

Abstract
In previous work, botryllamides discovered from the marine ascidian Botryllus tyreus were characterized as selective inhibitors of the ABCG2 multidrug transporter. However, the structural basis for this activity could not be established. In this study, botryllamide F, the core botryllamide structure, and botryllamide G, the most potent botryllamide ABCG2 inhibitor, were synthesized along with a series of structural variants for evaluation of structure–activity relationships. The biological activity of synthetic botryllamide analogs implied that the 2-methoxy-p-coumaric acid portion, and the degree of double bond conjugation within this group, were critical for inhibition of ABCG2. However, variations in the substituents on the two aryl groups did not appear to significantly impact the potency or degree of inhibition.
Keywords: Natural products, ABCG2 blocker, Structure-activity relationships, Synthesis
ABCG2 is a member of the ATP-binding cassette (ABC) family of transporters, which pump a wide range of chemically diverse substrates out of cells using energy from ATP hydrolysis1-3. The overexpression of ABCG2 in cancer cells may play an important role in exporting chemotherapeutic agents, such as mitoxantrone, topotecan, irinotecan, doxorubicin, SN-38, and flavopiridol, resulting in cellular resistance to these agents. Accordingly, compounds that inhibit ABCG2 function may prove to be useful therapeutic agents. Although a number of ABCG2 inhibitors have been described, they lack potency or specificity or have toxic effects, and none have been developed as clinical agents.
We recently developed a high-throughput cell-based assay4 to screen compounds for inhibition of ABCG2 activity by blocking efflux of the fluorescent substrate pheophorbide-a (PhA)5,6. A new structural class of ABCG2 inhibitors was characterized, botryllamides A–J from the Micronesian colonial ascidian Botryllus tyreus7. Two of these, I and J, were also novel structures. Despite only modest structural differences, the botryllamides showed variable activity against ABCG2 and variable specificity toward ABCG2 relative to other multidrug transporters [P-glycoprotein (P-gp) and MRP1]. For example, botryllamide G inhibited the efflux of PhA out of ABCG2-overexpressing cells with an IC50 value of 6.9 μM, but not the efflux of rhodamine out of P-gp-overexpressing cells at 50 μM. In contrast, botryllamide A was much less potent against ABCG2 (IC50 = 33 μM), but inhibited the export of rhodamine by P-gp. The difference in selectivity appears to be based on a very small structural difference (the presence or absence of an O-methyl group at C15). However, the structural basis for the range of potencies against ABCG2 could not be established based on the naturally occurring botryllamides that were obtained.
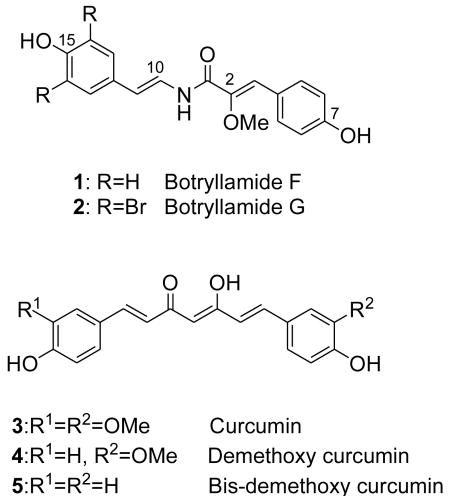
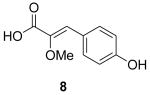
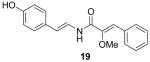
To explore structure-activity relationships (SAR) among the botryllamides with regard to ABCG2, additional structural analogs were synthesized. Given the relatively low yields that were obtained from natural product extracts, a synthetic approach to the botryllamides would also be useful for generation of sufficient material to support further biological characterization and in vivo testing of these compounds. Botryllamide F (1), the core botryllamide structure, was selected as the initial synthetic target. We utilized a strategy in which octopamine was condensed with the C1-C9 portion of 1 to generate an amide bond, and then dehydration of the adjacent hydroxyl group provided the key enamine amide functionality. The C1-C9 fragment, 2-methoxy-p-coumaric acid (8) 8, was synthesized from methylmethoxyacetate (7) and 4-hydroxybenzaldehyde (6). Then 8 was coupled with octopamine hydrochloride, and the three hydroxyl groups were acetylated. The hydroxyl group at C11 was dehydrated with a catalytic amount of K2CO3 to give 19,10. The 1H and 13C NMR spectrum of 1 showed good agreement with those of natural botryllamide F. Subsequently, we synthesized botryllamide G (2), which is the most potent and selective inhibitor of the ABCG2 transporter. The reactions for the synthesis of 2 were similar to those described for 1, apart from using bis-brominated octopamine11. The 1H and 13C NMR spectrum of 2 also showed good agreement with those of natural botryllamide G. Both synthetic botryllamides had comparable potency in the PhA assay as compared to the natural products (data not shown).
In selecting synthetic botryllamide analogs for an SAR study, we took into account the structure of the curcumins (3–5), which have also been reported to inhibit the transport of mitoxantrone and PhA out of ABCG2-overexpressing cells12. The curcumins and the botryllamides have a common biochemical profile of stimulating the ATPase activity of ABCG2. This effect is in contrast to many ABCG2 inhibitors, which reduce ATP hydrolysis by ABCG27,12. Curcumins are the major curcuminoids found in turmeric powder and their structures consist of two hydroxyl substituted phenyl rings joined by an acyclic linker. The general structural features of the botryllamides are quite similar to the curcumins, since they also have two aryl rings joined by an acyclic linker. Differences between these two families of compounds are seen in both the length and functional group composition of the linkers. Botryllamides have an enamine amide group conjugated to the enol ether of an α-keto group, while the cucurmins have a cross conjugated enolized β-diketone functionality. In addition, the cucurmins have a seven carbon long linker that is pseudosymmetrical due to enol tautomerization, while the botryllamides have a nonsymmetrical and shorter linker comprised of five carbons and one nitrogen. Differences in aryl group substitution patterns are also found between the botryllamides and the curcumins.
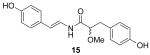
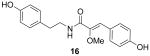
For the SAR study, we initially examined the relationship between the degree of conjugation in the linker portion of the molecules and their inhibitory activity toward ABCG2. We synthesized three analogs: 2,3-dihydro botryllamide F (15), 10,11-dihydro botryllamide F (16), and 2,3,10,11-tetrahydro botryllamide F (17). To synthesize 15, tyramine hydrochloride was coupled with 8 instead of octopamine hydrochloride, and 16 and 17 were prepared by the hydrogenation of 8 and 1, respectively.
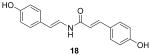
ABCG2 inhibitory activity was evaluated by the cellular accumulation of the ABCG2 specific substrate PhA in cells that over expressed the transporter, as described previously4,13. Potency (IC50) for each compound was estimated from dose-response curves and shown in Table 1. Although a high degree of conjugation in the linker is a common structural feature of both the botryllamides and curcumins, the activities of the three analogs indicated that only maintenance of the Δ2,3 double bond was essential to inhibit ABCG2. The desmethoxy analog (18) was inactive at 100 μM, suggesting that the methoxy group at C2 was also necessary for activity. Synthetic intermediates 8 and 9 were inactive, suggesting that the most significant moiety for inhibitory activity was the 2-methoxy-p-coumaric acid (C1–C9) portion; moreover, the C10-C17 half-styrene moiety in the botryllamides may play a role as an anchor.
Table 1.
ABCG2 inhibitory activity of analogs 8, 9 and 15-18.
| Structure | IC50 | SDa |
|---|---|---|
 |
> 100 μM | |
 |
25.2 μM | ± 1.7 |
 |
> 100 μM | |
 |
> 100 μM | |
 |
> 100 μM | |
 |
> 100 μM |
Standard deviation; n=3-4
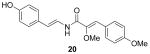
Next, we evaluated the effect of different functional group substitutions on the C4–C9 benzene ring, because the activity data suggested the 2-methoxy-p-coumaric acid portion of the botryllamides was important in interacting with the ABCG2 protein. While the natural botryllamides we isolated possessed a variety of substituents on the C12–C17 (left) aryl ring, the substitution pattern of the C4–C9 (right) aryl ring was generally conserved. Variations in the substituents on the C12–C17 ring did not correlate with ABCG2 inhibition, but they may contribute to specificity. Next, we modified the functional groups on the C4–C9 ring and tested their inhibitory activity against ABCG2.
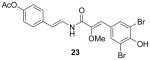
We used various starting materials instead of 4-hydroxybenzaldehyde to synthesize the analogs 21–26. The deshydroxyl derivative 21 retained inhibitory activity, indicating that the phenolic hydroxyl group on C7 was not a required functional group for activity. The observed inhibitory activity of the 6,7-dihydroxyl derivative 22 and the 6,8-dibromo derivative 23 suggested that the binding region on ABCG2 that the C4–C10 ring interacts with is large enough to accommodate additional functionalities. However, the O-benzyl group of compound 26 was too large for adequate binding to the protein, judging from its lack of inhibitory activity. Based on variations of the substituents on the C12-C17 aryl ring observed with the natural botryllamides, and their broad ABCG2 inhibitory activities, an ABCG2 binding site for this portion of the molecule would appear to be relatively large and structurally permissive.
In this study, we established a simple and readily scalable synthetic route to botryllamide F (1) and G (2), and prepared a series of botryllamide analogs. The ABCG2 inhibitory activities of the botryllamide analogs indicated that conjugation through C1 to C9 was necessary for the activity; however, extended conjugation through C10 to C17 was not required. Furthermore, the SAR study suggested that two distinct binding interactions occur with the ABCG2 protein. Thus, this synthetic study provides a means for the continued preclinical evaluation of the botryllamides and it may lead to the development of more potent and selective ABCG2 inhibitors as potential therapeutic agents.
Supplementary Material
Figure 1.

Strategy for the SAR study of botryllamides.
Scheme 1.

Reagents and conditions a) NaOMe, MeOH, reflux, overnight, 75%; b) Octopamine HCl (bis-brominated octopamine for 10), WSCI, HOBt, Et3N, r.t., 4h.; c) Ac2O, Pyridine, r.t., 2h, 2steps 58%; d) K2CO3, DMSO, 98°C, 2h. 45%
Table 2.
ABCG2 inhibitory activity of analogs 19-24.
| Structure | IC50 | SDa |
|---|---|---|
 |
15.4 μM | ± 1.0 |
 |
6.4 μM | ± 0.8 |
 |
18.8 μM | ± 1.2 |
 |
11.1 μM | ± 1.2 |
 |
23.2 μM | ± 2.5 |
 |
>100μM |
Standard deviation; n=3-4
Acknowledgments
We are grateful to Dr. Hiroshi Hirota, RIKEN, JAPAN, for helpful suggestions. This project has been partly supported by Comprehensive Support Programs for Creation of Regional Innovation from Japan Science and Technology Agency. This project has been also funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Supplementary data associated with this article can be found, in the online version, at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Dean M, Annilo T. Annu Rev Genom Human Genet. 2005;6:123. doi: 10.1146/annurev.genom.6.080604.162122. [DOI] [PubMed] [Google Scholar]
- 2.Robey RW, Polgar O, Deeken J, To KW, Bates SE. Cancer Metastasis Rev. 2007;26:39. doi: 10.1007/s10555-007-9042-6. [DOI] [PubMed] [Google Scholar]
- 3.Calcagno AM, Kim IW, Wu CP, Shukla S, Ambudkar SV. Curr Drug Deliv. 2007;4:324. doi: 10.2174/156720107782151241. [DOI] [PubMed] [Google Scholar]
- 4.Henrich CJ, Bokesch HR, Dean M, Bates SE, Robey R, Goncharova EI, Wilson JA, McMahon JB. J Biomol Screen. 2006;11:176. doi: 10.1177/1087057105284576. [DOI] [PubMed] [Google Scholar]
- 5.Jonker JW, Buitelaar M, Wagenaar E, van der Valk MA, Scheffer GL, Scheper RJ, Plosch T, Kuipers F, Oude Elferink RPJ, Rosing H, Beijnen JH, Schinkel AH. Proc Natl Acad Sci USA. 2002;99:15649. doi: 10.1073/pnas.202607599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abbott BL. Hematol Oncol. 2003;21:115. doi: 10.1002/hon.714. [DOI] [PubMed] [Google Scholar]
- 7.Henrich CJ, Robey R, Takada K, Bokesch HR, Bates SE, Shukla S, Ambudkar SV, McMahon JB, Gustafson KR. ACS Chem Biol. 2009;4:637. doi: 10.1021/cb900134c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fürstner A, Domostoj MM, Scheiper B. J Am Chem Soc. 2005;127:11620. doi: 10.1021/ja0541175. [DOI] [PubMed] [Google Scholar]
- 9.Snider BB, Song F, Foxman BM. J Org Chem. 2000;65:793. doi: 10.1021/jo991454l. [DOI] [PubMed] [Google Scholar]
- 10.Preparation of the C1-C9 fragment was accomplished by adding methylmethoxyacetate (7) drop-wise to a solution of 4-hydroxybenzaldehyde (6) and sodium methoxide in MeOH to afford 2-methoxy-p-coumaric acid (8)8. Then 8 was coupled with octopamine hydrochloride, and the three hydroxyl groups were acetylated. After the purification of the triacetate 11 on SiO2, the hydroxyl group at C11 was dehydrated with a catalytic amount of K2CO3 in DMSO at 95°C for 2 h9. The reaction mixture was purified by reversed-phase HPLC (Cosmosil ARII) to give 1.
- 11.Kotoku N, Tsujita H, Hiramatsu A, Mori C, Koizumi N, Kobayashi M. Tetrahedron. 2005;71:7211. [Google Scholar]
- 12.Chearwae W, Shukla S, Limtrakul P, Ambudkar SV. Mol Cancer Ther. 2006;5:1995. doi: 10.1158/1535-7163.MCT-06-0087. [DOI] [PubMed] [Google Scholar]
- 13.PhA was added to NCI-H460/MX20 cells, followed by the appropriate dilutions of synthetic test compound, and the cells were incubated. After removal of the medium and washing with PBS, intracellular fluorescence intensity was read and normalized to cells treated with the ABCG2 inhibitor fumitremorgin C (FTC).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.