

Abstract
Background/Objective:
Anti-inflammatory drugs may prevent Parkinson disease (PD) by inhibiting a putative underlying neuroinflammatory process. We tested the hypothesis that anti-inflammatory drugs reduce PD incidence and that there are differential effects by type of anti-inflammatory, duration of use, or intensity of use.
Methods:
MEDLINE and EMBASE were searched for studies that reported risk of PD associated with anti-inflammatory medications. Random-effects meta-analyses were used to pool results across studies for each type of anti-inflammatory drug. Stratified meta-analyses were used to assess duration- and intensity-response.
Results:
Seven studies were identified that met the inclusion criteria, all of which reported associations between nonaspirin nonsteroidal anti-inflammatory drugs (NSAIDs) and PD, 6 of which reported on aspirin, and 2 of which reported on acetaminophen. Overall, a 15% reduction in PD incidence was observed among users of nonaspirin NSAIDS (relative risk [RR] 0.85, 95% confidence interval [CI] 0.77–0.94), with a similar effect observed for ibuprofen use. The protective effect of nonaspirin NSAIDs was more pronounced among regular users (RR 0.71, 95% CI 0.58–0.89) and long-term users (RR 0.79, 95% CI 0.59–1.07). No protective effect was observed for aspirin (RR 1.08, 95% CI 0.92–1.27) or acetaminophen (RR 1.06, 95% CI 0.87–1.30). Sensitivity analyses found results to be robust.
Conclusions:
There may be a protective effect of nonaspirin nonsteroidal anti-inflammatory drug use on risk of Parkinson disease (PD) consistent with a possible neuroinflammatory pathway in PD pathogenesis.
GLOSSARY
- CI
= confidence interval;
- COX
= cyclooxygenase;
- NOS
= Newcastle-Ottawa Scale;
- NSAID
= nonsteroidal anti-inflammatory drug;
- OTC
= over-the-counter;
- PD
= Parkinson disease;
- RR
= relative risk.
Parkinson disease (PD) is a common neurodegenerative movement disorder associated with substantial morbidity and mortality,1 and the number of persons affected is expected to increase dramatically in coming years.2 Although its pathogenesis is not fully understood, neuroinflammatory mechanisms may be involved.3,4 If so, anti-inflammatory medications, including nonsteroidal anti-inflammatory drugs (NSAIDs), may confer protection against PD. While studies of animal models of PD support this hypothesis,5 the epidemiologic literature regarding the effect of anti-inflammatory drug use on PD is mixed. Several studies suggest a protective effect of anti-inflammatory medications on PD in humans,6,7 whereas others do not corroborate these findings.8,9
The purpose of this study was to quantify the effect of anti-inflammatory drug use on PD incidence by meta-analyzing existing studies and to examine the effect by type of anti-inflammatory (e.g., nonaspirin NSAIDs, aspirin, acetaminophen), duration of use, and intensity of use.
METHODS
Search strategy.
Our systematic review was conducted according to the Meta-analysis of Observational Studies in Epidemiology guidelines.10 Each author independently conducted a systematic search of MEDLINE and EMBASE from their commencement to April 2009. Separate search filters were constructed for each database and are provided in appendix e-1 on the Neurology® Web site at www.neurology.org. Initial searches were restricted to English-language publications and studies conducted in humans. A secondary search with no language restriction did not identify any additional relevant articles. Reference lists of articles identified for inclusion in the meta-analysis were examined to identify additional potentially relevant studies.
Eligibility criteria.
Abstracts of identified articles were screened to exclude studies that clearly did not meet the eligibility criteria. The full text of those selected for further review was obtained and evaluated. Studies were included if they met the following criteria: 1) presented original data from an epidemiologic study; 2) defined the outcome of interest as incident PD, based on clearly stated diagnostic criteria or identified through diagnostic codes with additional confirmation; 3) defined exposure as use of nonaspirin NSAIDs, aspirin, or acetaminophen; 4) ascertained exposure status during a period including time at least 1 year or more before PD diagnosis11; 5) described adjustment for potential confounding; 6) reported effect estimates with confidence intervals (CIs), standard errors, or sufficient information to calculate these; and 7) met at least 5 Newcastle-Ottawa Scale (NOS) criteria,12 a standard set in other studies.13,14 The NOS is an 8-item instrument, with up to 9 possible points, and is intended to assess the quality of observational studies to be included in systematic reviews and meta-analyses. It has been used in several recently published meta-analyses of observational studies.13–16
Data extraction.
Both authors independently extracted data from each study. Discrepancies were discussed and resolved by agreement. The following data were extracted from each study: study name, year of publication, setting, study design, number of participants, mean age, outcome definition, exposure definitions, methods for confounding adjustment and variables adjusted for, effect estimates and CIs or standard errors (or information required to compute these), and information required to complete the NOS questionnaire.
When multiple effect estimates were reported, maximally adjusted estimates were extracted. When results were presented with and without lag periods, with multiple lag periods, or with multiple periods of exposure ascertainment, the estimates based on the longest time between exposure and disease onset were used, given the insidious nature of PD.11
Data analysis.
For all analyses, we used random-effects models.17 Primary analyses compared exposed vs unexposed for each of the 3 anti-inflammatory exposures of interest: nonaspirin NSAIDs, aspirin, and acetaminophen. For studies that reported only stratified results (e.g., by sex), fixed-effect methods with Mantel-Haenszel weighting were used to summarize the stratified estimates into a single parameter for each study.
When possible, stratified analyses were conducted to examine differences by sex, duration of use, and intensity of use. To investigate differential effects by duration of use, results were stratified on short- vs long-term use. Short-term use was defined as the first duration stratum for each study that reported results stratified by exposure duration over several years. Long-term use was defined as exposure duration beyond the first stratum. When studies reported more than 1 stratum that qualified for long-term use, we combined the estimates with fixed-effects methods. To investigate differential effects by intensity of use, results were stratified by whether exposure definitions required demonstration of regular use.
Furthermore, we hypothesized that, in the case of a true effect of anti-inflammatory agents on PD, reliance on prescription drug data alone would lead to nondifferential misclassification of exposure status due to widespread use of over-the-counter (OTC) anti-inflammatory drugs. If present, such misclassification would, in expectation, result in a biased estimate of association that would be closer to unity than an estimate that considered both prescription and OTC drug use to define exposure. To assess the potential for and impact of this exposure misclassification, we conducted analyses stratified by whether exposure status was defined on the basis of prescription data alone or whether it also captured possible OTC use.
Sensitivity analyses were conducted to assess the robustness of the primary analyses for each exposure type. Studies excluded from the analysis because they did not meet our eligibility criteria were included in sensitivity analyses. We also conducted sensitivity analyses by repeating the original analyses while separately omitting 1 study at a time to assess whether single studies had undue influence on the results.
Measures of association were combined under the assumption that odds ratios were accurate approximations of relative risks (RRs). Presence of heterogeneity in effects was assessed using the Cochrane Q test and quantified using the I2 test.18 Publication bias was assessed via visual inspection of the Begg funnel plot.
RESULTS
Literature search.
The MEDLINE search yielded 16 potentially relevant articles and the EMBASE search yielded 49 articles, for a total of 55 unique citations (figure 1). Forty-three of these articles were excluded based on the abstract review because they were clearly irrelevant to the objectives of this meta-analysis. Thus, a total of 12 articles were obtained for full-text review. One of the 12 articles did not report on the association of interest and was excluded.19 Two studies were excluded from primary analyses because exposures were ascertained exclusively within 1 year of PD diagnosis,20,21 and 2 studies were excluded because exposure was defined as any anti-inflammatory drug without reporting of separate results for each drug type.22,23 Thus, 7 studies met our inclusion criteria and were included in the primary analyses. The 4 studies that were excluded on the basis of exposure definitions were included in sensitivity analyses.

Figure 1 Flow diagram of study selection
Study characteristics.
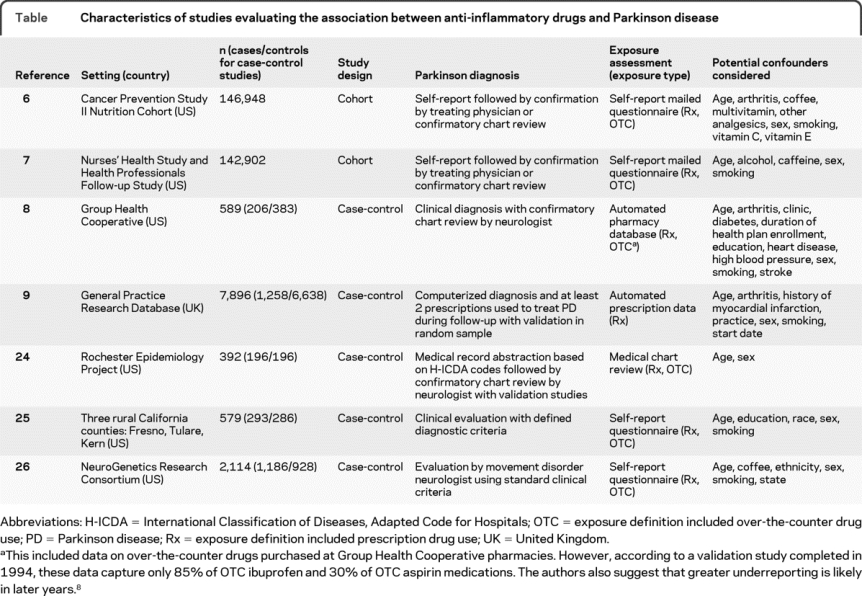
Five of the 7 studies included in the analysis were case-control studies,8,9,24–26 and 2 were cohort studies (table).6,7 All 7 of the studies met at least 5 NOS criteria. All 7 studies reported on the association between nonaspirin NSAIDs and PD, with 3 additionally reporting specifically on the association between ibuprofen and PD,6,8,9 6 reporting on the association between aspirin and PD,6–9,24,25 and 2 reporting on the association between acetaminophen and PD.6,9
Table Characteristics of studies evaluating the association between anti-inflammatory drugs and Parkinson disease
Nonaspirin NSAID use and PD risk.
The pooled estimate for the 7 studies that reported on the association between nonaspirin NSAIDs and PD was 0.85 (95% CI 0.77–0.94; figure 2). No heterogeneity was observed in these results (I2 = 0%, 95% CI 0%–71%). Analyses stratified by duration of use (short-term vs long-term) and intensity of use (nonregular use vs regular use) yielded results consistent with a dose-response relation (figure 3, A and B). Analyses stratified by type of exposure (prescription only vs both prescription and OTC) yielded results consistent with expected greater nondifferential exposure misclassification among studies considering only prescription nonaspirin NSAID exposure (figure 3C).

Figure 2 Relative risks (95% confidence intervals [CI]) from studies of nonaspirin nonsteroidal anti-inflammatory drug use and effect of Parkinson disease

Figure 3 Risk of Parkinson disease associated with nonaspirin nonsteroidal anti-inflammatory drug use stratified by duration, intensity, and type of use
*Rx only and Rx and OTC denote whether exposure definition included prescription (Rx) nonsteroidal anti-inflammatory drug (NSAID) use or both prescription and over-the-counter (OTC) NSAID use. CI = confidence interval.
Adding the 2 studies that were excluded from the analysis because exposure status was ascertained within a year of PD diagnosis20,21 slightly increased the primary estimate of effect (RR 0.87, 95% CI 0.77–0.97) and introduced some heterogeneity (I2 = 25%, 95% CI 0%–65%). In a separate sensitivity analysis, including the 2 studies that defined NSAID exposure as combination of aspirin or nonaspirin NSAIDs22,23 also slightly increased the effect estimate (RR 0.87, 95% CI 0.75–1.00) and introduced some heterogeneity (I2 = 35%, 95% CI 0%–70%). A sensitivity analysis including all 4 excluded studies yielded a higher effect estimate (RR 0.90, 95% CI 0.79–1.01) and involved greater heterogeneity (I2 = 39%, 95% CI 0%–70%). Sensitivity analyses omitting 1 study at a time from the original analysis found the main results to be robust.
Five studies reported results for nonaspirin NSAID use by sex.6,7,9,25,26 Sex-stratified meta-analyses yielded similar results among men (RR 0.80, 95% CI 0.70–0.92) and women (RR 0.75, 95% CI 0.52–1.08). Three studies reported results specifically for ibuprofen.6,8,9 The summary estimate for ibuprofen only was slightly stronger (RR 0.75, 95% CI 0.64–0.89) than the estimate for all nonaspirin NSAIDs. Given that data were available for only 3 studies, stratified and sensitivity analyses for ibuprofen use were not possible.
Aspirin use and PD risk.
One of the 6 studies that examined the risk of PD in relation to aspirin use reported only stratified results,6 which we combined into a single summary estimate. Pooling this estimate with the other 5 studies7–9,24,25 yielded an effect estimate of 1.08 (95% CI 0.92–1.27; figure 4). Some heterogeneity was observed in these results (I2 = 50%, 95% CI 0%–80%). Duration-response analyses produced similar results for both short-term use (RR 1.13, 95% CI 0.66–1.94) and long-term use (RR 0.93, 95% CI 0.58–1.50) but were limited by the number of studies (n = 2)9,25 that permitted such stratification. Intensity-response analyses also yielded similar results for nonregular use (RR 1.00, 95% CI 0.59–1.73) and regular use (RR 1.06, 95% CI 0.89–1.27). Analyses stratified by sex found an increased risk of PD associated with aspirin use among men (RR 1.22, 95% CI 1.03–1.44) but not women (RR 0.98, 95% CI 0.71–1.37). No substantial heterogeneity was observed in sex-stratified analyses.

Figure 4 Relative risks (95% confidence intervals [CI]) from studies of aspirin use and effect on Parkinson disease
Adding the results of the 2 studies that defined NSAID exposure as combination of aspirin or nonaspirin NSAIDs22,23 did not substantially change the effect estimate (RR 1.10, 95% CI 0.96–1.27) or the amount of heterogeneity (I2 = 43%, 95% CI 0%–75%). Sensitivity analyses omitting 1 study at a time from the original analysis found the primary results to be robust.
Acetaminophen use and PD risk.
One study suggested a small potential protective effect of acetaminophen on PD,6 whereas another reported a small adverse effect.9 Combining these studies yielded an effect estimate of 1.06 (95% CI 0.87–1.30). Stratified, dose-response, and sensitivity analyses were not possible given the availability of data from only 2 studies.
Publication bias.
Inspection of the Begg funnel plot did not suggest the presence of publication bias because the log RRs plotted against their standard errors yielded a rather symmetric distribution for studies that reported on nonaspirin NSAID use (figure e-1) and aspirin use (figure e-2). However, because these assessments are based on few studies, inference about publication bias should be made with caution. Assessment for publication bias for acetaminophen use or ibuprofen use was not meaningful because of the small number of studies that reported on these associations.
DISCUSSION
This analysis, which combined the results of several large epidemiologic studies, suggests that use of nonaspirin NSAIDs is associated with a 15% reduction in risk of PD. We also observed a greater reduction in risk with regular use (29% reduction) and long-term use (21% reduction), consistent with a dose-response relation. By contrast, no protective effect was observed with use of aspirin or acetaminophen.
The findings support a growing body of research that suggests the involvement of a neuroinflammatory process in PD pathogenesis. Postmortem and in vivo studies provide evidence of a neuroinflammatory response in patients with PD but do not differentiate whether such changes are a contributing cause or a result of the disorder.3,4,27 Animal models of PD suggest that neuroinflammation is part of the pathologic process and precedes dopaminergic cell death.3,4 Genetic studies show increased PD risk associated with polymorphisms in inflammatory genes, including tumor necrosis factor α and interleukin 1β, and high plasma concentrations of interleukin 6 have also been associated with increased PD risk.28,29
Furthermore, NSAIDs have been shown to exhibit neuroprotective effects in animal models of PD.30 However, the physiologic pathways on which these drugs act are many and varied,31 and the exact mechanism by which they may act to prevent or minimize neurodegeneration has not been definitively determined.3,30 Some theories postulate that inhibition of cyclooxygenase (COX)-2, COX-independent antioxidant effects, or modulation of the inflammatory response through impact on gene expression or cytokine production account for this neuroprotective effect.3,30 Moreover, no association was observed between acetaminophen use and PD, further supporting the involvement of an underlying neuroinflammatory process in PD because acetaminophen lacks overall anti-inflammatory ability.32
The findings of this meta-analysis support the idea that nonaspirin NSAIDs confer protection against PD, because bias is unlikely to account for these findings. Reverse causation is an unlikely explanation, because we required all studies to include ascertainment of exposure at least 1 year before PD diagnosis and those studies incorporating longer lags also exhibit a protective effect of nonaspirin NSAIDs. Selection bias is unlikely to account for our results, because both cohort and case-control studies exhibit similar findings, controls were appropriately selected in case-control studies, and loss to follow-up in cohort studies was minimal. Nevertheless, selection effects due to differential mortality cannot be excluded33; however, the magnitude of such bias is likely small. Recall bias is unlikely to affect the majority of the studies included in these analyses because, for most studies, exposure was recorded or assessed before PD ascertainment. However, nondifferential misclassification of exposure status may be present and could have biased the results toward the null in the original studies as well as in this meta-analysis. Thus, the primary analyses may be underestimates of the true effect of anti-inflammatory drugs on PD. Indeed, analyses restricted to studies that relied on prescription drug data alone, which may be vulnerable to substantial exposure misclassification by failing to account for OTC use, found a smaller protective effect of nonaspirin NSAIDs than studies incorporating information on both prescription and OTC use.
Bias due to unmeasured or residual confounding cannot be excluded, and any confounding in the original studies would have persisted to this meta-analysis. However, confounding is unlikely to fully account for the observed protective association of nonaspirin NSAIDs. Most studies controlled for the most influential known risk factors of PD, age and smoking, and those that further adjusted for other known or suspected risk factors, such as caffeine intake, alcohol consumption, or gout, found that these further adjustments did not materially change the effect estimates. For example, one study reported an age- and smoking-adjusted RR of 0.58 (95% CI 0.30–1.12) and found the estimate to be substantially unchanged after further adjustment for caffeine intake and alcohol consumption (RR 0.60, 95% CI 0.31–1.17).7 Additionally, adjustment for indications for nonaspirin NSAID use did not seem to substantially impact effect estimates.22
We observed a seemingly paradoxical harmful effect of aspirin use on PD risk among men. However, because men have historically been more likely to use aspirin for primary prevention of cardiovascular events,34–36 we suspect that this finding may be due to confounding or selection bias. Unmeasured confounding would exist if cardiovascular indications for aspirin use were not adjusted for in the original analyses and these indications are also risk factors for PD. Alternatively, selection bias due to differential mortality may explain the results because, among older individuals, those who do not use aspirin are more likely to die of cardiovascular disease than those who do use aspirin, and death from cardiovascular disease precludes diagnosis of PD that would have developed later in life. Thus, the association between aspirin use and PD could be noncausal, and further research is needed determine whether the observed association is real or is an artifact of these potential sources of bias.
Our conclusion that use of certain types of anti-inflammatory drugs are associated with decreased PD risk conflicts with the conclusion drawn by the authors of another recently published meta-analysis on this topic.37 Methodologic differences between the 2 meta-analyses account for the divergent findings and highlight the need to carefully consider the biology and pharmacology that govern the association of interest. First, whereas we considered nonaspirin NSAIDs and aspirin separately and observed a protective effect for nonaspirin NSAIDs but not for aspirin, the other authors considered these drugs as a single class and found no substantial overall effect (RR 0.95, 95% CI 0.80–1.12). Because these drugs act through different biologic pathways and exhibit different pharmacologic profiles,31,38 it is reasonable to expect a different effect of aspirin and nonaspirin NSAIDs on PD risk. Second, whereas we excluded studies that assessed exposure entirely within 1 year of PD diagnosis, the other authors included these studies. Because PD initiation likely occurs long before symptom onset, it is implausible that exposures ascertained within 1 year of PD diagnosis affect disease incidence.11
Additional well-designed and well-conducted epidemiologic studies can further our understanding of the relation between anti-inflammatory drugs and PD by investigating the relative effects of specific agents, doses, timing, and durations of use. In this analysis, we observed a stronger protective effect when examining ibuprofen alone vs all nonaspirin NSAIDs. Regular use and longer duration of use were also associated with stronger effects. However, optimal dose, timing, and duration of use are unknown. Moreover, the insult that initiates the hypothesized neuroinflammatory mechanism has not been conclusively identified.39 Unraveling the biologic mechanism of neuroinflammation in PD, along with the mediators this process, may aid in the identification of its causes.39 Although this meta-analysis suggests that use of nonaspirin NSAIDs may have a positive impact on PD risk, the prudent long-term public health strategy would focus on preventing exposure to the underlying causes of PD rather than relying on mitigating the consequences of such exposures through long-term NSAID administration.
AUTHOR CONTRIBUTIONS
Joshua J. Gagne (academic—Harvard School of Public Health) conducted the statistical analysis.
ACKNOWLEDGMENT
The authors thank Alberto Ascherio, MD, DrPH, from the Departments of Nutrition and Epidemiology, Harvard School of Public Health; Channing Laboratory, Department of Medicine, Brigham and Women's Hospital; and Harvard Medical School, Boston, MA, for review of a previous draft of the manuscript.
DISCLOSURE
Dr. Gagne is support by a National Institute on Aging training grant (T32 AG000158). Ms. Power is supported by a National Institute of Environmental Health Sciences training grant (T32 ES007069).
Supplementary Material
Address correspondence and reprint requests to Dr. Joshua J. Gagne, 1620 Tremont St., Suite 3030, Boston, MA 02120 jgagne1@partners.org
Supplemental data at www.neurology.org
Disclosure: Author disclosures are provided at the end of the article.
Received August 25, 2009. Accepted in final form January 4, 2010.
REFERENCES
- 1.Driver JA, Kurth T, Buring JE, Gaziano JM, Logroscino G. Parkinson disease and risk of mortality: a prospective comorbidity-matched cohort study. Neurology 2008;70(pt 2):1423–1430. [DOI] [PubMed] [Google Scholar]
- 2.Dorsey ER, Constantinescu R, Thompson JP, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 2007;68:384–386. [DOI] [PubMed] [Google Scholar]
- 3.Hirsch EC, Hunot S. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol 2009;8:382–397. [DOI] [PubMed] [Google Scholar]
- 4.Hunot S, Hirsch EC. Neuroinflammatory processes in Parkinson's disease. Ann Neurol 2003;53(suppl 3):S49–S58. [DOI] [PubMed] [Google Scholar]
- 5.Teismann P, Ferger B. Inhibition of the cyclooxygenase isoenzymes COX-1 and COX-2 provide neuroprotection in the MPTP-mouse model of Parkinson's disease. Synapse 2001;39:167–174. [DOI] [PubMed] [Google Scholar]
- 6.Chen H, Jacobs E, Schwarzschild MA, et al. Nonsteroidal antiinflammatory drug use and the risk for Parkinson's disease. Ann Neurol 2005;58:963–967. [DOI] [PubMed] [Google Scholar]
- 7.Chen H, Zhang SM, Hernán MA, et al. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch Neurol 2003;60:1059–1064. [DOI] [PubMed] [Google Scholar]
- 8.Ton TG, Heckbert SR, Longstreth WT Jr, et al. Nonsteroidal anti-inflammatory drugs and risk of Parkinson's disease. Mov Disord 2006;21:964–969. [DOI] [PubMed] [Google Scholar]
- 9.Hernán MA, Logroscino G, García Rodríguez LA. Nonsteroidal anti-inflammatory drugs and the incidence of Parkinson disease. Neurology 2006;66:1097–1099. [DOI] [PubMed] [Google Scholar]
- 10.Stroup DF, Berlin JA, Morton SC, et al. Meta-analysis of observational studies in epidemiology: a proposal for reporting. Meta-analysis of Observational Studies in Epidemiology (MOOSE) group. JAMA 2000;283:2008–2012. [DOI] [PubMed] [Google Scholar]
- 11.Ascherio A, Tanner CM. Use of antihypertensives and the risk of Parkinson disease. Neurology 2009;72:578–579. [DOI] [PubMed] [Google Scholar]
- 12.Wells G, Shea B, O'Connell D, et al. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomized studies in meta-analyses. Ottawa Health Research Institute. Available at: http://www.ohri.ca/programs/clinical_epidemiology/oxford.htm. Accessed March 4, 2009.
- 13.Millett GA, Flores SA, Marks G, Reed JB, Herbst JH. Circumcision status and risk of HIV and sexually transmitted infections among men who have sex with men: a meta-analysis. JAMA 2008;300:1674–1684. [DOI] [PubMed] [Google Scholar]
- 14.Aziz O, Constantinides V, Tekkis PP, et al. Laparoscopic versus open surgery for rectal cancer: a meta-analysis. Ann Surg Oncol 2006;13:413–424. [DOI] [PubMed] [Google Scholar]
- 15.Gagne JJ, Griesdale DE, Schneeweiss S. Aprotinin and the risk of death and renal dysfunction in patients undergoing cardiac surgery: a meta-analysis of epidemiologic studies. Pharmacoepidemiol Drug Saf 2009;18:259–268. [DOI] [PubMed] [Google Scholar]
- 16.Stead LG, Gilmore RM, Bellolio MF, Rabinstein AA, Decker WW. Percutaneous clot removal devices in acute ischemic stroke: a systematic review and meta-analysis. Arch Neurol 2008;65:1024–1030. [DOI] [PubMed] [Google Scholar]
- 17.DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials 1986;7:177–188. [DOI] [PubMed] [Google Scholar]
- 18.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med 2002;21:1539–1558. [DOI] [PubMed] [Google Scholar]
- 19.Hancock DB, Martin ER, Vance JM, Scott WK. Nitric oxide synthase genes and their interactions with environmental factors in Parkinson's disease. Neurogenetics 2008;9:249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Etminan M, Suissa S. NSAID use and the risk of Parkinson's disease. Curr Drug Saf 2006;1:223–225. [DOI] [PubMed] [Google Scholar]
- 21.Etminan M, Carleton BC, Samii A. Non-steroidal anti-inflammatory drug use and the risk of Parkinson disease: a retrospective cohort study. J Clin Neurosci 2008;15:576–577. [DOI] [PubMed] [Google Scholar]
- 22.Bornebroek M, de Lau LM, Haag MD, et al. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Neuroepidemiology 2007;28:193–196. [DOI] [PubMed] [Google Scholar]
- 23.Hancock DB, Martin ER, Stajich JM, et al. Smoking, caffeine, and nonsteroidal anti-inflammatory drugs in families with Parkinson disease. Arch Neurol 2007;64:576–580. [DOI] [PubMed] [Google Scholar]
- 24.Bower JH, Maraganore DM, Peterson BJ, Ahlskog JE, Rocca WA. Immunologic diseases, anti-inflammatory drugs, and Parkinson disease: a case-control study. Neurology 2006;67:494–496. [DOI] [PubMed] [Google Scholar]
- 25.Wahner AD, Bronstein JM, Bordelon YM, Ritz B. Nonsteroidal anti-inflammatory drugs may protect against Parkinson disease. Neurology 2007;69:1836–1842. [DOI] [PubMed] [Google Scholar]
- 26.Powers KM, Kay DM, Factor SA, et al. Combined effects of smoking, coffee, and NSAIDs on Parkinson's disease risk. Mov Disord 2008;23:88–95. [DOI] [PubMed] [Google Scholar]
- 27.Ouchi Y, Yoshikawa E, Sekine Y, et al. Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann Neurol 2005;57:168–175. [DOI] [PubMed] [Google Scholar]
- 28.Wahner AD, Sinsheimer JS, Bronstein JM, Ritz B. Inflammatory cytokine gene polymorphisms and increased risk of Parkinson disease. Arch Neurol 2007;64:836–840. [DOI] [PubMed] [Google Scholar]
- 29.Håkansson A, Westberg L, Nisson S, et al. Interaction of polymorphisms in the genes encoding interleukin-6 and estrogen receptor beta on the susceptibility to Parkinson's disease. Am J Med Genet B Neuropsychiat Genet 2005;133B:88–92. [DOI] [PubMed] [Google Scholar]
- 30.Esposito E, Di Matteo V, Benigno A, Pierucci M, Crescimanno G, Di Giovanni G. Non-steroidal anti-inflammatory drugs in Parkinson's disease. Exp Neurol 2007;205:295–312. [DOI] [PubMed] [Google Scholar]
- 31.Asanuma M, Miyazaki I. Common anti-inflammatory drugs are potentially therapeutic for Parkinson's disease? Exp Neurol 2007;206:172–178. [DOI] [PubMed] [Google Scholar]
- 32.Aronoff DM, Oates JA, Boutaud O. New insights into the mechanism of action of acetaminophen: its clinical pharmacologic characteristics reflect its inhibition of the two prostaglandin H2 synthases. Clin Pharmacol Ther 2006;79:9–19. [DOI] [PubMed] [Google Scholar]
- 33.Hernán MA, Alonso A, Logroscino G. Cigarette smoking and dementia: potential selection bias in the elderly. Epidemiology 2008;19:448–450. [DOI] [PubMed] [Google Scholar]
- 34.Chandra NC, Ziegelstein RC, Rogers WJ, et al. Observations of the treatment of women in the United States with myocardial infarction: a report from the National Registry of Myocardial Infarction-I. Arch Intern Med 1998;158:981–988. [DOI] [PubMed] [Google Scholar]
- 35.Gan SC, Beaver SK, Houck PM, MacLehose RF, Lawson HW, Chan L. Treatment of acute myocardial infarction and 30-day mortality among women and men. N Engl J Med 2000;343:8–15. [DOI] [PubMed] [Google Scholar]
- 36.Blomkalns AL, Chen AY, Hochman JS, et al; CRUSADE Investigators. Gender disparities in the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes: large-scale observations from the CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes With Early Implementation of the American College of Cardiology/American Heart Association Guidelines) National Quality Improvement Initiative. J Am Coll Cardiol 2005;45:832–837. [DOI] [PubMed] [Google Scholar]
- 37.Samii A, Etminan M, Wiens MO, Jafari S. NSAID use and the risk of Parkinson's disease: systematic review and meta-analysis of observational studies. Drugs Aging 2009;26:769–779. [DOI] [PubMed] [Google Scholar]
- 38.Paul-Clark MJ, Van Cao T, Moradi-Bidhendi N, Cooper D, Gilroy DW. 15-Epi-lipoxin A4-mediated induction of nitric oxide explains how aspirin inhibits acute inflammation. J Exp Med 2004;200:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu B, Gao HM, Hong JS. Parkinson's disease and exposure to infectious agents and pesticides and the occurrence of brain injuries: role of neuroinflammation. Environ Health Perspect 2003;111:1065–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.