extracted and RNA expression levels of type I IFNs: IFN-α1, -α2, -β1 and -ω1 were measured by quantitative PCR. Data shown are normalized to G6PDH and are representative of two independent experiments. (C) PBMCs from healthy donors were cultured in the absence or presence of Flu or CL097 for 3 h. Secretion was blocked by adding brefeldin A for a further 4 h. Intracellular IFN-α production was measured in HLA-DR+ BDCA4+ pDCs by flow cytometry. Dot plots show the percentage of IFN-α-producing cells among pDCs. Representative results from three different donors are shown. (D) GEN2.2 cells were cultured in the absence or presence of Flu or CL097 for 3 h. Cells were lyzed and proteins extracted. The different NFκB subunits were quantified in nuclear fractions using the TransAM kit. The mean OD and standard deviation from duplicate values of three independent experiments are shown. (E) GEN2.2 cells were cultured in the absence or presence of Flu or CL097 for 3 h. Cells were immunostained for IRF7 and evans blue colored. Immunofluorescence was assessed by microscopy. Representative images from three independent experiments are shown.
The transcription factors NFκB and IRF7 play essential roles in inducing the expression of pro-inflammatory cytokines and type I IFN, respectively. Therefore, we examined the translocation of these two factors into the nucleus following TLR7 triggering. Both Flu and CL097 induced similar nuclear translocation of p50 and p65 subunits of the canonical NFκB dimer (Figure 2.D). The quantities of p52 and RelB subunits of the noncanonical NFκB pathway were not modified during activation. Interestingly, the NFκB c-Rel subunit was only detectable in the nucleus of the CL097-stimulated GEN2.2 cells (Figure 2.D). In contrast, IRF7 nuclear translocation was only observed in Flu-activated GEN2.2 cells (Figure 2.E). This result corroborates the type I IFN production profile described above. These data suggest that, whereas both ligands induce inflammatory cytokine secretion via the NFκB pathway, Flu but not CL097 induces IFN-α secretion following IRF7 translocation.
IFN-inducible genes are expressed after TLR7 triggering in a type I IFN-independent manner
Secretion of type I IFNs by pDCs leads to the expression of a group of IFN-inducible genes including MxA, CXCL10 and TRAIL18–20. We next examined whether the differential type I IFN secretion observed after TLR7 triggering could induce differential expression of these IFN-inducible genes. IFN-α secretion was observed within 4 h after TLR7 stimulation by Flu, whereas no detectable IFN-α was measured at any time point (up to 24 h) after CL097 activation (Figure 3.A). Surprisingly, the IFN-inducible genes products were nevertheless detected within 4 h after activation by both TLR7 ligands (Figure 3. B–D). The expression profiles were identical whatever the ligand applied: expression peaked at 4 h and decreased at 24 h for MxA and TRAIL genes products (Figure 3.C and 3.D), whereas CXCL10 production continuously increased up to and beyond 24 h (Figure 3.B). This suggests that, in our model, the induction of MxA, CXCL10 and TRAIL could be independent of IFN-α.
Figure 3. TLR7 triggers expression of IFN-inducible genes in a type I IFN-independent manner.
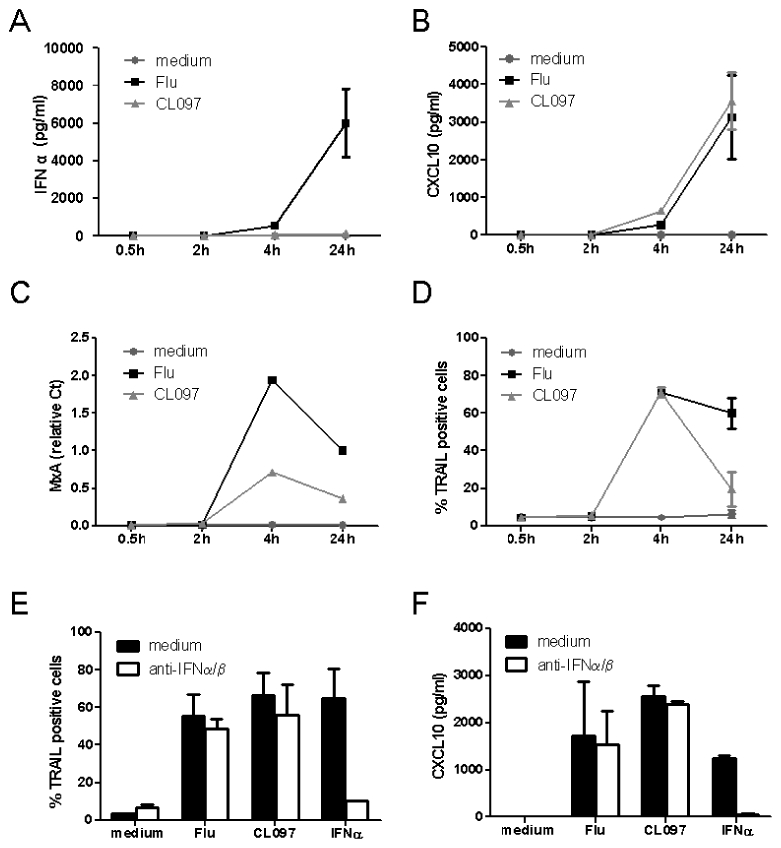
GEN2.2 cells were untreated or stimulated with Flu or CL097 for different times. Supernatants were collected for evaluation of (A) IFN-α and (B) CXCL10 production. (C) RNA was extracted for measurement of MxA transcription levels by real-time PCR. (D) TRAIL expression was evaluated by flow cytometry. GEN2.2 cells were untreated or stimulated with Flu or CL097 in the absence or presence of anti-IFN-α/β and anti-IFN-α/βR neutralizing antibodies for 4 h. (E) TRAIL expression was evaluated by flow cytometry and (F) CXCL10 production was measured in culture supernatants by CBA. Data shown are the means and standard deviation from duplicate values from two independent experiments except data in (C) which are representative of two independent experiments.
Very low levels of type I IFN could be responsible for inducing the expression of IFN-inducible genes following CL097 stimulation of GEN2.2 cells. To investigate this possibility, GEN2.2 cells were stimulated by the two TLR7 ligands in the presence or absence of neutralizing anti-IFN-α/β and anti-IFN-α/βR antibodies. In accordance with the absence of any detectable IFN-α production, the blocking of type I IFN signaling had no effect on TRAIL and CXCL10 expression induced by TLR7 ligands (Figure 3. E-F). The pre-incubation of cells with the neutralizing antibodies for 30 or 60 min prior to ligand exposure in order to exclude the involvement of any autocrine intracellular type I IFN pathway had no effect on these expressions (supplemental figure 1). However, the neutralizing antibodies could block both TRAIL and CXCL10 expression induced by exogenous IFNα/β. Altogether, these data demonstrate that expression of IFN-inducible genes could be triggered independently of type I IFN within 4 h after TLR7 triggering.
Expression of IFN-inducible genes is independent of the NFκB pathway and extracellular factors
In order to analyze whether NFκB was involved in the observed expression of IFN-inducible genes, we measured TRAIL expression in GEN2.2 cells 4 h post-activation by Flu and CL097 in the presence of the IκB kinase (IKK) inhibitors BAY11-708221 and BMS-34554122. Neither of these inhibitors altered the TRAIL expression profile (Figure 4.A), whereas they both abrogated NFκB-dependent CD40 upregulation (Figure 4.B). These results indicate that the expression of IFN-inducible genes following TLR7 triggering is independent of NFκB activation.
Figure 4. Expression of IFN-inducible genes is independent of the NFκB pathway and of extracellular factors.
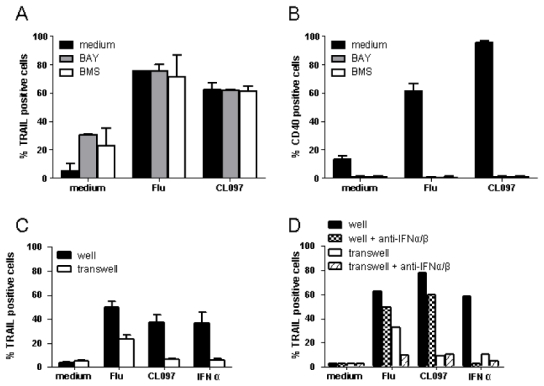
GEN2.2 cells were untreated or stimulated with Flu or CL097 in the absence or presence of the NFκB inhibitors BAY and BMS. TRAIL (A) and CD40 (B) expression were evaluated by flow cytometry after 4 h and 24 h of culture, respectively. The mean percentages and standard deviation from duplicate values of four independent experiments are shown. (C) GEN2.2 cells were untreated or stimulated for 2 h. After washing, cells were placed in 24-well plates; untreated cells were added in transwells. After 4 h of culture, TRAIL expression was evaluated by flow cytometry on cells in wells and transwells. Data shown are the means and standard deviation from duplicate values of three independent experiments. (D) GEN2.2 cells were untreated or stimulated for 2 h in the absence or presence of anti-IFN-α/β and anti-IFN-α/βR neutralizing antibodies. After washing, cells were placed in 24-well plates; untreated cells were added in transwells. After 4 h of culture, TRAIL expression was evaluated by flow cytometry on cells in wells and transwells. Data from one representative experiment are shown.
Extracellular factors could also contribute to the induction of expression of IFN-inducible genes. To rule out their involvement, TRAIL expression was measured on untreated GEN2.2 cells to which supernatants from TLR7-activated GEN2.2 cells were applied. No induction of TRAIL expression was observed in cells exposed to supernatants from CL097-activated cells (Figure 4.C). The expression of TRAIL induced by the supernatant from Flu-activated cells was due to the presence of secreted IFN-α in this supernatant. This expression could be blocked using neutralizing anti-IFN-α/β antibodies (Figure 4.D). These latter results demonstrate that expression of IFN-inducible genes following TLR7 triggering is due to a previously undescribed intracellular signaling pathway.
Phosphorylation of STAT1 in response to TLR7 triggering depends on the PI3K-p38MAPK pathway
Expression of IFN-inducible genes is regulated by the transcription factor IFN stimulated gene factor 3 (ISGF3)23. ISGF3 is in fact a complex of proteins including STAT1, STAT2 and IRF9. We next examined whether this complex was activated in response to TLR7 triggering. Activation of GEN2.2 cells by both Flu and CL097 induced phosphorylation of STAT1 within 2 h (Figure 5.A and B) at levels comparable to the classic type I IFN activation (Figure 5.B). Low-level phosphorylation of STAT2 was also observed (Figure 5.B). In these analyses, no variation in total STAT protein quantity was observed (data not shown). These results were further confirmed by flow cytometry analysis. Results showing that STAT1 was phosphorylated in one third of GEN2.2 cells within 2 h after TLR7 activation are shown in Figure 5.C. The presence of neutralizing anti-IFN-α/β and anti-IFN-α/βR antibodies, which prevented IFN-α-induced STAT1 activation, had no effect on the level of STAT1 phosphorylation induced subsequent to TLR7 triggering (Figure 5.D). Thus STAT1 activation could also be induced via TLR7 signaling in a type I IFN-independent manner.
Figure 5. STAT1 is phosphorylated independently of type I IFN after TLR7 triggering with either ligand.
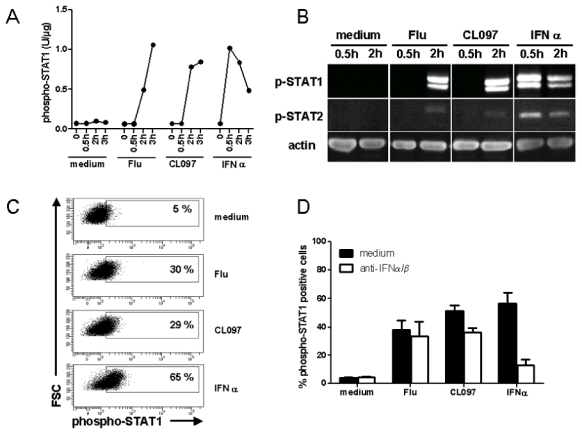
GEN2.2 cells were untreated or stimulated with Flu, CL097 or IFN-α for 30 min, 2 h and 3 h. Whole cell protein extracts were prepared. (A) Phospho-STAT1 (pY701) was quantified in the protein extracts by CBA. Data shown are representative of two independent experiments. (B) Western-blot analysis of phospho-STAT1 (pY701) and phospho-STAT2 (pY690) following activation of GEN2.2 cells. Data shown are representative of two independent experiments (C) After 2 h of stimulation, cells were fixed and permeabilised for phospho-STAT1 (pY701) analysis by flow cytometry. Representative dot plots of four independent experiments are shown. (D) Activation of GEN2.2 cells in the absence or presence of anti-IFN-α/β and anti-IFN-α/βR neutralizing antibodies for 2 h. phospho-STAT1 (pY701) was analysed by flow cytometry. Data shown are the means and standard deviation from duplicate values of two independent experiments.
Other groups have suggested that STAT1 phosphorylation in response to TLR9 triggering depends on p38MAPK activation24. To test this hypothesis in the TLR7 model, we analyzed the activation of p38MAPK in stimulated GEN2.2 cells. Phosphorylation of p38MAPK was observed within 30 min after stimulation of GEN2.2 cells by either Flu or CL097, but not after activation by type I IFN, as was expected (Figure 6.A). Thus, phosphorylation of p38MAPK is induced by TLR7 stimulation. We next investigated the possible pathway linking TLR7 via p38MAPK to STAT1. We found PI3K, which has previously been linked to the TLR7 pathway25, to have a role in this pathway. The PI3K inhibitor LY294002, in addition to altering the p38MAPK phosphorylation profile in response to TLR7 triggering (Figure 6.B), also impaired STAT1 phosphorylation and TRAIL expression by GEN2.2 cells. A similar effect was observed in the presence of the specific26 p38MAPK inhibitor SB203580 (Figure 6. C–D). These results were confirmed on blood-isolated pDCs after stimulation of cells with Flu or CL097 where the presence of LY or SB inhibited TRAIL expression (Figure 6. E) and CXCL10 production (Figure 6.F). Taken together, these data illustrate that both STAT1 activation and the expression of IFN-inducible genes following TLR7 triggering were dependent on the PI3K-p38MAPK pathway.
Figure 6. STAT1 phosphorylation and expression of IFN-inducible genes following TLR7 triggering depends on the PI3K-p38MAPK pathway.
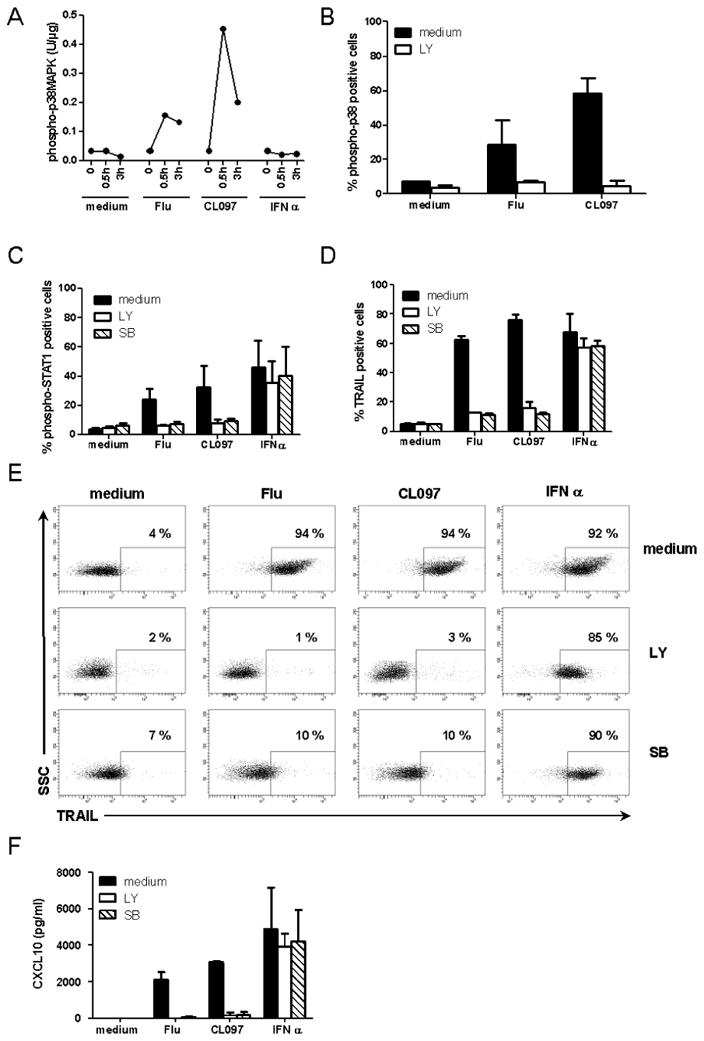
GEN2.2 cells were untreated or stimulated with Flu, CL097 or IFN-α for 30 min or 3 h. (A) phospho-p38MAPK (pT180/pY182) was quantified on whole-cell lysates by CBA. Data shown are representative of two independent experiments. (B) GEN2.2 cells were untreated or stimulated with Flu, CL097 or IFN-α for 30 min in the absence or presence of the PI3K inhibitor LY. Cells were fixed and permeabilised for phosphop38MAPK (pT180/pY182) analysis by flow cytometry. The mean percentages and standard deviation from duplicate values of two independent experiments are shown. (C) phospho-STAT1 (pY701) and (D) TRAIL expression were analysed by flow cytometry after 2 h or 4 h of stimulation of GEN2.2 cells with Flu, CL097 or IFN-α in the absence or presence of LY and the specific p38MAPK inhibitor SB203580 (SB). The mean percentages and standard deviation from duplicate values of three independent experiments are shown. Blood-isolated pDCs were unstimulated or stimulated with Flu or CL097 for 4 h in the absence or presence of LY and SB. (E) Expression of TRAIL was evaluated by flow cytometry. Dot plots representative of two experiments are shown. The percentage of TRAIL positive cells is indicated on each plot. (F) CXCL10 production was measured in cell culture supernatants by CBA. The mean and standard deviation from duplicate values of two experiments performed with two different donors are shown.
Discussion
Many studies have suggested that the effects triggered by TLR7 ligands as adjuvants in cancer immunotherapy could be attributed to the activation of pDCs and their subsequent type I IFN production7,27,28. In order to understand how agonists for the same receptor lead to different activated pDC phenotypes we initiated a study of the signaling pathways induced in pDCs by two different TLR7 ligands. In this article we show that, in contrast to Flu activation, CL097 stimulation of pDCs does not induce production of high levels of IFN-α. However, we were able to demonstrate that both TLR7 ligands led to rapid expression IFN-inducible genes via a type I IFN independent pathway.
The data presented here reveal that Flu and CL097 both trigger human TLR7 but induce differential cytokine production. Using stably shRNA-transfected cells, GENshTLR7, we could demonstrate that human pDC activation by inactivated Influenza virus required TLR7, as had been previously described in a mouse model 17. Type I IFN production after CL097 activation was found to be null or extremely low compared to the massive amounts of IFN-α produced in the presence of Flu. This result corroborated those concerning IRF7 translocation into the nucleus of pDCs following stimulation with the different ligands. This differential type I IFN production is reminiscent of results obtained with TLR9 ligands29. Kerkmann et al. demonstrate that TLR9 triggering with CpG A ODNs leads to a high production of type I IFN while CpG B ODNs, although they bind TLR9, are unable to induce IFN secretion. The mechanism responsible for the differential behaviour of pDCs following TLR9 triggering has been suggested to be due to the spatiotemporal location of the ligand within the cell and its higher order structure30. Indeed, multimeric CpG A ODNs are retained within endosomes whereas monomeric CpG B ODNs traffic to lysosomal compartments where they trigger the activation of different signaling factors31,32. Similarly, Heil et al. observed that synthetic TLR7 ligands induce higher IFN-α production by PBMCs when complexed to the cationic lipid DOTAP33. Thus, Flu’s RNAs may follow the same cell trafficking as multimeric CpG A ODNs whereas CL097 may rather stay in a monomeric structure like CpG B, leading to a lysosomal location and absence of type I IFN production.
MxA, CXCL10 and TRAIL, through their role in the inhibition of virus replication, in the chemotaxis of immune cells and in the apoptosis of transformed and infected cells, are key players in countering infection and cancers34. Here, we demonstrate that these genes, which have been described to be tightly regulated by IFNs in various haematopoietic cells23,35, are rapidly expressed in TLR7-activated pDCs in the absence of type I IFN production, even when type I IFN-receptor signaling was blocked with neutralizing anti-IFN-α/β and anti-IFN-α/βR antibodies. While expression of the “IFN-inducible” genes was independent of IFN, it remained dependent on STAT1 phosphorylation on Tyr-701. This phosphorylation was found to be p38MAPK-dependent in our model, a similar signaling pathway was also described in CpG A-activated pDCs24. However, since p38MAPK is a serine kinase, known to phosphorylate STAT1 on Ser-72736, this tyrosine phosphorylation of STAT1 must be mediated by an intermediate tyrosine kinase whose activation is dependent on p38MAPK. Both the Janus kinases (JAK) 1 and 2 and c-Src are able to phosphorylate STAT1 on Tyr-70123,37 but their activation is generally associated with cytokine receptors38. In our model, we demonstrated that no soluble factors were involved. Therefore, the intermediate kinase responsible for STAT1 phosphorylation in response to TLR7 stimulation remains to be identified.
Here, we have shown that PI3K is involved in the activation of p38MAPK while other groups have demonstrated its implication in IRF7 translocation25,39. Moreover, the NFκB-dependent expression of CD80 and cytokines production were also impaired in the presence of LY following TLR7 triggering (data not shown), suggesting that PI3K is a key early signaling factor in the TLR7 pathway.
Altogether, exhaustive characterization of the signaling pathway downstream of TLR7 in pDCs led us to describe the expression of IFN-inducible genes in the absence of cytokine receptor engagement. This originality may confer on pDCs the capacity to respond more rapidly to viral infection, reinforcing their role in the early phases of the immune response. Finally, the work described here may help to define new therapeutic targets which could favor pDC activation in tumor and viral immunotherapy or to dampen it in pDC-driven autoimmunity.
Supplementary Material
Acknowledgments
This work was supported by two grants from the Institut National du Cancer and the Cancéropôle Lyon- Auvergne Rhône-Alpes (ACI-63-04 and Cancéropôle 2004-5) J. D. D.’s PhD was financed by the Ministère de l’Education Nationale de la Recherche et de la Technologie. A. B. was recipient grant from EFS. M. G-G. received a Young Researcher’s grant from Institut National de la Santé et de la Recherche Médicale.
Footnotes
Contribution: J. D. D. performed the experiments, analyzed the results, made the figures and wrote the paper; A. B. and J-P. M. performed a part of the experiments; M. G-G., L. C., and J. P. did proof-reading; J. D. D., L. C. and J. P. designed the research.
Conflict-of-interest disclosure: The authors declare no conflicting financial interests.
References
- 1.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 2.Ito T, Wang YH, Liu YJ. Plasmacytoid dendritic cell precursors/type I interferon-producing cells sense viral infection by Toll-like receptor (TLR) 7 and TLR9. Springer Semin Immunopathol. 2005;26:221–229. doi: 10.1007/s00281-004-0180-4. [DOI] [PubMed] [Google Scholar]
- 3.Gibson SJ, Lindh JM, Riter TR, et al. Plasmacytoid dendritic cells produce cytokines and mature in response to the TLR7 agonists, imiquimod and resiquimod. Cell Immunol. 2002;218:74–86. doi: 10.1016/s0008-8749(02)00517-8. [DOI] [PubMed] [Google Scholar]
- 4.Asselin-Paturel C, Trinchieri G. Production of type I interferons: plasmacytoid dendritic cells and beyond. J Exp Med. 2005;202:461–465. doi: 10.1084/jem.20051395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fonteneau JF, Gilliet M, Larsson M, et al. Activation of influenza virus-specific CD4+ and CD8+ T cells: a new role for plasmacytoid dendritic cells in adaptive immunity. Blood. 2003;101:3520–3526. doi: 10.1182/blood-2002-10-3063. [DOI] [PubMed] [Google Scholar]
- 6.Chaperot L, Blum A, Manches O, et al. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. J Immunol. 2006;176:248–255. doi: 10.4049/jimmunol.176.1.248. [DOI] [PubMed] [Google Scholar]
- 7.Stary G, Bangert C, Tauber M, Strohal R, Kopp T, Stingl G. Tumoricidal activity of TLR7/8-activated inflammatory dendritic cells. J Exp Med. 2007;204:1441–1451. doi: 10.1084/jem.20070021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garland SM, Waddell R, Mindel A, Denham IM, McCloskey JC. An open-label phase II pilot study investigating the optimal duration of imiquimod 5% cream for the treatment of external genital warts in women. Int J STD AIDS. 2006;17:448–452. doi: 10.1258/095646206777689161. [DOI] [PubMed] [Google Scholar]
- 9.Gollnick H, Barona CG, Frank RG, et al. Recurrence rate of superficial basal cell carcinoma following treatment with imiquimod 5% cream: conclusion of a 5-year long-term follow-up study in Europe. Eur J Dermatol. 2008;18:677–682. doi: 10.1684/ejd.2008.0519. [DOI] [PubMed] [Google Scholar]
- 10.Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8:594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 11.Birmachu W, Gleason RM, Bulbulian BJ, et al. Transcriptional networks in plasmacytoid dendritic cells stimulated with synthetic TLR 7 agonists. BMC Immunol. 2007;8:26. doi: 10.1186/1471-2172-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gautier G, Humbert M, Deauvieau F, et al. A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med. 2005;201:1435–1446. doi: 10.1084/jem.20041964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaperot L, Bendriss N, Manches O, et al. Identification of a leukemic counterpart of the plasmacytoid dendritic cells. Blood. 2001;97:3210–3217. doi: 10.1182/blood.v97.10.3210. [DOI] [PubMed] [Google Scholar]
- 14.Chaperot L, Perrot I, Jacob MC, et al. Leukemic plasmacytoid dendritic cells share phenotypic and functional features with their normal counterparts. Eur J Immunol. 2004;34:418–426. doi: 10.1002/eji.200324531. [DOI] [PubMed] [Google Scholar]
- 15.Jurk M, Heil F, Vollmer J, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol. 2002;3:499. doi: 10.1038/ni0602-499. [DOI] [PubMed] [Google Scholar]
- 16.Lee J, Chuang TH, Redecke V, et al. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor 7. Proc Natl Acad Sci U S A. 2003;100:6646–6651. doi: 10.1073/pnas.0631696100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 18.Gurney KB, Colantonio AD, Blom B, Spits H, Uittenbogaart CH. Endogenous IFN-alpha production by plasmacytoid dendritic cells exerts an antiviral effect on thymic HIV-1 infection. J Immunol. 2004;173:7269–7276. doi: 10.4049/jimmunol.173.12.7269. [DOI] [PubMed] [Google Scholar]
- 19.Megjugorac NJ, Young HA, Amrute SB, Olshalsky SL, Fitzgerald-Bocarsly P. Virally stimulated plasmacytoid dendritic cells produce chemokines and induce migration of T and NK cells. J Leukoc Biol. 2004;75:504–514. doi: 10.1189/jlb.0603291. [DOI] [PubMed] [Google Scholar]
- 20.Kemp TJ, Elzey BD, Griffith TS. Plasmacytoid dendritic cell-derived IFN-alpha induces TNF-related apoptosis-inducing ligand/Apo-2L-mediated antitumor activity by human monocytes following CpG oligodeoxynucleotide stimulation. J Immunol. 2003;171:212–218. doi: 10.4049/jimmunol.171.1.212. [DOI] [PubMed] [Google Scholar]
- 21.Pierce JW, Schoenleber R, Jesmok G, et al. Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- 22.Burke JR, Pattoli MA, Gregor KR, et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J Biol Chem. 2003;278:1450–1456. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]
- 23.Stark GR. How cells respond to interferons revisited: from early history to current complexity. Cytokine Growth Factor Rev. 2007;18:419–423. doi: 10.1016/j.cytogfr.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takauji R, Iho S, Takatsuka H, et al. CpG-DNA-induced IFN-alpha production involves p38 MAPK-dependent STAT1 phosphorylation in human plasmacytoid dendritic cell precursors. J Leukoc Biol. 2002;72:1011–1019. [PubMed] [Google Scholar]
- 25.Guiducci C, Ghirelli C, Marloie-Provost MA, et al. PI3K is critical for the nuclear translocation of IRF-7 and type I IFN production by human plasmacytoid predendritic cells in response to TLR activation. J Exp Med. 2008;205:315–322. doi: 10.1084/jem.20070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inglefield JR, Dumitru CD, Alkan SS, et al. TLR7 agonist 852A inhibition of tumor cell proliferation is dependent on plasmacytoid dendritic cells and type I IFN. J Interferon Cytokine Res. 2008;28:253–263. doi: 10.1089/jir.2007.0097. [DOI] [PubMed] [Google Scholar]
- 28.Smits EL, Ponsaerts P, Berneman ZN, Van Tendeloo VF. The use of TLR7 and TLR8 ligands for the enhancement of cancer immunotherapy. Oncologist. 2008;13:859–875. doi: 10.1634/theoncologist.2008-0097. [DOI] [PubMed] [Google Scholar]
- 29.Kerkmann M, Rothenfusser S, Hornung V, et al. Activation with CpG-A and CpG-B oligonucleotides reveals two distinct regulatory pathways of type I IFN synthesis in human plasmacytoid dendritic cells. J Immunol. 2003;170:4465–4474. doi: 10.4049/jimmunol.170.9.4465. [DOI] [PubMed] [Google Scholar]
- 30.Honda K, Ohba Y, Yanai H, et al. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- 31.Guiducci C, Ott G, Chan JH, et al. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J Exp Med. 2006;203:1999–2008. doi: 10.1084/jem.20060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haas T, Schmitz F, Heit A, Wagner H. Sequence independent interferon-alpha induction by multimerized phosphodiester DNA depends on spatial regulation of Toll-like receptor-9 activation in plasmacytoid dendritic cells. Immunology. 2009;126:290–298. doi: 10.1111/j.1365-2567.2008.02897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heil F, Hemmi H, Hochrein H, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 34.Chawla-Sarkar M, Lindner DJ, Liu YF, et al. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8:237–249. doi: 10.1023/a:1023668705040. [DOI] [PubMed] [Google Scholar]
- 35.van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 36.Kovarik P, Stoiber D, Eyers PA, et al. Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated protein kinase whereas IFN-gamma uses a different signaling pathway. Proc Natl Acad Sci U S A. 1999;96:13956–13961. doi: 10.1073/pnas.96.24.13956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang YJ, Holtzman MJ, Chen CC. Differential role of Janus family kinases (JAKs) in interferon-gamma-induced lung epithelial ICAM-1 expression: involving protein interactions between JAKs, phospholipase Cgamma, c-Src, and STAT1. Mol Pharmacol. 2004;65:589–598. doi: 10.1124/mol.65.3.589. [DOI] [PubMed] [Google Scholar]
- 38.Baker SJ, Rane SG, Reddy EP. Hematopoietic cytokine receptor signaling. Oncogene. 2007;26:6724–6737. doi: 10.1038/sj.onc.1210757. [DOI] [PubMed] [Google Scholar]
- 39.Cao W, Manicassamy S, Tang H, et al. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat Immunol. 2008;9:1157–1164. doi: 10.1038/ni.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.