

Abstract
DNA methylation patterns are set up early in mammalian development and are then copied during the division of somatic cells. A long-established model for the maintenance of these patterns explains some, but not all, of the data that is now available. We propose a new model, which suggests that maintenance of DNA methylation relies not only on the recognition of hemi-methylated DNA by the methyltransferase DNMT1 but also on the localization of the DNMT3A and DNMT3B enzymes to specific chromatin regions containing methylated DNA.
DNA Methylation as a Somatically Heritable Epigenetic Mark
The methylation of cytosine residues within the CpG dinucleotide in animals (and other contexts in plants) has profound effects on gene expression and is essential for mammalian development. Distribution of CpG dinucleotides is asymmetric in the human genome, with CpG rich and CpG poor regions, and it is the only dinucleotide that occurs less frequently than expected in DNA. Not all CpG sites are methylated and DNA methylation patterns are tissue specific and are involved in several key physiological processes including X-chromosome inactivation, imprinting, and silencing of germ line specific genes and repetitive elements. Patterns of methylation are perturbed in important human diseases, such as imprinting disorders and cancer, so understanding how these patterns are set up and maintained is of great importance. It is generally thought that patterns are established during embryonic development and then are faithfully inherited in somatic cells by a “maintenance” mechanism1,2.
DNA methylation is unique among epigenetic marks, such as histone modifications, in that the known biochemistry of the process can be used to partially explain somatic inheritance of epigenetic states. The mechanisms by which DNA methylation patterns could be inherited through generations of somatic cells were first hypothesized more than 30 years ago in two seminal papers published by Riggs3 and Holliday and Pugh4. The main hypotheses annunciated in those papers were that DNA methylation could alter gene expression profiles by influencing the binding affinities of transcription factors or other proteins to DNA, that patterns of DNA methylation would exist and that these patterns would vary in different cell types. The key to somatic inheritance was proposed to be the existence of enzymes that preferentially catalyze the methylation of hemi-methylated DNA that is generated during DNA replication. That is, when a methylated CpG dinucleotide is replicated the C on the nascent strand is initially unmethylated and such enzymes would replicate the parental pattern of methylation. This prediction led to the idea of “maintenance DNA methyltransferases” which could ensure somatic inheritance by copying patterns established in the early embryo by “de novo methyltransferases” or so-called “switch enzymes”4.
This suggestion that eukaryotes might have two classes of DNA methyltransferases (maintenance and de novo methyltransferases) implied that DNA methylation in eukaryotes is different to that in prokaryotes; prokaryotes have only one DNA methyltransferase per strain when they contain modified bases in their DNA. Fewer sites (cytosines or adenines) are modified in prokaryotes because the sequence that is recognized as a substrate for methylation is generally more complex; it often involves a palindrome containing four or more nucleotides and therefore occurs less frequently than the CpG used in eukaryotes. Also, prokaryotes do not have patterns of methylated and unmethylated sites because all the recognition sequences are modified on both strands of the palindrome. Prokaryotes are capable of maintaining DNA methylation because their methyltransferases are equally effective on unmethylated and hemi-methylated DNA5. As a result, the daughter strand is rapidly methylated after DNA synthesis. The key difference then between eukaryotes and prokaryotes was suggested to be a DNA methyltransferase with a preference for hemi-methylated DNA.
Although a model of a maintenance methyltransferase reproducing the pattern of methylation after each round of replication (which will be described in more detail later) has served us well for many years, experimental observations have been accumulating that do not fit with this simple model. For example, mouse embryonic stem cells (ES) containing the “maintenance” enzyme Dnmt1 as the sole DNA methyltransferase show virtually no methylation of imprinted genes and repeats and gradually lose methylation of other sequences with increasing numbers of divisions6-8. Although ES cells may not be an ideal model for studying methylation, it seems timely to reassess the data on the mechanism of DNA methylation inheritance. Here we identify where current data does not fit with the established model and propose a revised model. The focus of this discussion is mammalian DNA methylation; plants have more complicated systems to ensure stability of methylation patterns.
Evidence for the established model
Much experimental evidence has accumulated to support the original proposals3,4 for the existence of de novo and maintenance methyltransferases. The two so-called de novo methyltransferases, Dnmt3a and 3b, were cloned by Okano et al.8, and are thought to be responsible for establishing the pattern of methylation in embryonic development (Fig. 1). Just like prokaryotic methyltransferases, these enzymes show equal activities on hemi- and unmethylated DNA. They are highly expressed in embryonic stem cells and down regulated in differentiated cells. This is consistent with the need for them to set up methylation patterns early on and with the assumption that they are then dispensable. However, even though they are down regulated, they are still expressed in somatic cells. The exception is the DNMT3A2 isoform, which can be silenced in somatic cells by de novo methylation of its own promoter (unpublished observations). Dnmt3a and 3b are both needed for the embryonic or neonatal viability of mice and mutations in DNMT3B are responsible for the rare human disease ICF syndrome9,10.
Figure 1. Current model for the establishment and inheritance of DNA methylation patterns.
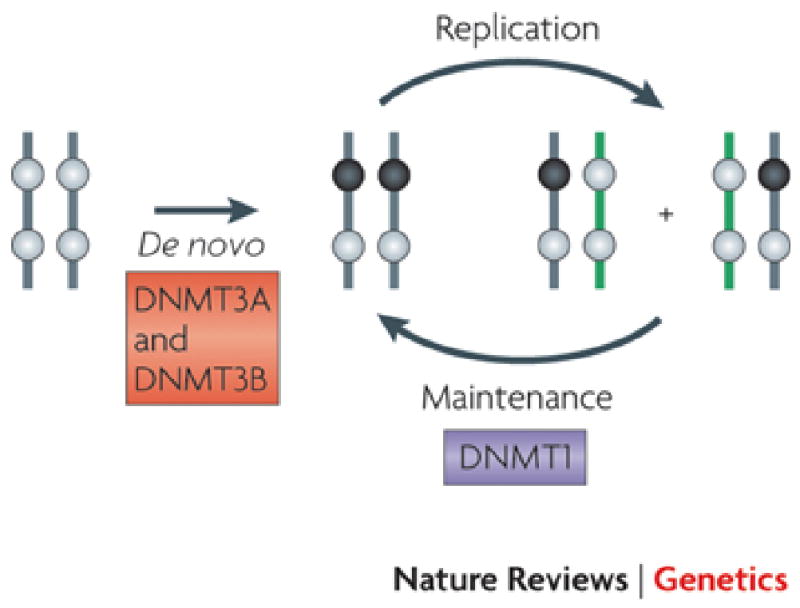
The current model for the establishment and inheritance of DNA methylation patterns relies heavily on the original hypotheses of Riggs3 and Holliday and Pugh4 as exemplified in several recent reviews1,2. The basis of this model is that DNA methylation patterns are established in germ cells and in developing embryos by the activity of de novo DNA methyltransferases DNMT3A and DNMT3B. Subsequently, methylation patterns (● methylated CpG site; ○ unmethylated CpG site) are inherited after DNA replication primarily due to the activity of DNMT1 which has a preference for hemi-methylated sites generated through DNA synthesis. The concept is that the enzyme is copying a pattern which is present on naked DNA. The model seems adequate in principle, however its failure to incorporate a correction mechanism or the constraints of chromatin structure suggests that additional factors are involved in copying DNA methylation patterns in mammalian cells.
The postulated “maintenance enzyme” Dnmt1 was cloned by Bestor et al.11 and shows a marked preference for hemi-methylated DNA12,13, although it also has de novo methyltransferase activity14. DNMT1 has domains that can interact with the DNA polymerase processivity factor PCNA15, which could ensure its localization to the replication fork. It is also possible that the ubiquitin-like PHD and RING finger domain containing protein UHRF116-20 could fulfill a similar function (the role of which is discussed below). DNMT1 is transcribed mostly during the S-phase of the cell cycle21, when it is most needed to methylate newly generated hemi-methylated sites. Knockout experiments have shown that this enzyme is responsible for the bulk of methylation in mouse cells and it is essential for embryonic development22. Interestingly, however, the enzyme is dispensable for the growth of embryonic stem cells, although differentiated cells immediately die, via a p53-associated process, in DNMT1 knockouts21. DNMT1 also appears to be essential for the viability of cancer cells since complete knockout of the enzyme results in HCT116 cell death23.
The biochemical and genetic evidence therefore favor the hypotheses that the de novo enzymes establish methylation patterns and that the maintenance DNMT1 is mostly responsible for copying the patterns and that the two types of enzymes have essentially non-overlapping functions. Recent excellent reviews1,2 have once again emphasized this division of function between the two enzyme types in establishing and maintaining DNA methylation patterns.
Problems with the established model
Proofreading
Although the bulk of the evidence is consistent with the original hypotheses regarding establishing and maintaining eukaryotic DNA methylation patterns, there are several key facts which do not fit. Foremost among these is a conceptual one regarding the lack of a component for error correction. All replicating systems, such as DNA synthesis, need some proofreading mechanism to ensure that the fidelity of patterns is maintained. Thus it is unlikely a pattern established early in development could be maintained simply by copying, without some mechanism to ensure that methylated sites remain methylated and unmethylated sites remain unmethylated. It is therefore doubtful that the genome can be kept methylated by the DNMT1 enzyme alone.
Levels of hemi-methylation
The established model predicts that hemi-methylated sites should be found in low abundance because the majority of them would be immediately converted into sites methylated on both DNA strands after replication. Classic experiments by Bird24 showed that the number of hemi-methylated sites was indeed small, thus providing the first experimental evidence for the establishment and maintenance model. However, using more sensitive approaches, we have found that hemi-methylation can be observed in a measurable proportion of CpG sites in single copy sequences but more so in repeat sequences7. Indeed, if the two de novo enzymes (Dnmt3A and 3B) are knocked out, then up to 30% of CpG sites in mouse repeats are hemi-methylated, suggesting that both Dnmt3 enzymes might help to keep these sites fully methylated. Furthermore, as mentioned earlier, Dnmt1 alone cannot maintain methylation by itself and a gradual loss of methylation occurs with division in ES cells lacking Dnmt3a and 3b6,7. The observations strongly suggest that ongoing methylation by the de novo enzymes is required, otherwise these sequences would eventually lose their methylation as cells divide.
Methylation fidelity
Since a given CpG site can either be methylated or not it might be expected that methylation levels would be 0% or 100% for a given allele or 50% if one allele was methylated and the other not. However quantitation of methylation levels of specific CpG sites, which measures the average of all the DNA molecules in a tissue seldom gives such clear-cut values. Instead, stable inheritance of average methylation levels of specific sites is found in mouse tissues and cell lines25. These results, and findings of Pfeifer et al.26, led Riggs and Xiong27 to suggest that the average state is maintained by a stochastic process requiring on-going de novo methylation. The data exclude the possibility that it is simply the faithful copying of a series of heterogeneous patterns set up in development which is causing the heterogeneity. Studies with hairpin-bisulfite PCR to measure methylation in dividing human lymphocytes have clearly demonstrated on-going de novo methylation and molecule to molecule variation in patterns28.
CpG islands
The presence of CpG islands in mammalian DNA also raises several questions with regard to the maintenance of methylation status. Although CpG islands make up less than 1% of the total genomic DNA they have received a great deal of attention because of their preferential localization to the start sites of >50% of human genes. Most CpG islands remain unmethylated in somatic cells, independently of the state of gene expression, with the exception of genes on the inactive X-chromosome, germ cell specific genes and a minority of other genes29.
Although CpG islands on active or inactive genes remain unmethylated in somatic cells there is nothing inherently “unmethylatable” about them, so how do they remain methylation-free over many cell divisions? It is often hypothesized that they are somehow “protected” from methylation yet they are readily accessible to exogenously added soluble DNA methylases. For example, the unmethylated CpG islands in the p16, MLH1 or GRP78 genes can be rapidly and efficiently methylated in a chromatin content by the Sss1 methylase which methylates CpG sites30-32. This observation suggests that if there were free endogenous DNA methylases within the cell then many CpG islands would become de novo methylated with time. The fact that they do not, suggests that the DNA methyltransferases within eukaryotic cells are not free, but are sequestered with other nuclear components so that they are compartmentalized in order to fulfill their specific functions in the nucleus.
Active demethylation could also be a potential mechanism to “clean up” aberrant methylation inadvertently acquired during cell division. To date, there have been no clear demonstrations of demethylase enzymes33. However recent discoveries of 5-hydroxymethylcytosine in mammalian DNA34 and the demonstration that TET proteins can oxidize 5-methylcytosine to the 5′-hydroxymethylcytosine35 provide potential routes to active demethylation. Nevertheless, it remains to be seen whether these pathways play roles in maintaining the methylation-free state or are potentially involved in gene activation.
However, when a CpG island does become methylated, it might present a particular challenge to the cell in terms of methylation maintenance, because of the high density of methylated sites in comparison to the rest of the genome28. The profound asymmetry of the genome with respect to the distribution of CpG-rich and -poor regions may therefore necessitate the existence of mechanisms in addition to DNMT1 to maintain the high-densities of 5-methylcytosines in methylated CpG islands. Close scrutiny of published genomic sequences of heavily (but not completely) methylated CpG islands shows that the patterns of methylation present on individual DNA molecules are quite heterogeneous7,36 Similarly, heterogeneous patterns of hemimethylated sites have been detected by hairpin PCR in the heavily methylated FMR CpG island in human lymphocytes yet the hypermethylated island remains methylated after many cell division28. These data do not support the idea that there is a strict copying of the pattern from parent DNA molecule to daughter molecule. Rather, they suggest that the methylation “state” is being maintained by a mechanism that includes factors in addition to DNMT1.
Role of Accessory Proteins and Chromatin in DNA Methylation
The original model for methylation maintenance (Fig. 1) was constructed without consideration of the fact that most DNA does not exist as a naked molecule within eukaryotes, but is wrapped up in nucleosomes. Also, as mentioned above, the DNA methyltransferases are apparently not available as soluble enzymes in the nucleus, but are compartmentalized by interaction with nuclear components such as nucleosomes37 Thus it is likely that chromatin structure and the enzymes that modify it will have an important impact on the maintenance of methylation. In other words, to ensure correct maintenance of DNA methylation it might be necessary to copy components of the chromatin structure, in addition to “reading” the DNA methylation mark.
Accessory proteins
The roles of the different DNMTs and several chromatin associated proteins in directing and maintaining DNA methylation have been extensively studied by genetic approaches (Table 1). Many knockout studies in germ cells and embryos have demonstrated the central roles of Dnmt3a, 3b and 3L in setting up the patterns of specific and sometimes overlapping sequence classes and the severe phenotypic consequences of deleting these functions. Some examples of chromatin proteins such as G9a, Lsh and Lsd1 among others are shown in Table 1 although the details of how these interactions effect methylation remain sketchy. As mentioned above, Dnmt1 can interact with PCNA and associate with the replication fork during DNA replication15. Immunofluorescence studies have confirmed that PCNA and Dnmt1 co-localize at replication forks and this model of PCNA bringing Dnmt1 to the site of replication still receives considerable attention2 although it has been questioned by studies showing that disruption of the PCNA-DNMT interaction causes only small changes in methylation38,39. Recently it has been shown that UHRF1 (also known as NP95) is also essential for maintaining DNA methylation in plant and mammalian cells17-20. Since UHRF1 preferentially binds to hemi-methylated sites Dnmt1 might be recruited to DNA replication foci18,20 and to hemi-methylated sites present in bulk DNA even after replication.
Table 1.
Genetic Approaches to Study Mechanisms of DNA Methylation
| Species | Disrupted Gene(s) | Change in DNA Methylation | Sequence Specificity | Somatic Phenotype | Reference |
|---|---|---|---|---|---|
| Mouse Germ Cells | Dnmt3a | ↓ | H19, Dlk1/Gtl2, and SineB1 also Rasgrf1, IAPs and Line 1 | 63 | |
| Dnmt3b | ↓ | Satellites, IAPs, Line 1 and Rasgrf1 | 63 | ||
| Dnmt3L | ↓ | All of above sequences | 63,64 | ||
| Mouse Embryonic Stem Cells and Embryos | Dnmt 1 | ∼80% ↓ | Non specific | Lethal; (p53-dependent apoptosis in somatic cells) | 8,22,65 |
| Dnmt 3a | No change | N/A | Lethal (postnatal) | 8 | |
| Dnmt 3b | Not dramatic | Centromeric repeats ↓ | Lethal | 8 | |
| Dnmt 3a & 3b | 20% ↓ and ↑ hemi-methylation | Imprined genes↓ Repeats ↓ | Lethal | 7,8 | |
| Dnmt 1/3a/3b | No DNA methylation | All CpG sites | N/A | 66 | |
| Dnmt3L | Altered | Imprinted genes ↓ | Sterile in both sexes | 67 | |
| Dnmt 2 | No change | tRNA methylation | None | 68,69 | |
| G9a | Dramatic ↓ | Repeats ↓ | Lethal | 43,70 | |
| UHRF1 (Np95) | ∼80% ↓ | Hemi-methylated sites ↑ | Lethal (gastrulation) | 20 | |
| Lsd1 | ∼40% ↓ | Non specific | Lethal (gastrulation) | 71 | |
| Lsh | 45-67% ↓ | Non specific | Lethal (perinatal) | 53 | |
| Human Somatic Cells | DNMT1 | ↑ Hemi-methylation | CpG poor ↓ | Lethal in somatic cells | 38,72,73 |
| DNMT3B (HCT116) | Not dramatic | Non specific | ICF | 74 | |
| DNMT1 (hypomorphs) and 3B | Dramatic ↓ | Non specific | N/A | 74 |
Chromatin context
Recent genome-wide studies have clearly shown that certain histone variants and histone tail modifications are refractory or counter-distributed with DNA methylation40. For example, the histone variant H2A.Z, which is preferentially located at the start sites of genes, is anti-correlated with the distribution of DNA methylation41. Likewise trimethylation of lysine 4 on histone H3 (H3K4me3) is associated with active genes and is therefore also anti-correlated with DNA methylation1. Perhaps one of the most definitive biochemical demonstrations of the potential mechanisms for de novo methylation during germ cell development came from the seminal work of Ooi et al.42 who showed that the Dnmt3a variant, Dnmt3a2 and the non-catalytic Dnmt3L, which are both expressed in germ cells, form a cage-like structure that can accommodate a nucleosome. This structure is possibly responsible for de novo methylation during development. A key observation was that the presence of H3K4me1, me2 or me3 prevents the association of this complex with the nucleosome. This suggested that DNA in a nucleosome bearing specific histone modifications might be differentially susceptible to methylation and suggested how methylation patterns could be set up around genes but excluded from the start sites containing this active mark.
Several chromatin marks or enzymes, such as the histone methyltransferases EZH2 (a component of the polycomb repressive complex 2) and G9a, that are associated with transcriptional silencing have been suggested to recruit DNMT3A/3B to particular regions of DNA. There is good evidence that G9a recruits Dnmt3a/3b43,44, but recruitment of DNMT1 and DNMT3A by EZH2 is not as well substantiated45-47. Several other repressive proteins such as heterochromatin protein 1 (HP1)48,49 and histone deacetylase (HDAC)50 have also been suggested to play a role in recruiting DNA methyltransferases to particular regions of silent chromatin.
Since DNA in a nucleosomal complex has been shown to be refractory to induced DNA methylation in the test tube51,52 it might well be that nucleosomes have to be moved or altered in order to allow methylation to occur. In this regard it is particularly interesting that the chromatin remodeling factor Lsh has been found to be implicated in establishment and maintenance of methylation patterns in developing mice53.
The data presented above suggest maintenance of DNA methylation might be more complex than described by existing models. Therefore we argue that some modifications of the existing models for maintenance methylation are necessary.
A Revised Model for Maintenance of DNA Methylation
We propose an updated model for DNA methylation maintenance which takes account of the existing information and can provide a framework for future experimentation to define the mechanism of this key physiological and pathological process (Fig. 2). The localization of DNMT1 to the replication fork, the interaction of DNMT3A and 3B with nucleosomes bearing specific modifications, and cooperation between different methyltransferase enzymes are all needed for the maintenance of DNA methylation especially in repeats and imprinted genes6-8. We propose that the bulk of DNA methylation in dividing cells is indeed maintained by Dnmt1 - the most abundant DNMT in the cell54 – in conjunction with UHRF1. With its marked preference for hemi-methylated sites DNMT1 is ideally suited for the maintenance of most DNA methylation. In this sense, the enzyme fulfills the original ideas put forward in the classic papers of 1975 (Fig. 1). It is important to emphasize that the main role of DNMT1 is in reading the DNA sequence and applying methyl groups opposite to newly replicated hemi-methylated CpG sites. In other words the enzyme “reads” the modifications on the DNA without regard for the chromatin configuration in which that particular piece of DNA is located.
Figure 2. Revised model for the maintenance of DNA methylation patterns.

In this revised model the DNMT3A and 3B enzymes remain bound to chromatin in somatic cells, and in particular to nucleosomes that contain methylated CpG sites (A). When DNA replicates (B) the bulk of methylation copying is achieved by DNMT1, which is the predominant DNA methylase in the cell and is localized to the replication fork by PCNA, and possibly by UHFR1. Soon after DNA replication we propose that DNMT3A and 3B complete the methylation process and correct errors left by the DNMT1 enzyme. These enzymes do not “read” the parental strand for DNA methylation patterns but rather methylate newly replicated CpG sites which are unmethylated because the are compartmentalized to the chromatin region containing methylated DNA37. In this way, the enzymes function similarly to other chromatin modifying enzymes such as EZH2 which are also localized to their product after methyl transfer. As described in the text, DNMT3A and 3B seem to be compartmentalized to CpG islands and repetitive elements whereas DNMT1 has a preferential ability to maintain the majority of DNA methylation in the cell which is found at non-CpG islands. UHFR1 might help locate sites missed during the replication process and be responsible for the delayed methylation seen after the DNA has left the replication fork.
In addition to its action at the replication fork, there is also some evidence that DNMT1 is able to perform error correction. Tagging of newly replicated DNA with bromodeoxyuridine followed by methylation analysis shows that complete methylation of the CpG sites within the newly replicated DNA can be accomplished after the DNA has left the replication fork in mouse embryonic stem cells which have Dnmt1 as the sole known active DNA methyltransferase7. Similar conclusions were reached by Schermelleh et al.39 using a different approach in mouse embryonic stem cells. Furthermore, in human cancer cell lines in which the PCNA binding site of DNMT1 has been disabled by genetic disruption, DNMT1 is able to complete the conversion of some of the hemi-methylated sites into fully methylated sites, as a function of time after the DNA has left the replication fork38,55. UHRF1 might be involved in recruiting DNMT1 to hemi-methylated sites away from the replication fork but this has not yet been shown.
It seems possible that the high frequency of occurrence of 5-methylcytosines in methylated CpG islands and repeats might represent a challenge to the maintenance process because of the rapid generation of hemi-methylated sites during DNA synthesis. We argue that cooperativity between different DNMT enzymes might be required in mammalian cells to maintain DNA methylation of these densely methylated regions. Cooperativity might also be responsible for maintaining the methylation of repetitive elements such as Alus and LINES7 which, as mentioned previously, contain a high level of hemi-methylation in ES cells lacking Dnmt3a and 3b. We propose that Dnmt3a and 3b are associated with specific regions of DNA that need to be maintained as highly methylated through recruitment to specific nucleosomal contexts. For example, G9a which is associated with maintaining H3K9me3 and is frequently found at repeats might be responsible for recruiting DNMT3A and 3B to complete methylation after the DNA has left the replication fork. Indeed Schlesinger et al.56 have shown that DNMT3A is associated with methylated CpG islands. We have confirmed their results with regard to CpG islands and also with repeat elements7,37. Our data also show the strong anchoring of both DNMT3A and DNMT3B but not DNMT1 to nucleosomes37 which presumably ensures their effective compartmentalization to methylated regions and does not allow for “free” enzymes to be present in the nucleus.
The DNMT3 enzymes anchored to nucleosomes containing methylated DNA37 do not “read” the methylation on the existing DNA sequence but are proposed rather to methylate sites missed by DNMT1 activity at the replication fork. In this sense the DNMT3 enzymes act like prokaryotic enzymes which are capable of maintaining methylation without regard for whether sites are hemi-methylated or completely unmethylated. If regions of DNA that are methylated remain associated with the DNMT3A and B after replication, this would ensure that methylation state of a region is maintained, rather than a specific methylation pattern. The evidence for maintenance of state rather than an exact CpG-by-CpG pattern comes from the observation that most methylated CpG islands do not have consistent DNA methylation patterns across all molecules7,36 and that unmethylated sites within methylated CpG islands have a high probability of undergoing de novo methylation28. This is a key conceptual difference from the standard model of methylation maintenance; in our revised model which is similar to that proposed by Riggs and Xiong27 the methylation state in general is being copied rather than the specific patterns of methylated sites.
The key to the supportive roles for DNMT3A and 3B in DNA methylation maintenance is the continued association of the enzymes with their products after the enzymatic reaction has occurred. Indeed there are several examples with respect to chromatin modifying enzymes such as the EZH2 enzyme which remains associated with its H3-K27me3 product suggesting that this mechanism is quite common and may be responsible for the inheritance of epigenetic states57,58.
The revised model lessens the earlier emphasis on “de novo” and “maintenance” DNMTs with non-overlapping functions. It emphasizes the need for continual cooperativity between the DNMTs to maintain DNA methylation patterns after the DNA has left the replication fork. It also brings together the two separate pathways which have been suggested to be necessary for inheritance of DNA methylation and histone modification patterns respectively.59
Future perspectives
The initial description of the DNA helix immediately and famously gave insights into the probable mechanisms for genetic inheritance60. It has been far more difficult to decipher the mechanisms of epigenetic inheritance in somatic cells and indeed there is still discussion as to what is encompassed by the term “epigenetics”61. We think that the model proposed here - a hybrid between the semi-conservative reading of the pre-existing methylated groups on the DNA3,4 and interpretation of chromatin states – provides an explanation for the current data on DNA methylation inheritance as has been suggested for propagation of other epigenetic marks57. Unanswered questions include the biochemical mechanisms responsible for the compartmentalization of DNMT3s to nucleosomes, the proportion of methylation performed at the replication fork and also immediately after in the context of reassembly of nucleosomes. The exact roles of chromatin modifications and accessory proteins also need to be deciphered. Literally thousands of publications have dealt with the mechanisms by which proofreading and repair of genetic information occurs during DNA replication. With the current interest in epigenetics and epigenomics62, it seems that more attention should be paid to understanding the biochemistry of epigenetic inheritance in somatic cells. The details of how DNA methylation patterns are maintained are critical to the structure of the human epigenome in normal and pathological conditions.
Acknowledgments
This work was supported by the National Institutes of Health grants R37 CA082422 (PAJ), 5R01 CA083867 (PAJ) and 5 R01 CA124518 (GL).
Contributor Information
Peter A. Jones, Email: jones_p@ccnt.usc.edu.
Gangning Liang, Email: gliang@usc.edu.
References
- 1.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 2.Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol. 2009;10:192–206. doi: 10.1038/nrm2640. [DOI] [PubMed] [Google Scholar]
- 3.Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975;14:9–25. doi: 10.1159/000130315. [DOI] [PubMed] [Google Scholar]
- 4.Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–232. [PubMed] [Google Scholar]
- 5.Smith HO, Kelly SV. Methylases of the Type II Restriction-Modification Systems. In: Razin A, Cedar H, Riggs AD, editors. DNA Methylation Biochemistry and Biological Significance. Springer-Verlag; New York: 1984. pp. 39–71. [Google Scholar]
- 6.Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol. 2003;23:5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang G, et al. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol Cell Biol. 2002;22:480–491. doi: 10.1128/MCB.22.2.480-491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 9.Hansen RS, et al. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci USA. 1999;96:14412–14417. doi: 10.1073/pnas.96.25.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu GL, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- 11.Bestor T, Laudano A, Mattaliano R, Ingram V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzyme is related to bacterial restriction methyltransferases. J Mol Biol. 1988;203:971–983. doi: 10.1016/0022-2836(88)90122-2. [DOI] [PubMed] [Google Scholar]
- 12.Bestor TH, Ingram VM. Two DNA methyltransferases from murine erythroleukemia cells: purification, sequence specificity, and mode of interaction with DNA. Proc Natl Acad Sci USA. 1983;80:5559–5563. doi: 10.1073/pnas.80.18.5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hermann A, Goyal R, Jeltsch A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem. 2004;279:48350–48359. doi: 10.1074/jbc.M403427200. [DOI] [PubMed] [Google Scholar]
- 14.Pradhan S, Bacolla A, Wells RD, Roberts RJ. Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J Biol Chem. 1999;274:33002–33010. doi: 10.1074/jbc.274.46.33002. [DOI] [PubMed] [Google Scholar]
- 15.Chuang LS, et al. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science. 1997;277:1996–2000. doi: 10.1126/science.277.5334.1996. [DOI] [PubMed] [Google Scholar]
- 16.Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818–821. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- 17.Avvakumov GV, et al. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008;455:822–825. doi: 10.1038/nature07273. [DOI] [PubMed] [Google Scholar]
- 18.Bostick M, et al. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–1764. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- 19.Hashimoto H, et al. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008;455:826–829. doi: 10.1038/nature07280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharif J, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–912. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- 21.Robertson KD, Keyomarsi K, Gonzales FA, Velicescu M, Jones PA. Differential mRNA expression of the human DNA methyltransferases (DNMTs) 1, 3a and 3b during the G0/G1 to S phase transition in normal and tumor cells. Nucleic Acids Res. 2000;28:2108–2113. doi: 10.1093/nar/28.10.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 23.Chen T, et al. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 24.Bird AP. Use of restriction enzymes to study eukaryotic DNA methylation: II. The symmetry of methylated sites supports semi-conservative copying of the methylation pattern. J Mol Biol. 1978;118:49–60. doi: 10.1016/0022-2836(78)90243-7. [DOI] [PubMed] [Google Scholar]
- 25.Turker MS, Swisshelm K, Smith AC, Martin GM. A partial methylation profile for a CpG site is stably maintained in mammalian tissues and cultured cell lines. J Biol Chem. 1989;264:11632–11636. [PubMed] [Google Scholar]
- 26.Pfeifer GP, Steigerwald SD, Hansen RS, Gartler SM, Riggs AD. Polymerase chain reaction-aided genomic sequencing of an X chromosome-linked CpG island: methylation patterns suggest clonal inheritance, CpG site autonomy, and an explanation of activity state stability. Proc Natl Acad Sci USA. 1990;87:8252–8256. doi: 10.1073/pnas.87.21.8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riggs AD, Xiong Z. Methylation and epigenetic fidelity. Proc Natl Acad Sci USA. 2004;101:4–5. doi: 10.1073/pnas.0307781100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laird CD, et al. Hairpin-bisulfite PCR: assessing epigenetic methylation patterns on complementary strands of individual DNA molecules. Proc Natl Acad Sci USA. 2004;101:204–209. doi: 10.1073/pnas.2536758100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Illingworth RS, Bird AP. CpG islands--‘a rough guide’. FEBS Lett. 2009;583:1713–1720. doi: 10.1016/j.febslet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 30.Fatemi M, et al. Footprinting of mammalian promoters: use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res. 2005;33:e176. doi: 10.1093/nar/gni180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gal-Yam EN, et al. Constitutive nucleosome depletion and ordered factor assembly at the GRP78 promoter revealed by single molecule footprinting. PLoS Genet. 2006;2:e160. doi: 10.1371/journal.pgen.0020160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin JC, et al. Role of nucleosomal occupancy in the epigenetic silencing of the MLH1 CpG island. Cancer Cell. 2007;12:432–444. doi: 10.1016/j.ccr.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ooi SK, Bestor TH. The colorful history of active DNA demethylation. Cell. 2008;133:1145–1148. doi: 10.1016/j.cell.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 34.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalgo ML, et al. The role of DNA methylation in expression of the p19/p16 locus in human bladder cancer cell lines. Cancer Res. 1998;58:1245–1252. [PubMed] [Google Scholar]
- 37.Jeong S, et al. Selective anchoring of DNA methyltransferases 3A/3B to nucleosomes containing methylated DNA. Mol Cell Biol. doi: 10.1128/MCB.00484-09. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Egger G, et al. Identification of DNMT1 (DNA methyltransferase 1) hypomorphs in somatic knockouts suggests an essential role for DNMT1 in cell survival. Proc Natl Acad Sci USA. 2006;103:14080–14085. doi: 10.1073/pnas.0604602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schermelleh L, et al. Dynamics of Dnmt1 interaction with the replication machinery and its role in postreplicative maintenance of DNA methylation. Nucleic Acids Res. 2007;35:4301–4312. doi: 10.1093/nar/gkm432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 41.Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature. 2008;456:125–129. doi: 10.1038/nature07324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ooi SK, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong KB, et al. DNA methylation in ES cells requires the lysine methyltransferase G9a but not its catalytic activity. EMBO J. 2008;27:2691–2701. doi: 10.1038/emboj.2008.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Epsztejn-Litman S, et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol. 2008;15:1176–1183. doi: 10.1038/nsmb.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smallwood A, Esteve PO, Pradhan S, Carey M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007;21:1169–1178. doi: 10.1101/gad.1536807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vire E, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 47.Kondo Y, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet. 2008;40:741–750. doi: 10.1038/ng.159. [DOI] [PubMed] [Google Scholar]
- 48.Fuks F, Hurd PJ, Deplus R, Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31:2305–2312. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Honda S, Selker EU. Direct interaction between DNA methyltransferase DIM-2 and HP1 is required for DNA methylation in Neurospora crassa. Mol Cell Biol. 2008;28:6044–6055. doi: 10.1128/MCB.00823-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robertson KD, et al. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25:338–342. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- 51.Robertson AK, Geiman TM, Sankpal UT, Hager GL, Robertson KD. Effects of chromatin structure on the enzymatic and DNA binding functions of DNA methyltransferases DNMT1 and Dnmt3a in vitro. Biochem Biophys Res Commun. 2004;322:110–118. doi: 10.1016/j.bbrc.2004.07.083. [DOI] [PubMed] [Google Scholar]
- 52.Gowher H, et al. De novo methylation of nucleosomal DNA by the mammalian Dnmt1 and Dnmt3A DNA methyltransferases. Biochemistry. 2005;44:9899–9904. doi: 10.1021/bi047634t. [DOI] [PubMed] [Google Scholar]
- 53.Dennis K, Fan T, Geiman T, Yan Q, Muegge K. Lsh, a member of the SNF2 family, is required for genome-wide methylation. Genes Dev. 2001;15:2940–2944. doi: 10.1101/gad.929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walsh CP, Bestor TH. Cytosine methylation and mammalian development. Genes Dev. 1999;13:26–34. doi: 10.1101/gad.13.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spada F, et al. DNMT1 but not its interaction with the replication machinery is required for maintenance of DNA methylation in human cells. J Cell Biol. 2007;176:565–571. doi: 10.1083/jcb.200610062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schlesinger Y, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–236. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 57.Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- 58.Hansen KH, et al. A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol. 2008;10:1291–1300. doi: 10.1038/ncb1787. [DOI] [PubMed] [Google Scholar]
- 59.Felsenfeld G. A Brief History of Epigenetics. In: Allis CD, Jenuwein T, Reinberg D, editors. Epigenetics. Cold Spring Harbor Laboratory Press; New York: 2007. pp. 15–22. [Google Scholar]
- 60.Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 61.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moving AHEAD with an international human epigenome project. Nature. 2008;454:711–715. doi: 10.1038/454711a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kato Y, et al. Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum Mol Genet. 2007;16:2272–2280. doi: 10.1093/hmg/ddm179. [DOI] [PubMed] [Google Scholar]
- 64.La Salle S, et al. Loss of spermatogonia and wide-spread DNA methylation defects in newborn male mice deficient in DNMT3L. BMC Dev Biol. 2007;7:104. doi: 10.1186/1471-213X-7-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jackson-Grusby L, et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat Genet. 2001;27:31–39. doi: 10.1038/83730. [DOI] [PubMed] [Google Scholar]
- 66.Tsumura A, et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells. 2006;11:805–814. doi: 10.1111/j.1365-2443.2006.00984.x. [DOI] [PubMed] [Google Scholar]
- 67.Bourc'his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–2539. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- 68.Goll MG, et al. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science. 2006;311:395–398. doi: 10.1126/science.1120976. [DOI] [PubMed] [Google Scholar]
- 69.Okano M, Xie S, Li E. Dnmt2 is not required for de novo and maintenance methylation of viral DNA in embryonic stem cells. Nucleic Acids Res. 1998;26:2536–2540. doi: 10.1093/nar/26.11.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tachibana M, et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002;16:1779–1791. doi: 10.1101/gad.989402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang J, et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41:125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 72.Chen T, Li E. Establishment and maintenance of DNA methylation patterns in mammals. Curr Top Microbiol Immunol. 2006;301:179–201. doi: 10.1007/3-540-31390-7_6. [DOI] [PubMed] [Google Scholar]
- 73.Rhee I, et al. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature. 2000;404:1003–1007. doi: 10.1038/35010000. [DOI] [PubMed] [Google Scholar]
- 74.Rhee I, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552–556. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]