

Abstract
Background
The angiotensin II (AngII) type 1 receptor (AT1) regulates cardiovascular function by activating various signal pathways. The purpose of this study was to evaluate the effects of a mutant AT1 receptor on AngII responding blood pressure and cardiac hypertrophy in conjunction with altered AngII activation of RhoA and Akt.
Methods
A mutant AT1 receptor was constructed and overexpressed in FVB mice using a ubiquitous-expression vector pCAGGS. The phenotype and signal transduction of the transgenic mice were compared with the wild type (WT) mice.
Results
The transgenic mice showed a similar baseline phenotype as WT mice, but their blood pressure in response to continuous AngII infusion was significantly lower, as measured on day 3, 4, 7 and 14, with a difference of 20 mmHg by day 14. There was also a significantly larger heart to total body weight ratio in the WT mice, whose heart weight was 0.441 ± 0.008 % of total body weight compared to the transgenic mice at 0.416 ± 0.008 %. Aortic endothelial cells isolated from these transgenic mice displayed an altered signaling profile, such as diminished activation of Akt and RhoA in response to AngII. In contrast, Gαq coupling and ERK/JNK activation did not change.
Conclusion
The expression of an AT1 mutant receptor in the presence of WT receptor can effectively modulate AngII effected signaling. Furthermore, the elimination of Akt and RhoA activation by AngII significantly reduces but does not eliminate its hypertensive effect.
INTRODUCTION
Angiotensin II (AngII), acting through its type I receptor (AT1), is a primary contributor to cardiovascular disease including hypertension 1. Inhibition of AT1 receptor action is crucial in the treatment of cardiovascular disease 2. Bradykinin acting mostly via its B2 receptor may also play a role in cardiovascular regulation3. In fact ACE inhibitors which are used for regulating hypertension not only diminish the conversion of Angiotensin I to AngII they also block the destruction of bradykinin 4,5.
We have been identifying and manipulating AT1 receptor structures such that we can regulate the signal transductions associated with this receptor in response to AngII. In the process we successfully replaced major portions of the signaling segments within the intracellular face of the AT1 receptor 6. In the communication we showed that the hybrids possess a number of signaling characteristics of the BKB2 receptor. Some hybrids demonstrated BK as opposed to AngII like phosphorylation of Akt6. Our results also suggested that the Lys-Ser-Arg motif in the second intracellular loop of AT1 receptor is crucial for Gαq coupling. The third and C-terminal tail are the regions determining the non-G protein coupled signaling in the BKB2 and AT1 receptors. One hybrid receptor, namely AB3T, where the third intracellular loop and C-terminus of the AT1R were replaced with corresponding regions of the BKB2R, demonstrated a reduced phosphorylation of Akt and activation of RhoA compared to WT AT1R. Since both Akt and RhoA have been shown to affect the function of the cardiovascular system 7,8, we postulated that the expression of this AB3T receptor will modulate the in vivo hypertrophic and hypertensive effects of AngII.
In this study we generated transgenic (TG) mice expressing this AB3T receptor mutant. The signaling profile of this TG mouse model in response to AngII was analyzed in cultures of aortic endothelial cells obtained from these mice and compared to those isolated from WT mice. The cardiovascular phenotype at baseline and response to a two-week continuous AngII infusion, including the blood pressure and cardiac hypertrophy, was also compared between the WT and AB3T transgenic mice. The phenotype of these mice showed a marked reduction in angiotensin II generated hypertension and hypertrophy.
METHODS
Generation of AB3T hybrid receptor
To construct the AB3T mutant, the AT1 third intracellular loop was first replaced with the IC3 of the BKB2R: the pcDNA3.1-AT1R cassette6 was digested with SpeI (position 214) and EcoRV (position 237). The corresponding sense and anti-sense oligonucleotides for the IC3 region of the BKB2 receptor (sense: 5' - CTAGTTATACCCTTATCATGCAGGTGCTGAGGAACAACGAGATGAAGAAGTTCAAGGAGGTCCAGACGGAGAAG -3'; antisense: 5' –CTTCTCCGTCTGGACCTCCTTGAACTTCTTCATCTCGTTGTTCCTCAGCACCTGCATGATAAGGGTATAA-3') were then annealed at equal molar ratio and ligated into the SpeI/EcoRV digested vector cassette. The entire C-terminus AT1R/BKB2R hybrid was then constructed by PCR to amplify the C-terminus of BKB2R using the rat BKB2R cDNA as a template. After obtaining the single loop mutants AB3 and ABT, the AB3T was constructed by exchanging the EcoRI - XhoI fragments containing the 3rd loop and C-tail with rest of the receptor. The AB3T mutant was sequenced by an in-house facility.
Generation transgenic mice expressing the AB3T gene
The cDNA of the AB3T was cloned into the pCAGGS vector, an expression vector containing the CMV/chicken β-actin enhancer/promoter. Heterozygous lines of transgenic mice were generated at the Transgenic/Knockout Core Facility at Boston University . Expression of the AB3T receptor was determined with reverse transcription-PCR in the major AT1-expressing tissues (liver, heart, kidney, and the aorta). Primers (forward 5’-CAACGAGATGAAGAAGTTCAAGGA reverse 5'-AAGCACAATCGCCATAATTATCC-3') were designed so that one matched the AT1 receptor and the other the BKB2 receptor. The genetic background of the experimental group of mice and the control group of mice was FVB. The animals were housed in the animal quarters with 12:12 hours light-dark cycle and all experiments were conducted in the accordance with the "Guidelines for the Care and Use of Animals" approved by the Boston University School of Medicine.
AngII infusion and blood pressure/ heart rate measurement
Two groups of adult male littermates weighing 25 to 40g, one AB3T TG and one WT (n =17 per group) were used in the experiment. An additional 10 mice per group were weighed and sacrificed at baseline in order to obtain the body to heart weight ratio. For five days the experimental mice were trained to measure blood pressure via the computerized noninvasive tail cuff system (Model BP 2000, Visitech systems). The blood pressure system was configured to measure eight preliminary readings which were discarded followed by up to 15 readings to report an average reading. A baseline reading was determined using reading from measurements recorded across 3 days. Both group of mice received continuous AngII infusion (0.9µg/h) via subcutaneous osmotic minipumps (Alzet model 2004) for 14 days. The minipumps were implanted in the scapular area under anesthesia with ketamine/xylazine (100mg/kg/ 10mg/kg). The animals were allowed a 48 hour recovery period. Systolic blood pressure and heart rates were measured daily using the non invasive tail cuff method. Tail pulse was detected by passage of the tail through a tail cuff sensor attached to the amplifier. BP measurements were made by the automated inflation of the tail cuff pressure to greater than 200 mmHg causing obstruction of the blood flow followed by release of the pressure. This cycle was repeated 15 times to give an average blood pressure value for a sitting. At the end of the 14 days the mice were sacrificed by CO2 inhalation and cervical dislocation. They were weighed and the hearts were carefully extracted and washed in PBS. After washing away the blood in the heart, the hearts were gently dried and weighed.
Mouse aortic endothelial cells (MAEC) isolation and culture
Isolation of primary MAEC cultures followed the procedure of Dong et al with some modification 9,10. Aortae were dissected, washed and digested with 200 units/ml collagenase at 37° C for 1 hour with intermittent agitation. The digestion mixture was diluted by addition of an equal volume of DMEM containing 20% FBS and subjected to centrifugation at 400 × g for 10 minutes at 4° C. The cell pellet was resuspended in 3 ml of cold PBS and incubated with 30 µl (per 3–5 aortae) magnetic beads (Dynal, Invitrogen) that have been pre-coated with rat anti-mouse PECAM-1 antibody (BD Pharmingen, San Jose, California) for 10 minutes at room temperature. Bead-bound cells were collected from the mixture in a high-field-strength magnet assembly and washed four times in PBS containing 0.1% FBS and 2mM EDTA. Cells were washed again in DMEM with 20% FBS and harvested as before by centrifugation. Cells were then resuspended again in DMEM with 20% FBS and plated in collagen-coated T25 flasks in complete medium supplemented with 100 µg/ml endothelial cell mitogen (BTI, Stoughton, MA). The confluent cells were harvested by brief trypsinization and subjected to a second round of selection with beads pre-coated with rat anti-mouse ICAM-2 (BD Pharmingen), following the procedure of the first selection round.
Ligand binding
AngII binding studies in intact aortic endothelial cells were carried out as described by Yu et al 11.
Phosphoinositide (PI) turnover
The aortic endothelial cells from the WT or AB3T TG mice were incubated with 1µCi/ml myo-[3H]inositol (60 Ci/mmol, Perkin Elmer, Boston, MA) in 1ml of growth medium and the levels of inositol phosphates (IPs) determined 24h later as described by Prado et al 12.
Western blot analysis
Western blot analysis for ERK, JNK and Akt was performed following a 5 min incubation of WT and AB3T mouse derived endothelial cells with 100nM AngII as described by Yu et al 6. Antibodies were purchased from Cell Signaling Technologies (Beverly, MA). RhoA activation was measured with the Rho Activation Kit from Upstate Cell Signaling according to the manufacturer's instructions (Lake Placid, NY, #17-294).
Statistical analysis and data analysis
The Kd, Bmax of AngII binding to endothelial cells and the EC50, and maximal response of PI turnover were estimated using the SigmaPlot® 8 Program Pharmacology Module (SPSS Inc). Statistical evaluation of the data was carried out using the student t-test. Probability values less than 0.05 were considered significant.
RESULTS
Generation of TG mice expressing mutant AT1 receptor
To understand the in vivo cardiovascular and signaling actions of the altered AT1 receptor (AB3T) we generated AB3T overexpressing TG mice using the pCAGGS, an expression vector containing the CMV/chicken β-actin enhancer/promoter 13. Genomic DNA from tail biopsies was used to screen the AB3T expressing mice. The global expression of the AB3T receptor was initially confirmed by reverse transcription PCR with RNA extracted from the kidney, liver, aorta, lung, and heart of the TG mice. The results are illustrated in Fig. 1. The AB3T gene was expressed in those organs from TG mice, but not WT mice.
Figure 1. AB3T mutant receptor expression in aorta, heart and lung by reverse transcription PCR.
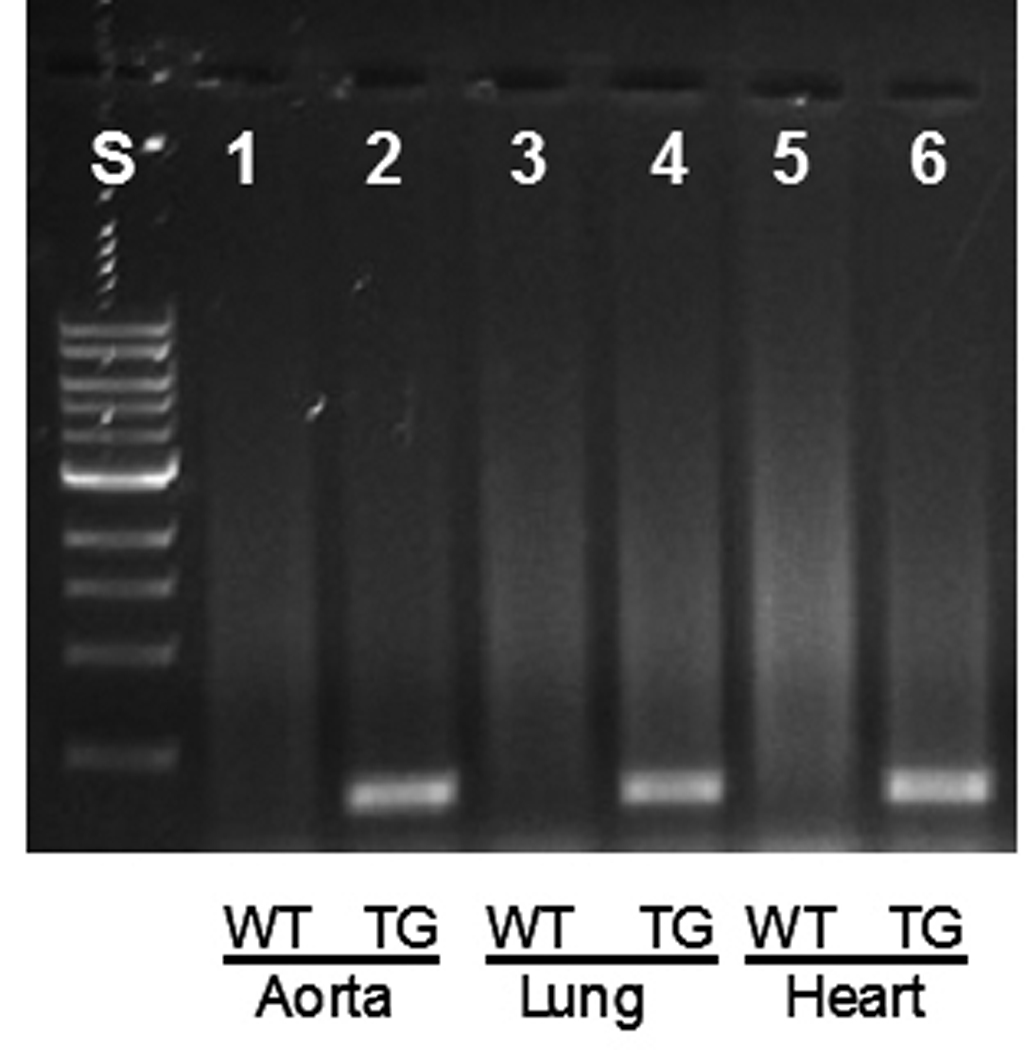
S: Molecular Weight Standard; Lane 1, WT mouse aorta; 2, AB3T TG mouse aorta; 3, WT mouse lung; 4. AB3T TG mouse lung; 5, WT mouse heart; 6, AB3T TG mouse heart.
The AB3T expression in the primary mice aortic endothelial cells (MAECs) was then confirmed with [3H] labeled AngII binding at the protein level. The AB3T aortic endothelial cells demonstrated markedly increased (7.5 fold) binding (Fig. 2). The Bmax of the combined AT1 receptors increased to 5.61 fmole/mg protein in AB3T cells compare with 0.75 fmole/mg protein in the WT cells.
Figure 2. [3H]-AngII binding assay.
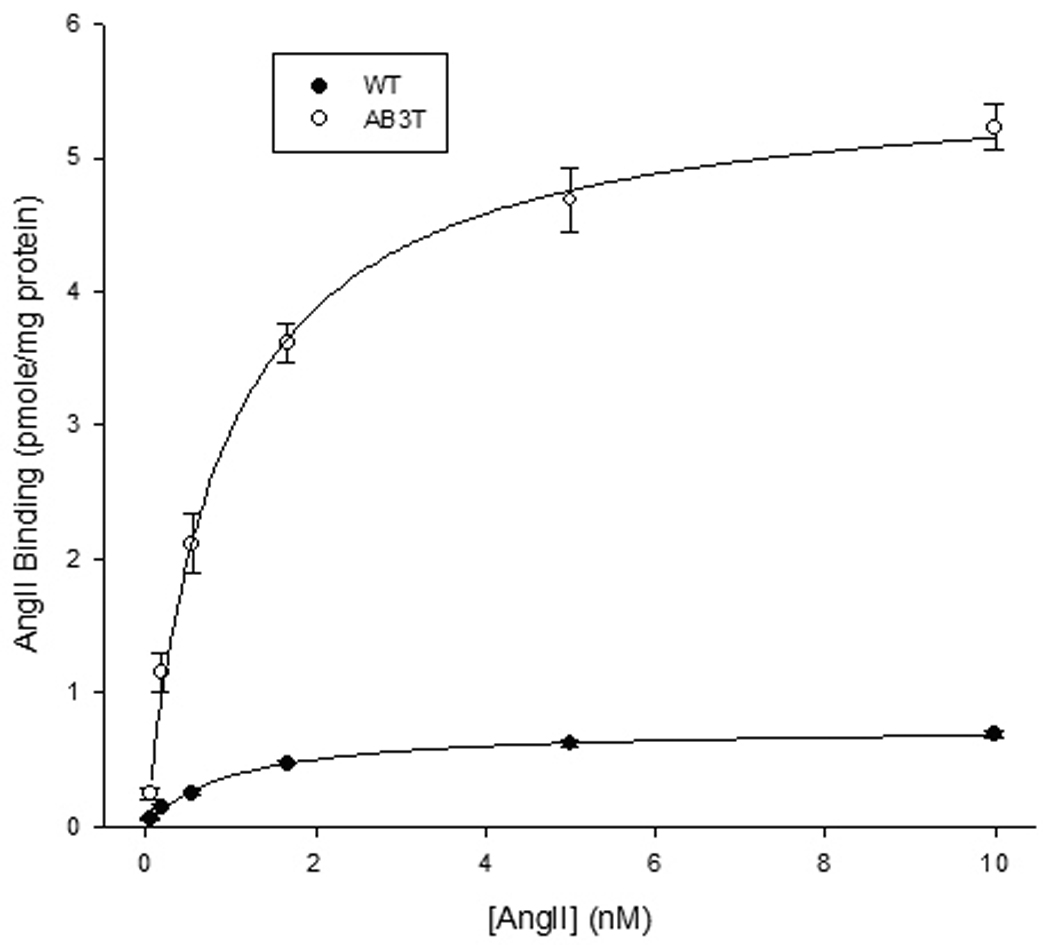
Ligand binding experiments were carried out using [3H]AngII on wild type and AB3T aortic endothelial cells at concentrations ranging from 0.04 to 10nM. The dissociation constant (Kd) and maximal binding (Bmax) were calculated using the SigmaPlot® 8 Program Pharmacology Module (SPSS Inc.). Each Point is the average of cells from triplicate wells ± S.E. from a representative experiment of three experiments. The Bmax for WT and AB3T was 0.75 ± 0.02 and 5.61 ± 0.17 fmole/mg protein respectively and the difference is significant (p< 0.001). The Kd for WT and AB3T was 0.98 ± 0.11 and 0.90 ± 0.11 nM respectively. There is no significant difference between the Kd of WT and AB3T cells (p > 0.05).
Protein kinase activation
We evaluated the activation of protein kinases (Akt/PKB, ERK and JNK) in the aortic endothelial cells isolated from the AB3T mice and WT mice. AngII increased the phosphorylation of Akt at Ser 473 in the WT cells. However in the cells from AB3T mice, AngII had an opposite effect. It reduced the presence of phospho-Akt (Fig. 3A). The activation of ERK and JNK by AngII remained unchanged in the AB3T cells compared to WT cells (Fig. 3B).
Figure 3.
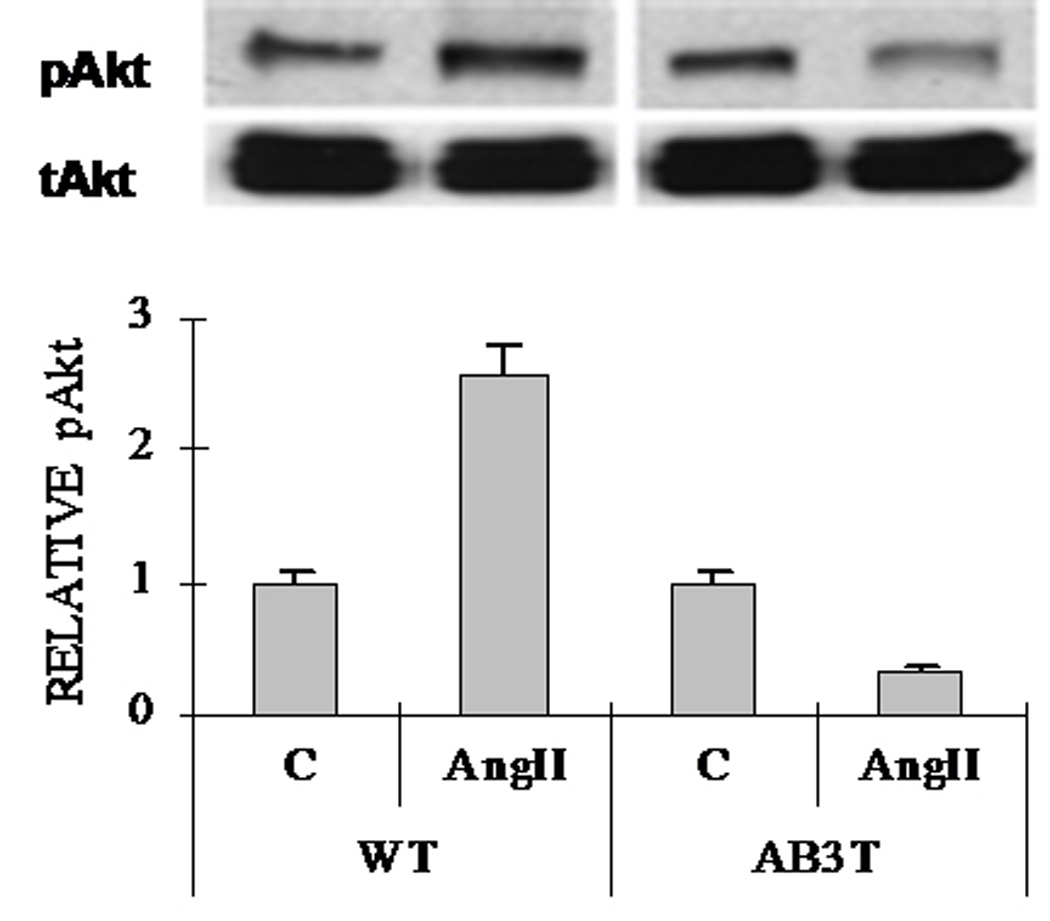
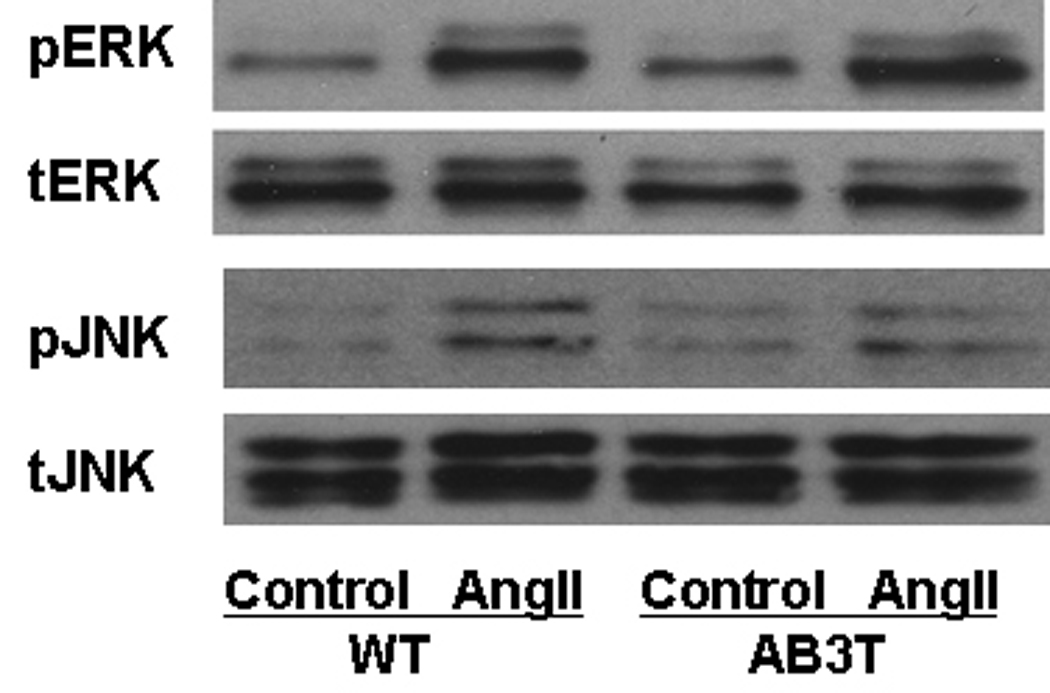
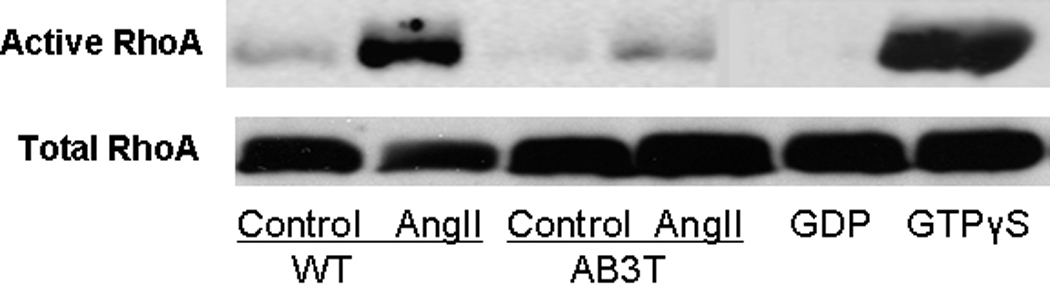
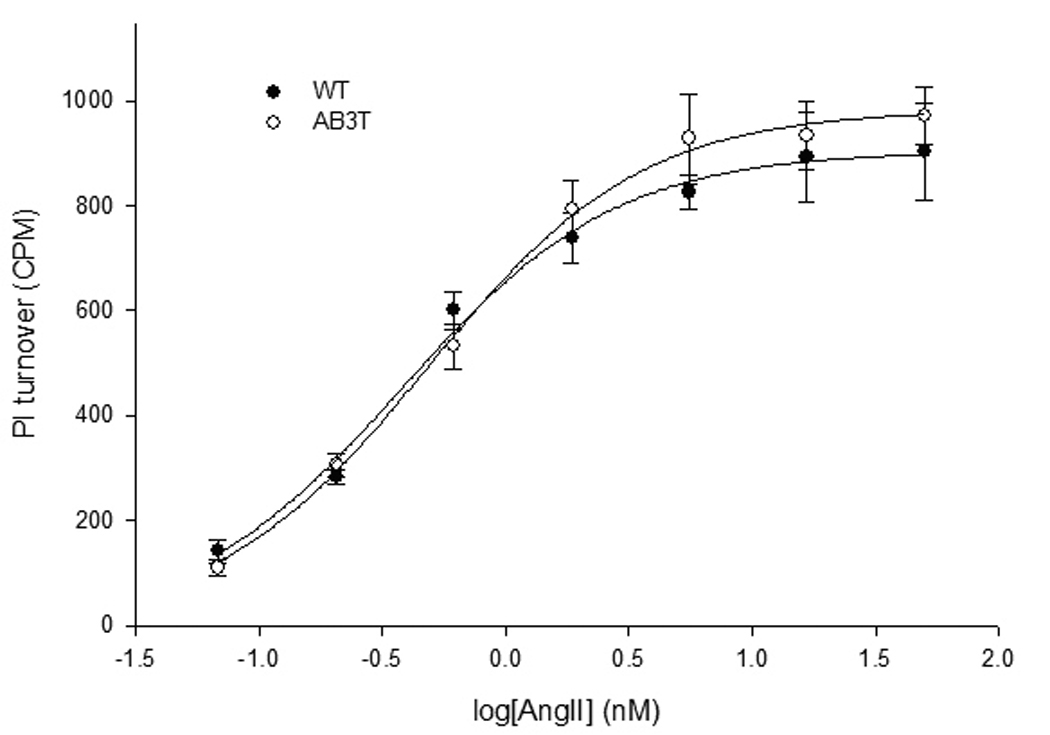
Figure 3A. Akt activation. Western blot analysis of cell lysates of the aortic endothelial cells from wild type and AB3T mice. Cells were incubated with or without 100nM AngII for 5 minutes. The upper bands are phospho-Akt at Serine 473 and the lower bands are total Akt. The results are representative of at least three experiments. The quantitation of the western was illustrated as a graph which shows fold change in spot intensity of the bands with basal level as 1. The blots shown are representatives of three experiments.
Figure 3B. ERK and JNK activation. Same procedure was followed as in Figure 3A.
Figure 3C. RhoA activation. WT or AB3T MAECs either untreated (control) or 100nM AngII treated for 5 minutes. Rhotekin Rho-binding peptide pulldown assays were performed on cell lysates in order to measure RhoA activity. The pull-down samples from cell lysates were immunoblotted with a Rho specific antibody for visualization of the active protein levels. Detergent soluble lysates were also immunoblotted with either a Rho specific antibody for total Rho protein levels. The GDP and GTPγS lanes were used as negative and positive control respectively.
Figure 3D. Concentration-response curve of AngII-stimulated nositol phosphate production in the WT and AB3T endothelial cells. PI turnover was measured in myo-[3H] inositol-labeled cells as described in Methods sections. Results are presented as AngII stimulated PI turnover (cpm: counts per minute) minus basal IP as measured by the scintillation counter. Each point is the average of cells from triplicate wells ± S.E. from three experiments. The EC50 and Emax (maximal response) of PI turnover were calculated using the SigmaPlot® 8 Program Pharmacology Module (SPSS Inc.). Each Point is the average of cells from triplicate wells ± S.E. from a representative experiment of three experiments. The Emax for WT and AB3T was 904 ± 32 and 983 ± 32 cpm respectively. The EC50 for WT and AB3T was 0.4 ± 0.1 and 0.5 ± 0.1 nM respectively. There is no significant difference of EC50 and Bmax between WT and AB3T cells (p > 0.05).
Activation of small G-protein RhoA
Small G-protein RhoA and its downstream kinase Rho kinase (ROCK), play deleterious roles in both blood pressure regulation and vascular smooth muscle function 8,14. As shown in Fig. 3C, AngII strongly activated RhoA after 5 minutes exposure in the WT endothelial cells. In the AB3T cells, AngII only activated RhoA marginally.
AngII induced Gαq Activation
We measured phosphatidyl inositol turnover to illustrate the receptor Gαq coupling capacity 6. The aortic endothelial cells from both WT and AB3T TG mice displayed similar dose-response curves in response to AngII (Fig. 3D). The maximal response and the EC50 are similar between WT and AB3T cells.
Blood pressure in response to AngII infusion
The AB3T mice (n=17) and WT mice (n=17) showed similar baseline (resting) blood pressure (WT: 122 ± 3 mmHg vs. AB3T: 121 ± 3 mmHg, p > 0.05), and heart rate (WT: 698 ± 20 vs. AB3T: 671 ± 7 beats per minute, p > 0.05). Despite these baseline similarities, compared to WT mice, the AB3T mice displayed a delayed onset of hypertension during the initial 3 days of AngII infusion and a markedly reduced increase in blood pressure thereafter (Fig. 4A). The pressures were significantly different on day 3, 4, 7 and 14 of the continuous AngII infusion, with a difference of 20 mmHg by day 14 (173 ± 8mmHg vs. 151 ± 6 mmHg, p < 0.05).
Figure 4.


Figure 4A. Systolic blood pressure upon AngII infusion. The systolic blood pressures of WT mice were represented as unfilled circles, and those of AB3T mice as filled circles. *: significant difference between WT mice and AB3T TG mice (p< 0.05).
Figure 4B. Heart weight/body weight ratio upon AngII infusion. Ten wild type mice and ten transgenic mice were sacrificed at baseline, to get the heart weight. The Heart weight/body weight ratios of WT mice were represented as white bars, and those of AB3T mice as grey bars. *: significant difference of heart weight/ body weigh ratio between WT mice and AB3T mice after AngII infusion (p< 0.05).
Cardiac hypertrophy in WT and TG mice following AngII infusion
Heart weight (HW) to total body weight (BW) ratios were generated to assess myocardial hypertrophy. There was no significant difference in the heart weight (WT: 144 ± 6 mg vs. TG: 136 ± 7 mg, n=10, p > 0.05) and body weight (WT: 34.5 ± 3.2 g vs. TG: 33.4 ± 3.7g, n=10, p > 0.05) between the WT and TG mice before AngII infusion. Continuous infusion of AngII for 2 weeks led to a significant increase in the HW/BW ratio in both WT and TG mice. However a statistically significant lower ratio increase was observed in the TG mice (WT: 4.41 ± 0.08% vs. TG: 4.16 ± 0.08%, n = 17, p = 0.036) (Fig. 4B). The heart rate didn’t change after AngII infusion in either the WT or TG mice (WT: 672 ± 18 vs. TG: 678 ± 11 beats per minute, n=17, p > 0.05).
DISCUSSION
The AT1 receptor has been established as a focal determinant in the progressive dysfunction of the cardiovascular system. The AT1 knock-out mouse has demonstrated that it is required for vascular and hemodynamic responses to AngII, and that altered expression of the AT1 gene has marked effects on blood pressure 15. However, the specific receptor generated signaling molecules acting in this process remain obscure. There is evidence that the activation of RhoA and Akt are important factors in cardiac hypertrophy and in hypertension. For example, the inhibition of Rho Kinase (ROCK) provides cardiovascular benefits 8. An increased RhoA expression and an enhanced RhoA activity have been observed in aortas of hypertensive rats. Jin et al. showed that the Rho-kinase inhibitor, Y-27632 inhibits spontaneous tone in aortic rings from AngII-treated rats 16. Other studies show that in vascular endothelial cells the RhoA/Rho-kinase pathway downregulates eNOS gene expression through post-transcriptional mRNA destabilization 17. RhoA/ROCK also inhibits the activity of eNOS by phosphorylating eNOS at Thr495 18. eNOS produced NO which is a powerful dilator and antiproliferative factor 19. In addition, Rho-kinase inhibitors have been shown to inhibit formation of hypertensive vascular lesions 20. Activation of Akt, has been linked to smooth muscle cell proliferation and vascular disorders 21. This activation is markedly inhibited by the calcium channel blocker lercanidipine 22 as well as the renin inhibitor ithaliskiren 23. Because of the importance of these two signal elements in the cardiovascular pathology, we proceeded to develop transgenic mice which overexpress a mutant AT1 receptor which fails to activate either RhoA or Akt in vitro in response to AngII 6.
Our aim in the current series of experiments was to determine whether the expression of this receptor will attenuate AngII- induced hypertension and cardiac hypertrophy. The pCAGGS vector 13 was used to construct the AB3T transgene. This vector contains a CAG-promoter (Cytomegalovirus enhancer + chicken β-actin promoter) and rabbit β-globin polyadenylation sites. These cis-acting elements produce high, ubiquitious systemic expression of the transgene 24. In our model the AB3T receptor was clearly expressed in lung, heart, kidney and liver. We also determined that the AB3T receptor is expressed in the aorta stripped of its adventitia.
Hypertension is characterized by increased peripheral vascular resistance and/or vascular structural remodeling. Endothelial cells, the primary barrier to the circulating blood, release vasoactive factors, such as NO and endothelin-1 and prostacyclin, which then regulate vascular tone and the pathophysiology of hypertension 25. Impairment of endothelium-dependent vasodilatation has been demonstrated in hypertension patients 26,27. For these reasons we combined our in vivo work with studies on aortic endothelial cells in primary cell cultures isolated from WT and the AB3T TG expressing mice. This enabled us to determine the potential endothelial signaling in both WT and AB3T expressing cells. The homogeneity of the MAEC cultures after a secondary selection was confirmed by immunohistochemistry using PECAM-1 antibodies. PECAM-1 is a specific marker for vascular endothelium, with a very limited expression in other cell types 28. Also, the cultures exhibited typical cobblestone morphology. Thus although a 100% endothelial purity is most likely not attainable in any primary culture signal contribution by smooth muscle cells would be minimal at most. The endothelial cells from the AB3T mice demonstrate that the normal (WT) AngII induced RhoA and Akt activation is not taking place while AngII induced Gαq and ERK/JNK signaling continue. Both these signaling junctions have been linked to hypertension. Thus our results suggest that preventing AngII activation of RhoA and Akt is at least in part responsible for the attenuated AngII induced hypertension and cardiac hypertrophy in the TG mice. Interestingly, the AB3T mice have similar baseline cardiovascular characteristics such as blood pressure, resting heart rate and heart weight/body weight ratio as WT mice.
It is important to note that the level of expression of the WT or mutant AT1 receptor could modulate the phenotype. For example, Zhai et al showed that both ventricles and atria of the heart were enlarged in cardiac AT1 over-expressed transgenic mice compared with WT mice 29,30. Furthermore Hein et al. showed that overexpression of the angiotensin AT1 receptor transgene in the mouse myocardium produced a lethal phenotype associated with myocyte hyperplasia and heart block 31. AT1 transgenic mice generated by Paradis et al. also exhibited premature death with cardiac hypertrophy and fibrosis 32. Deleterious effects of AT1 overexpression are also seen in the kidney. Transgenic rats overexpressing the WT AT1 receptor in kidney podocytes exhibit glomerulosclerosis 33. All the damaging physiology by an over-expressed AT1 receptor in sum is contrary to that found in the AB3T expressing mice which demonstrate an unchanged baseline phenotype and a lower hypertensive response to angiotensin compared to WT mice. However, the mutant receptor described here was not tissue specifically targeted. Instead it was expressed ubiquitously. Interestingly, transgenic mice overexpressing the WT AT1 receptor ubiquitously have, to our knowledge, not been reported in the literature. This void may be due to a lethal condition. However, clearly if these mice were successfully generated they would more accurately as control to test the effectiveness of the mutant receptor.
Continuous AngII infusion for two weeks in mice leads to elevation in blood pressure, which mimics many characteristics of essential hypertension in humans 34. We found that AngII infusion had a very different effect on blood pressure and cardiac hypertrophy in the WT and in the AB3T mice. AngII induced hypertension and cardiac hypertrophy were attenuated but not eliminated in the AB3T mice. Clearly, other factor(s) in addition to Akt and RhoA are also involved in this process.
It should also be noted that while we showed that endothelial cells derived from the aorta express the AB3T receptor in culture we did not directly illustrate this expression in the endothelium in situ. We did show AB3T expression by the entire aorta devoid of the adventitia. Thus, it is possible that the observed changes in Akt and RhoA activation by the AB3T receptor in the aorta are taking place in either smooth muscle or endothelium or both. Of course, alterations in RhoA and Akt signaling in such tissue as kidney in response to AngII may also be crucial to the regulation of AngII promoted hypertension.
ACKNOWLEDGMENTS
The authors thank Dr. Peter Polgar and Dr. Linda Taylor for the scientific discussion and technical support.
This work was supported by NIH Grant number HL25776 and fund from the Department of Biochemistry at Boston University School of Medicine.
Footnotes
Conflict of Interest: None
DISCLOSURE
None.
REFERENCES
- 1.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 2.Gavras H, Brunner HR. Role of Angiotensin and Its Inhibition in Hypertension, Ischemic Heart Disease, and Heart Failure. Hypertension. 2001;37:342–345. doi: 10.1161/01.hyp.37.2.342. [DOI] [PubMed] [Google Scholar]
- 3.Duka A, Duka I, Gao G, Shenouda S, Gavras I, Gavras H. Role of bradykinin B1 and B2 receptors in normal blood pressure regulation. Am J Physiol Endocrinol Metab. 2006;291:E268–E274. doi: 10.1152/ajpendo.00382.2005. [DOI] [PubMed] [Google Scholar]
- 4.Murphey L, Vaughan D, Brown N. Contribution of bradykinin to the cardioprotective effects of ACE inhibitors. Eur Heart J Suppl. 2003;5:A37–A41. [Google Scholar]
- 5.Hornig B, Kohler C, Drexler H. Role of Bradykinin in Mediating Vascular Effects of Angiotensin-Converting Enzyme Inhibitors in Humans. Circulation. 1997;95:1115–1118. doi: 10.1161/01.cir.95.5.1115. [DOI] [PubMed] [Google Scholar]
- 6.Yu J, Lubinsky D, Tsomaia N, Huang Z, Taylor L, Mierke D, Navarro J, Miraz O, Polgar P. Activation of ERK, JNK, Akt, and G-protein coupled signaling by hybrid angiotensin II AT1/bradykinin B2 receptors expressed in HEK-293 cells. J Cell Biochem. 2007;101:192–204. doi: 10.1002/jcb.21161. [DOI] [PubMed] [Google Scholar]
- 7.Shiojima I, Walsh K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006;20:3347–3365. doi: 10.1101/gad.1492806. [DOI] [PubMed] [Google Scholar]
- 8.Noma K, Oyama N, Liao JK. Physiological role of ROCKs in the cardiovascular system. Am J Physiol Cell Physiol. 2006;290:C661–C668. doi: 10.1152/ajpcell.00459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim Y-C, Garcia-Cardena G, Allport JR, Zervoglos M, Connolly AJ, Gimbrone MA, Jr, Luscinskas FW. Heterogeneity of Endothelial Cells from Different Organ Sites in T-Cell Subset Recruitment. Am J Pathol. 2003;162:1591–1601. doi: 10.1016/S0002-9440(10)64293-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong QG, Bernasconi S, Lostaglio S, Wainstok De Calmanovici R, Martin-Padura I, Breviario F, Garlanda C, Ramponi S, Mantovani A, Vecchi A. A General Strategy for Isolation of Endothelial Cells From Murine Tissues: Characterization of Two Endothelial Cell Lines From the Murine Lung and Subcutaneous Sponge Implants. Arterioscler Thromb Vasc Biol. 1997;17:1599–1604. doi: 10.1161/01.atv.17.8.1599. [DOI] [PubMed] [Google Scholar]
- 11.Yu J, Polgar P, Lubinsky D, Gupta M, Wang L, Mierke D, Taylor L. Coulombic and hydrophobic interactions in the first intracellular loop are vital for bradykinin B2 receptor ligand binding and consequent signal transduction. Biochemistry. 2005;44:5295–5306. doi: 10.1021/bi048288i. [DOI] [PubMed] [Google Scholar]
- 12.Prado GN, Taylor L, Polgar P. Effects of intracellular tyrosine residue mutation and carboxyl terminus truncation on signal transduction and internalization of the rat bradykinin B2 receptor. J Biol Chem. 1997;272:14638–14642. doi: 10.1074/jbc.272.23.14638. [DOI] [PubMed] [Google Scholar]
- 13.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 14.Rikitake Y, Liao JK. Rho GTPases, Statins, and Nitric Oxide. Circ Res. 2005;97:1232–1235. doi: 10.1161/01.RES.0000196564.18314.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito M, Oliverio MI, Mannon PJ, Best CF, Maeda N, Smithies O, Coffman TM. Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:3521–3525. doi: 10.1073/pnas.92.8.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin L, Ying Z, Hilgers RHP, Yin J, Zhao X, Imig JD, Webb RC. Increased RhoA/Rho-Kinase Signaling Mediates Spontaneous Tone in Aorta from Angiotensin II-Induced Hypertensive Rats. J Pharmacol Exp Ther. 2006;318:288–295. doi: 10.1124/jpet.105.100735. [DOI] [PubMed] [Google Scholar]
- 17.Laufs U, Liao JK. Post-transcriptional Regulation of Endothelial Nitric Oxide Synthase mRNA Stability by Rho GTPase. J. Biol. Chem. 1998;273:24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 18.Sugimoto M, Nakayama M, Goto TM, Amano M, Komori K, Kaibuchi K. Rho-kinase phosphorylates eNOS at threonine 495 in endothelial cells. Biochemical and Biophysical Research Communications. 2007;361:462. doi: 10.1016/j.bbrc.2007.07.030. [DOI] [PubMed] [Google Scholar]
- 19.Yetik-Anacak G, Catravas JD. Nitric oxide and the endothelium: History and impact on cardiovascular disease. Vascular Pharmacology. 2006;45:268. doi: 10.1016/j.vph.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 20.Hirooka Y, Shimokawa H, Takeshita A. Rho-kinase, a potential therapeutic target for the treatment of hypertension. Drug News Perspect. 2004;17:523–527. doi: 10.1358/dnp.2004.17.8.863696. [DOI] [PubMed] [Google Scholar]
- 21.Sandirasegarane L, Kester M. Enhanced Stimulation of Akt-3/Protein Kinase B-[gamma] in Human Aortic Smooth Muscle Cells. Biochemical and Biophysical Research Communications. 2001;283:158. doi: 10.1006/bbrc.2001.4739. [DOI] [PubMed] [Google Scholar]
- 22.Wu JR, Liou SF, Lin SW, Chai CY, Dai ZK, Liang JC, Chen IJ, Yeh JL. Lercanidipine inhibits vascular smooth muscle cell proliferation and neointimal formation via reducing intracellular reactive oxygen species and inactivating Ras-ERK1/2 signaling. Pharmacol Res. 2008 doi: 10.1016/j.phrs.2008.09.015. In press. [DOI] [PubMed] [Google Scholar]
- 23.Westermann D, Riad A, Lettau O, Roks A, Savvatis K, Becher PM, Escher F, Jan Danser AH, Schultheiss H-P, Tschope C. Renin Inhibition Improves Cardiac Function and Remodeling After Myocardial Infarction Independent of Blood Pressure. Hypertension. 2008;52:1068–1075. doi: 10.1161/HYPERTENSIONAHA.108.116350. [DOI] [PubMed] [Google Scholar]
- 24.Tamás IO, Ágota A, Andrea N, Nóra V, Virág K, Anita S, Kornélia S, Zsuzsa E, György V, Éva K, László H, Katalin N, Elen G, Csaba M, Lajos M, Zoltán I, Zsuzsanna I, Balázs S. Applying a ldquoDouble-Featurerdquo Promoter to Identify Cardiomyocytes Differentiated from Human Embryonic Stem Cells Following Transposon-Based Gene Delivery. Stem Cells. 2009;27:1077–1087. doi: 10.1002/stem.45. [DOI] [PubMed] [Google Scholar]
- 25.Brocq ML, Leslie SJ, Milliken P, Megson IL. Endothelial Dysfunction: From Molecular Mechanisms to Measurement, Clinical Implications, and Therapeutic Opportunities. Antioxidants & Redox Signaling. 2008;10:1631–1674. doi: 10.1089/ars.2007.2013. [DOI] [PubMed] [Google Scholar]
- 26.Higashi Y, Sasaki S, Nakagawa K, Matsuura H, Chayama K, Oshima T. Effect of obesity on endothelium-dependent, nitric oxide-mediated vasodilation in normotensive individuals and patients with essential hypertension. Am J Hypertens. 2001;14:1038. doi: 10.1016/s0895-7061(01)02191-4. [DOI] [PubMed] [Google Scholar]
- 27.Li L-J, Geng S-R, Yu C-M. Endothelial Dysfunction in Normotensive Chinese with a Family History of Essential Hypertension. Clinical and Experimental Hypertension. 2005;27:1–8. doi: 10.1081/ceh-200044242. [DOI] [PubMed] [Google Scholar]
- 28.Vecchi A, Garlanda C, Lampugnani MG, Resnati M, Matteucci C, Stoppacciaro A, Schnurch H, Risau W, Ruco L, Mantovani A, Dejana E. Monoclonal antibodies specific for endothelial cells of mouse blood vessels. Their application in the identification of adult and embryonic endothelium. Eur J Cell Biol. 1994;63:247–254. [PubMed] [Google Scholar]
- 29.Zhai P, Yamamoto M, Galeotti J, Liu J, Masurekar M, Thaisz J, Irie K, Holle E, Yu X, Kupershmidt S, Roden DM, Wagner T, Yatani A, Vatner DE, Vatner SF, Sadoshima J. Cardiac-specific overexpression of AT1 receptor mutant lacking Gαq/Gαi coupling causes hypertrophy and bradycardia in transgenic mice. J. Clin. Invest. 2005;115:3045–3056. doi: 10.1172/JCI25330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhai P, Galeotti J, Liu J, Holle E, Yu X, Wagner T, Sadoshima J. An Angiotensin II Type 1 Receptor Mutant Lacking Epidermal Growth Factor Receptor Transactivation Does Not Induce Angiotensin II-Mediated Cardiac Hypertrophy. Circ Res. 2006;99:528–536. doi: 10.1161/01.RES.0000240147.49390.61. [DOI] [PubMed] [Google Scholar]
- 31.Hein L, Stevens ME, Barsh GS, Pratt RE, Kobilka BK, Dzau VJ. Overexpression of angiotensin AT1 receptor transgene in the mouse myocardium produces a lethal phenotype associated with myocyte hyperplasia and heart block. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:6391–6396. doi: 10.1073/pnas.94.12.6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paradis P, Dali-Youcef N, Paradis FoW, Thibault Gt, Nemer M. Overexpression of angiotensin II type I receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:931–936. doi: 10.1073/pnas.97.2.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffmann S, Podlich D, Hahnel B, Kriz W, Gretz N. Angiotensin II Type 1 Receptor Overexpression in Podocytes Induces Glomerulosclerosis in Transgenic Rats. J Am Soc Nephrol. 2004;15:1475–1487. doi: 10.1097/01.asn.0000127988.42710.a7. [DOI] [PubMed] [Google Scholar]
- 34.Reckelhoff JF, Romero JC. Role of oxidative stress in angiotensin-induced hypertension. Am J Physiol Regul Integr Comp Physiol. 2003;284:R893–R912. doi: 10.1152/ajpregu.00491.2002. [DOI] [PubMed] [Google Scholar]