

Abstract
Background:
Several missense mutations of CACNA1S and SCN4A genes occur in hypokalemic periodic paralysis. These mutations affect arginine residues in the S4 voltage sensors of the channel. Approximately 20% of cases remain genetically undefined.
Methods:
We undertook direct automated DNA sequencing of the S4 regions of CACNA1S and SCN4A in 83 cases of hypokalemic periodic paralysis.
Results:
We identified reported CACNA1S mutations in 64 cases. In the remaining 19 cases, mutations in SCN4A or other CACNA1S S4 segments were found in 10, including three novel changes and the first mutations in channel domains I (SCN4A) and III (CACNA1S).
Conclusions:
All mutations affected arginine residues, consistent with the gating pore cation leak hypothesis of hypokalemic periodic paralysis. Arginine mutations in S4 segments underlie 90% of hypokalemic periodic paralysis cases.
GLOSSARY
- HypoPP
= hypokalemic periodic paralysis.
Hypokalemic periodic paralysis (HypoPP) is an important autosomal dominant muscle channelopathy with onset in the first or second decade. It is characterized by episodes of flaccid paralysis in association with low serum potassium. Typically, attacks occur during the night or in the early morning and can last from several hours to days. Point mutations in CACNA1S or SCN4A, which encode the skeletal muscle voltage-gated calcium and sodium channels, associate with HypoPP.1–10 However, in most studies at least 20% of cases remain genetically undefined.8,11,12
Sodium and calcium channels have homologous pore-forming α subunits, each containing four domains containing six transmembrane segments. Two common mutations in CACNA1S and several additional mutations in SCN4A have been reported in HypoPP. All of these mutations affect arginine residues in S4 segments that contribute to voltage sensing. Functional characterization of known mutations suggests HypoPP is associated with loss of normal channel function. However, evidence also exists that at least some of these mutations allow an abnormal cation flux to pass through the aqueous omega pore that lines the voltage sensor.13,14 The full spectrum of HypoPP mutations has not been defined. In particular, it remains to be determined whether the gating pore cation leak hypothesis accounts for all cases. We therefore undertook an extensive analysis of all voltage sensors in SCN4A and CACNA1S in 83 clinically definite HypoPP cases.
METHODS
We examined 83 cases referred to our national patient referral center for skeletal muscle channelopathies with a firm clinical diagnosis of HypoPP. All patients gave written informed consent for the DNA analysis in this study, which had ethics committee approval from the National Hospital for Neurology and Neurosurgery & Institute of Neurology Joint Research Ethics Committee. We first undertook direct automated sequencing of CACNA1S for the common mutations described at amino acid positions R528 and R1239. For cases who were negative for these mutations we performed direct automated DNA sequencing of all regions of CACNA1S and SCN4A coding for the remaining S4 segments (exons 4 and 21 of CACNA1S and exons 5, 12, 13, 18, and 24 of SCN4A).
PCR reactions to amplify each exon of CACNA1S using the following reagents and conditions were performed: a 25 μL reaction contained 200 ng genomic DNA, 5 μL of 10 × PCR buffer without MgCl2 (Applied Biosystems), 4 μL of 25 mM MgCl2, 5 μL of 2 mM dNTPs, 15 pmol of each primer (forward and reverse), and 2.5 units of AmpliTaq Gold polymerase (Applied Biosystems). PCR reactions to amplify each exon of SCN4A using the following reagents and conditions were additionally performed: a 50 μL reaction contained 200 ng genomic DNA, 10 μL of 5 x PCR buffer (Promega), 5 μL of 2 mM dNTPs, 10 pmol of each primer (forward and reverse), and 1.0 unit of Go-Taq polymerase (Promega).
Cycling conditions consisted of an initial denaturing step of 95°C for 10 minutes followed by 30 cycles of 95°C for 30 seconds, 60°C for 30 seconds, 72°C for 30 seconds, and a final extension step of 72°C for 7 minutes. Samples were sequenced (bidirectionally) using the ABI Big Dye Terminator Sequencing Kit version 1.1, M13 universal primers, and an ABI Model 3730xl Automated DNA Sequencer. Data were analyzed using version 2.5 SeqScape Analysis software (ABI). See supplemental data on the Neurology® Web site at www.neurology.org for oligonucleotide primer sequences.
RESULTS
In 64 out of 83 cases of HypoPP, we identified previously reported mutations: either R528G/H (25 cases) or R1239G/H (39 cases) in CACNA1S. Of the remaining 19 cases, 10 harbored additional mutations in other S4 segments of CACNA1S or SCN4A. Six of these were positive for mutations that have been reported previously, namely the substitutions R672C/H/S and R1132Q in SCN4A. In the other four cases, we found three novel mutations, all of which neutralized arginine residues in S4 segments. These mutations were absent from 240 control chromosomes. One mutation was in the S4 segment of domain III (DIII/S4) of CACNA1S: c.2700G>T; p.R900S (figure 1). The other two mutations neutralized arginine residues in S4 segments of SCN4A; the first in DI c.664C>T; p.R222W and the second in DIII c.3404G>A; p.R1135H (see figure 2 and the table for details and figure e-1 for electropherograms). R222W was found in two apparently unrelated kindreds.

Figure 1 Diagrammatic representation of the alpha subunit of CACNA1S with S4 segments and arginine residues known to be mutated in hypokalemic periodic paralysis highlighted
The novel arginine substitution reported here is boxed in gray. + indicates the number of positively charged residues in each S4 segment.

Figure 2 Diagrammatic representation of the alpha subunit of SCN4A with S4 segments and arginine residues known to be mutated highlighted
Novel arginine mutations reported here are boxed in gray. + indicates the number of positively charged residues in each S4 segment. *Note substitutions of R1448 cause a phenotype of paramyotonia congenita.
Table Details of all reported S4 segment mutations in CACNA1S and SCN4A
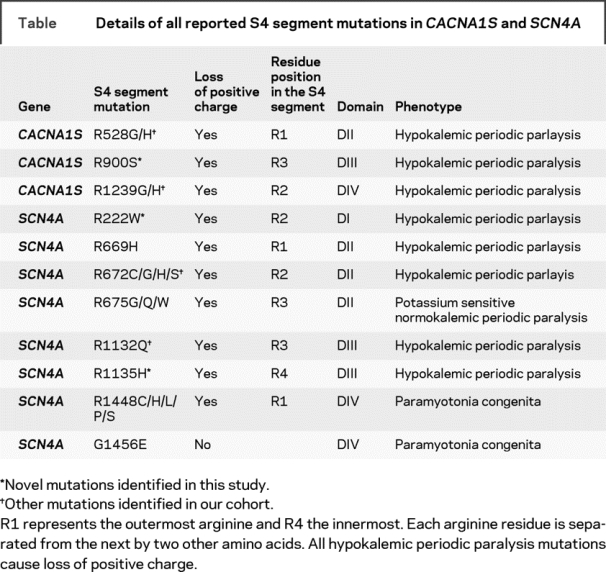
All four individuals with the new mutations had typical HypoPP phenotype. Age at onset of attacks of muscle paralysis was in the second decade, with attacks usually occurring at night or in the early morning, and associated with low serum potassium levels or with provocative factors that would induce low serum potassium. The frequency and severity of attacks were not different from those in individuals with previously reported mutations. To our knowledge, these individuals received no pharmacologic therapy or potassium supplementation only.
DISCUSSION
Our results expand the spectrum of S4 segment arginine mutations which cause HypoPP. These data add new genetic evidence to support the hypothesis that loss of positive charge in S4 voltage sensors is important in the molecular pathogenesis of this muscle channelopathy. We identified new point mutations in domain III of CACNA1S and in domains I and III of SCN4A. There are no previous reports of mutations in domain III of CACNA1S or in domain I of SCN4A in HypoPP. The present findings indicate that loss of positively charged residues in S4 segments in these domains can also cause HypoPP. Overall, the 74 genetically characterized HypoPP cases reported here argue that arginine mutations in the voltage sensors of both channels are the overwhelmingly most important cause of HypoPP. The present results, furthermore, suggest that screening for arginine mutations in S4 segments should yield a genetic diagnosis in approximately 90% of cases of HypoPP. We propose that analysis for S4 substitutions in domains II and IV of CACNA1S should be followed by analysis of the S4 regions in domains I, II, and III of SCN4A and in domain III of CACNA1S.
It remains unclear how S4 mutations lead to the HypoPP phenotype. Previous functional studies of the effect of the CACNA1S/SCN4A S4 mutations on the gating properties of the main pore all pointed to a loss of function defect. However, such a loss of function mechanism does not readily explain the prolonged depolarization of the sarcolemmal membrane associated with attacks of paralysis15 or the episodic occurrence of hypokalemia. The individual contribution of each S4 segment to channel gating is not yet fully understood. Current evidence suggests that the S4 segments of DIII and IV of Nav1.4 play a more significant role in fast inactivation.16 It is therefore of interest to note that replacement of the outermost arginine in DIV/S4 of SCN4A does not produce a HypoPP phenotype but rather one of paramyotonia congenita.17 In contrast, replacement of the third outermost arginine in DII/S4 of SCN4A produces a potassium-sensitive periodic paralysis.18 Only one non-arginine substitution has been described in an S4 segment of either channel, G1456E19 [SCN4A] also in domain IV, which resulted in a phenotype of paramyotonia congenita (table). Our genetic evidence indicates that loss of positive charge in the voltage sensor in domains I, II, and III of SCN4A and in domains II, III, and IV in CACNA1S is important in the pathogenesis of HypoPP.
Two recent studies have suggested that a gating pore current may be important in the pathogenesis of HypoPP,13,14 and our data are fully consistent with this hypothesis. In response to depolarizing voltages, S4 segments undergo a conformational change that moves these segments outwards through a gating pore (omega pore) which leads to the opening of the central pore of the channel (alpha pore). The outermost arginines in S4 occupy and occlude the narrowest part of the gating pore at the resting membrane potential while internal arginine residues occlude it at depolarized potentials. Recently, it was shown that in addition to disrupting the gating of the main pore (alpha pore), mutations that neutralize the two outermost arginines (R669/R672) in DII/S4 of Nav1.4 also generate a monovalent cation leak through the gating pore at hyperpolarized potentials.13 A histidine substitution at R672 produces a proton specific pore leak whereas other amino acid substitutions at this position cause a nonselective cation leak. The leak current is thought to be mainly mediated by protons and could contribute to the pathophysiology of HypoPP, possibly by an accumulation of intracellular protons and disruption of the intracellular homeostasis of pH.14 Consistent with the role of arginines in occluding the gating pore, a 10-fold larger leak current occurs with a glycine substitution compared with the histidine substitution at R663 in the rat isoform of NaV1.4 (comparable to R669 in the human isoform).14 Our finding of additional mutations that affect S4 arginines adds to the possibility that these arginines play a central role in HypoPP. Furthermore, our findings expand the number of channel domains and the range of arginines affected, notably including arginines buried more deeply in the channel. Interestingly, in the Shaker potassium channel, replacing more intracellular arginine residues with histidine residues has been shown to produce proton leak currents at depolarized potentials in contrast to substitutions of outer arginines that generate leak currents at hyperpolarized potentials.20 If the intracellular arginines that we have found lead to similar changes in SCN4A, then patients with mutations affecting these residues may have leak currents at depolarized potentials rather than at rest. Functional characterization of our new mutations will be important for determining the pathophysiologic effect of these more intracellular arginine residues.
ACKNOWLEDGMENT
The authors thank all clinical colleagues who have referred patients with muscle channel disorders to the UK national channelopathy service supported by the Department of Health (NCG) and the Neurogenetic Unit at the National Hospital for Neurology and Neurosurgery.
Supplementary Material
Address correspondence and reprint requests to Prof. M.G. Hanna, Medical Research Council Centre for Neuromuscular Diseases, Department of Molecular Neuroscience, Institute of Neurology and National Hospital for Neurology and Neurosurgery, Queen Square, London, WC1N 3BG, UK m.hanna@ion.ucl.ac.uk
Supplemental data at www.neurology.org
Editorial, page 1540
e-Pub ahead of print on December 31, 2008, at www.neurology.org.
Supported by the Medical Research Council UK through an MRC Centre grant (G0601943) and by the National Center for Research Resources (Grant No. 5U54 RR019498-05) through the NIH-Consortium for Clinical Investigation of Neurologic Channelopathies and through the Brain Research Trust. This work was undertaken at University College London Hospitals/University College London, which received a proportion of funding from the Department of Health’s National Institute for Health Research Biomedical Research Centres funding scheme. P.F.C. is a Wellcome Trust Senior Fellow in Clinical Science.
Disclosure: The authors report no disclosures.
Received July 23, 2008. Accepted in final form November 12, 2008.
REFERENCES
- 1.Fontaine B, Vale-Santos J, Jurkat-Rott K, et al. Mapping of the hypokalaemic periodic paralysis (HypoPP) locus to chromosome 1q31-32 in three European families. Nat Genet 1994;6:267–272. [DOI] [PubMed] [Google Scholar]
- 2.Ptacek LJ, Tawil R, Griggs RC, et al. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell 1994;77:863–868. [DOI] [PubMed] [Google Scholar]
- 3.Jurkat-Rott K, Lehmann-Horn F, Elbaz A, et al. A calcium channel mutation causing hypokalemic periodic paralysis. Hum Mol Genet 1994;3:1415–1419. [DOI] [PubMed] [Google Scholar]
- 4.Wang Q, Liu M, Xu C, et al. Novel CACNA1S mutation causes autosomal dominant hypokalemic periodic paralysis in a Chinese family. J Mol Med 2005;83:203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bulman DE, Scoggan KA, van O, et al. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology 1999;53:1932–1936. [DOI] [PubMed] [Google Scholar]
- 6.Jurkat-Rott K, Mitrovic N, Hang C, et al. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci USA 2000;97:9549–9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bendahhou S, Cummins TR, Griggs RC, Fu YH, Ptacek LJ. Sodium channel inactivation defects are associated with acetazolamide-exacerbated hypokalemic periodic paralysis. Ann Neurol 2001;50:417–420. [DOI] [PubMed] [Google Scholar]
- 8.Sternberg D, Maisonobe T, Jurkat-Rott K, et al. Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain 2001;124(Pt 6):1091–1099. [DOI] [PubMed] [Google Scholar]
- 9.Davies NP, Eunson LH, Samuel M, Hanna MG. Sodium channel gene mutations in hypokalemic periodic paralysis: an uncommon cause in the UK. Neurology 2001;57:1323–1325. [DOI] [PubMed] [Google Scholar]
- 10.Kim MK, Lee SH, Park MS, et al. Mutation screening in Korean hypokalemic periodic paralysis patients: a novel SCN4A Arg672Cys mutation. Neuromuscul Disord 2004;14:727–731. [DOI] [PubMed] [Google Scholar]
- 11.Fouad G, Dalakas M, Servidei S, et al. Genotype-phenotype correlations of DHP receptor alpha 1-subunit gene mutations causing hypokalemic periodic paralysis. Neuromuscul Disord 1997;7:33–38. [DOI] [PubMed] [Google Scholar]
- 12.Miller TM, Dias da Silva MR, Miller HA, et al. Correlating phenotype and genotype in the periodic paralyses. Neurology 2004;63:1647–1655. [DOI] [PubMed] [Google Scholar]
- 13.Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature 2007;446:76–78. [DOI] [PubMed] [Google Scholar]
- 14.Struyk AF, Cannon SC. A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J Gen Physiol 2007;130:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cannon SC. Pathomechanisms in channelopathies of skeletal muscle and brain. Annu Rev Neurosci 2006;29:387–415. [DOI] [PubMed] [Google Scholar]
- 16.Cha A, Ruben PC, George AL, Jr., Fujimoto E, Bezanilla F. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron 1999;22:73–87. [DOI] [PubMed] [Google Scholar]
- 17.Ptacek LJ, George AL, Jr., Barchi RL, et al. Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenita. Neuron 1992;8:891–897. [DOI] [PubMed] [Google Scholar]
- 18.Vicart S, Sternberg D, Fournier E, et al. New mutations of SCN4A cause a potassium-sensitive normokalemic periodic paralysis. Neurology 2004;63:2120–2127. [DOI] [PubMed] [Google Scholar]
- 19.Sasaki R, Takano H, Kamakura K, et al. A novel mutation in the gene for the adult skeletal muscle sodium channel alpha-subunit (SCN4A) that causes paramyotonia congenita of von Eulenburg. Arch Neurol 1999;56:692–696. [DOI] [PubMed] [Google Scholar]
- 20.Starace DM, Bezanilla F. Histidine scanning mutagenesis of basic residues of the S4 segment of the shaker k+ channel. J Gen Physiol 2001;117:469–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.