

Abstract
CE is the extract from Coeloglossum viride var.bracteatum, a plant widely used as a traditional medicine in the Southwest China, such as Tibet. The effects of CE against amyloid toxicity in cell culture were examined in the present study. The results indicate that CE can protect against amyloid β (Aβ)25-35-induced cytotoxicity in rat primary prefrontal cortex neurons. Bcl2 and Csp3 activation may be involved in CE protection. CE could be a promising candidate for amyloid-based Alzheimer's disease (AD) prevention or therapy.
Keywords: CE, Alzheimer's disease, Amyloid β, Coeloglossum viride var.bracteatum, traditional Chinese medicine
Introduction
Neuronal loss in the cerebral cortex and the hippocampus is a hallmark feature of Alzheimer's disease (AD). Stereological cell counting shows that densities of neurons in the AD cerebral cortex, the entorhinal cortex, the association cortex, the basal nucleus of Meynert, the locus coeruleus and the dorsal raphe decrease significantly compared to the age-matched non-AD controls [1-6]. Neuronal cell loss is one of the first events during AD development. In mild AD patient brains, remarkable neuronal cell loss of more than 40% is seen in the entorhinal cortex [5, 6]. Even in mild cognitive impairment patient brains, significant neuronal loss is also observed in the entorhinal cortex [6]. Furthermore, the degree of neuronal loss correlates better with the clinical dementia level in AD than other pathology. The loss of cholinergic neurons in AD is widely studied. The hippocampus and cortex receive major cholinergic input from the basal forebrain nuclei [7]. Decrease of choline acetyltransferase activity and acetylcholine synthesis correlate well with the degree of cognitive impairmentin AD patients [7-9]. Cholinergic neuronal lesion can be detected in the patients that have showed clinical memory loss symptoms for less than oneyear [10-13]. However, markers for dopamine, γ-aminobutyric acid (GABA), or somatostatin are not altered [10, 11, 13]. These results suggest that synaptic and neuronal loss is probably one of the early events in AD. One of the most remarkable pathological features of AD is extracellular deposition of senile plaques (SPs) containing amyloid b (Ab) peptide aggregates derived from amyloid precursor protein (APP). Aβ aggregations are toxic to neurons and are thought to contribute to neuronal loss in AD development [14].
CE is an extract from plant Coeloglossum viride var. bracteatum, a orchidaceae family plant widely used for medicine in the Northwest of China, especially in Tibet, Inner Mongolia, Gansu, Qinghai and Shanxi Provinces [15]. Described by Chinese traditional medicine, Coeloglossum viride var. bracteatum can increase vital energy, body fluid production, benefit to memory and tranquilize [15]. CE is a mixture of 4 chemicals identified in 2004 [16] (Figure 1). Although it is reported that CE reduces toxicity induced by scopolamine, cycloheximide and alcohol in rodents with impaired memory [15], there are not many controlled studies on CE implications as a potential therapeutic drug.
Figure 1.
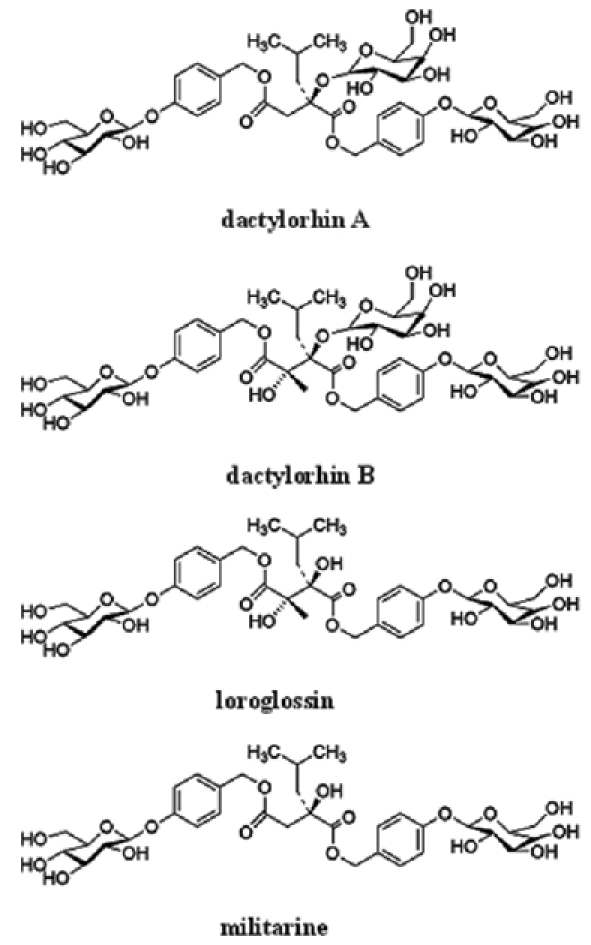
Chemical structure of CE components.
In the present study, we address if CE is protective against Aβ25-35 toxicity. Our data show that CE significantly prevents cell death induced by Aβ25-35 in cultured rat prefrontal cortex neurons. CE protection may be mediated through regulation of anti-apoptotic or apoptotic proteins Bcl2 and caspase-3 (Csp3).
Methods and materials
Primary cell culture and treatments
The prefrontal cortex tissues were dissected from the whole brain tissues of newborn SD rats (Experimental Animal Center of Peking University Health Science Center, Beijing, China) in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA). The tissues were dissociated mechanically and then with 0.25% trypsin (Invitrogen, Carlsbad, CA) for 30 minutes at 37°C. The mixture was then filtered through a nylon mesh to obtain homogenous suspension. After filtering the mixture through 70 mm sterilized filters, the flow-through was centri-fuged to pellet cells. Cells were sedimented and resuspended in DMEM with 10% fetal bovine serum (FBS), 2 g/l HEPES, penicillin G (100U/ml), and 100ug/ml streptomycin (all from Invitrogen, Carlsbad, CA). Cells were plated in poly-L-lysine-coated coverslips or petri-dishes and maintained in a humidified incubator with 5% CO2 and 95% O2 at 37°C. Cytosine arabinoside (10 μM; Sigma, St. Louis, Missouri) was supplemented after plating for 2-4 days to inhibit glia cell growth. Cells were treated at 7-8 days in culture. To make Aβ peptides into aggregates, Aβ25-35 (Sigma, MO) stock solution (2 mM in water) and Aβ1-42 (Sigma, MO) stock solution (2 mM in water) were aged for 5 days in a humid chamber at 37°C before use. Both Aβ25-35 and Aβ1-42 were aged in their stock solutions without adding any other incubation buffer. CE (a gift from Dr. Li Tang, Department of Pharmacology, Minzu University of China) and Bcl2 inhibitor HA14-1 (Tocris, UK) were added freshly into culture medium during treatments.
Immunocytochemistry
Treated cultured primary rat prefrontal cortex neurons were washed with 0.01 M phosphate buffered saline (PBS) before fixed with 4% paraformaldehyde. Cells were then washed with PBS and blocked using PBS with 10% donkey serum and 0.3% triton X-100. The primary antibody, anti-tubulin III (a kind gift from Daniel Lee, GSK) was diluted in 3% serum-PBS and incubated at 4°C overnight. After washing with PBS, cells were incubated with Cy3-conjugated secondary antibody (Jackson Immuno-Research Laboratories, Inc., PA) for 1 hour.
Cell viability assays
The viability of cells after various treatments was estimated by their ability to reduce the dye methyl thiazolyl tetrazolium (MTT, Sigma, MO) to blue formazan crystal. Cells cultured in 96-well plate for 7 days were gently washed with 0.01 M PBS. After wash, 90 ml of medium with 10 ml of 5 mg/ml MTT solution was added to each well and the plate was maintained at 37°C for 2-4 hours. Then the reactions were dissolved in DMSO for quantification by measuring the absorption at 570 nm using a microplate spectro-photometer (Bio-Rad, CA), representing relative cell viability.
Cell death was examined after treatments by fixing cells in fresh 4% paraformaldehyde, 4% sucrose in PBS for 20 minutes at room temperature and permeablized in 0.1% triton X-100, 0.1% sodium citrate in PBS for 2 minutes on ice. Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining was performed using the in situ cell death detection kit I as described by the manufacturer (Roche, Quebec, Canada). The coverslips were then washed once in distilled water for 5 minutes and mounted on glass slides to be observed under a fluorescence microscope. The percentage of cell death was determined by the ratio of the number of TUNEL-positive cells over the total of 100 cells in one count. The average of 5 counts was calculated as the percentage of neuronal cell death in a certain treatment.
Western blots
The neurons cultured in 6-well plates were washed three times with 0.01M PBS, 100 μl of cell lysis buffer with 1% phenylmethanesulfonyl fluoride (PMSF) was added into each well and cells were harvested with cell scrapers. The extracts were iced for 30 minutes and centrifuged at 14,800 g for 15 minutes, and the supernatant was harvested. Denatured protein samples diluted with loading buffer were loaded equally to each lane and separated by 10% SDS -PAGE and then blotted onto a polyvinylidene fluioride (PVDF, Millipore, MA) membrane. The membrane was then incubated for 1 hour in blocking buffer (tris-buffered saline containing 5% no-fat milk powder) at room temperature. The membrane was incubated at 4°C with the primary antibodies, washed with tris-buffered saline Tween-20 (TBST) and incubated again with horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson ImmunoRe-search Laboratories, Inc., PA) followed by washing. The primary antibodies used include: purified polyclonal anti-b-actin antibody (Santa Cruz, CA), polyclonal anti-activated caspase-3 antibody (Santa Cruz, CA) and polyclonal anti-Bcl2 antibody (Santa Cruz, CA). Immunoblots were developed in the presence of enhanced chemiluminescence reagents, and the images detected in X-ray films were quantified by densitometric scanning using Gel Imaging Analysis System Gel-Pro 4400 (Media Cybernetics, MD).
Statistical analysis
All data are presented as means ± S.E.M. Statistical significance (*p<0.05 or **p<0.01) among groups was determined by two-way analysis of variance (ANOVA) and two-tailed Student's t-test. The Sheffé's test was applied as a post hoc for the significant difference shown by ANOVAs.
Results
CE protects against Aβ25-35-induced cytoxicity
Cultured rat prefrontal cortex contains about 90% neurons labeled with tubulin III (Figure 2A). CE (10 mg/ml) effectively protects these neurons from Aβ25-35 (20 μM) toxicity showing by tubulin III labeled morphology (Figure 2B). MTT assays demonstrate CE at 10 mg/ml increases cell viability by about 15-20% compared with Ab25-35 treatment, although lower concentrations of CE do not protect (Figure 2C). CE itself does not induce significant cytotoxicity (Figure 2C). Data from TUNEL assays show that CE (10 mg/ml) reduces cell death induced by Aβ25-35 by nearly 40-50% (Figure 2D). To compare with the naturally occurring Aβ species, Aβ1-42 (20 μM) was applied with or without CE treatment. The data show that CE also reduces cell death induced by Aβ1-42 (Figure 2D). Taken together, our data suggest that CE at 10 mg/ml concentration significantly decreases Aβ25-35-induced cytoxicity in rat primary prefrontal cortex neurons.
Figure 2.
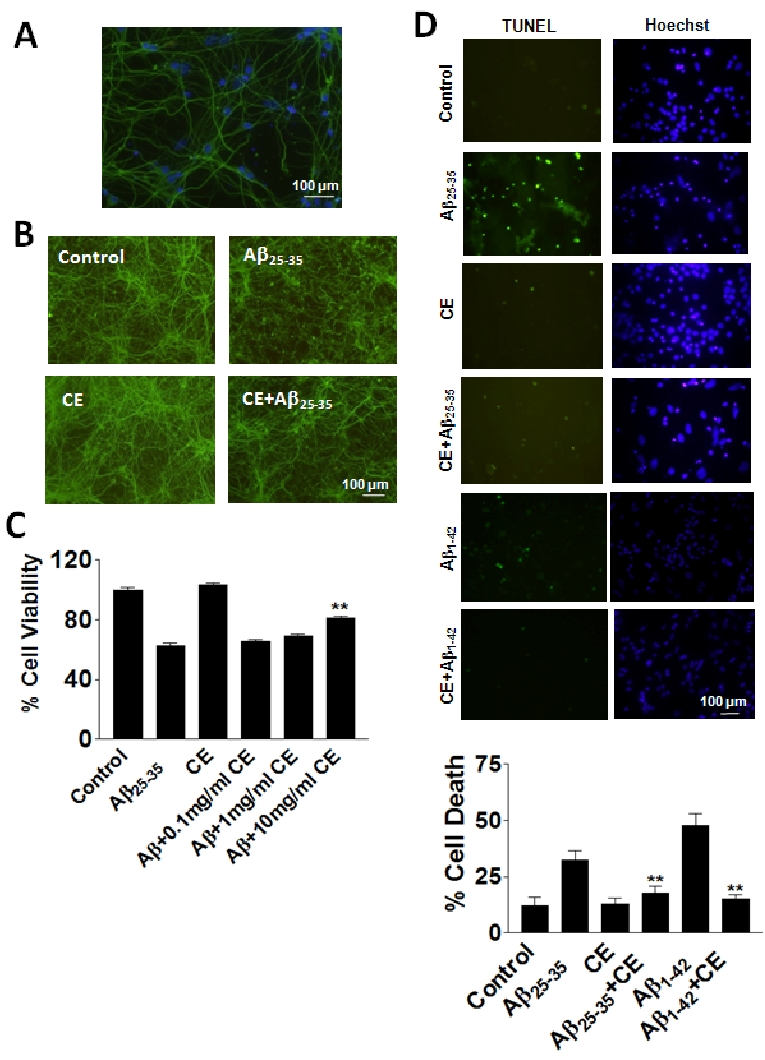
CE protects against Aβ toxicity in rat primary neurons. A. There are 90% neurons in rat primary prefrontal tissue cultures. Green: specific neuronal marker tubulin III; blue: Hoechst. B. Tubulin III staining shows the morphology of neurons under treatments. Aβ25-35 induces significant deform of neuronal morphology after 24 hours of treatment. CE (10 mg/ml) reverses the morphological changes induced by Aβ25-35. C. MTT assays show that CE (10 mg/ ml) increases cell viability compared with Aβ treatment. D. TUNEL assays show that CE (10 mg/ml) decreases cell death induced by Aβ. Data represent mean+SE (n=3). **P<0.01 compared to Aβ25-35 or Aβ1-42 group. Scale bar: 100 mm.
Bcl2 and Csp3 activation may be involved in CE protection
Anti-apoptotic factor Bcl2 decreases during Aβ25-35 treatment, while CE (10 mg/ml) reverses the change induced by Aβ25-35 treatment (Figure 3A). Cell death assays show that Bcl2 inhibitor HA14-1 (20 mM) effectively blocks the protection of CE suggesting that Bcl2 is involved in CE protection against Aβ25-35 toxicity (Figure 3B). Activation of an apoptotic protein Csp3 was also examined. CE (10 mg/ml) downregulates the increase of activated Csp3 by Aβ25-35 treatment remarkably (Figure 3C), suggesting that both Bcl2 and caspase-3 activation may be involved in CE protection.
Figure 3.
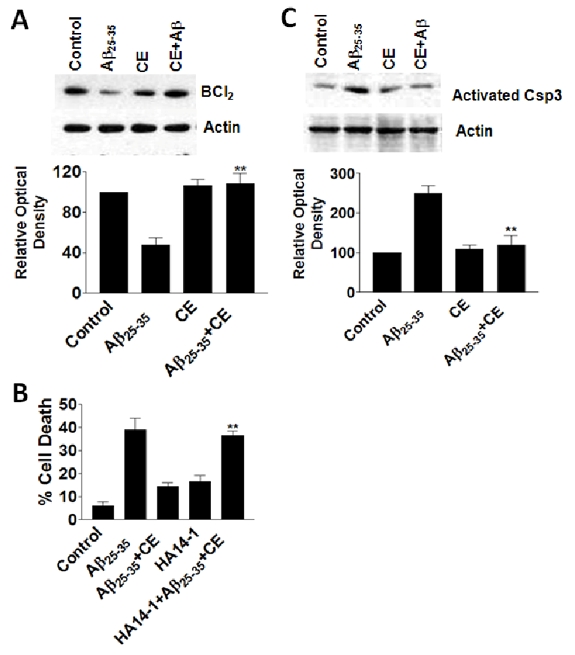
CE upregulates anti-apoptotic protein Bcl2 and downregulates apoptotic protein activated Csp3. A. CE (10 mg/ml) increases levels of Bcl2. B. Bcl2 inhibitor HA14-1 blocks CE protection against Aβ25-35 toxicity. Data represent mean±SE (n=3). **P<0.01 compared to Aβ25-35+CE group. C. CE (10 mg/ml) decreases levels of activated Csp3. Data represent mean±SE (n=3). **P<0.01 compared to Aβ25-35 group.
Discussions
The results of the present study indicate that CE can protect against Aβ25-35-induced cytoxicity in rat primary prefrontal cortex neurons. Bcl2 and Csp3 activation may be involved in CE protection. The beneficial effects of CE are consistent with other observations that CE reduces toxicity induced by scopolamine, cycloheximide and alcohol in rodents with impaired memory [15].
Aβ25-35 is shown toxic to SH-SY5Y cells and isolated mitochondria from rat brains [17]. Dactylorhin B, one of components of CE, protects against Aβ25-35 induced toxicity in the above systems [17]. There are 4 components in CE: dactylorhin A, dactylorhin B, loroglossin and militarine [16]. The studies on the implications of each component and the interaction of components will make CE a potential drug candidate for Aβ hypothesis-based therapy.
Evidence has shown apoptosis involvement in AD. Overexpression of familial AD (FAD)-related mutations causes apoptosis in the transfected cell lines, cultured neurons and transgenic mice. For example, overexpression of FAD mutations of APPV642I, APPV642F and APPV642G in COS or F11 cells increases number of apoptotic cells as determined by TUNEL staining assay, which can be inhibited by anti-apoptotic protein Bcl2 overexpression [18]. These results support the role of FAD mutations in inducing apoptosis. Similarly, the data from transgenic mice confirm the above observations. Transgenic mice over-expressing FAD mutant APPV717F develop neuritic dystrophy similar to some pathological features in AD patients [19, 20]. The degenerating neurons in these mice also show typical apoptotic features, such as chromatin segmentation and condensation, and positive TUNEL staining [21]. However, in these studies, the FAD mutant proteins are normally overexpressed far beyond the physiological levels. However, FAD neurons do not necessarily undergo apoptosis. In the mutant PS1 expressing neurons, increased apoptosis is not reported [22]. Also, there is no neuronal loss found in mutant PS1 transgenic mice [23].
Our results show that CE upregulates Bcl2 level and downregulates activated Csp3 level. These might be the mechanisms of CE protection against Aβ toxicity. In our previous study, we also find that curcumin protection is mediated by regulation of Csp3 and Bcl2 levels in rat neurons [24]. In APPswe/PSDE9 transgenic mice, activated Csp3 and Bcl2 are reported to be related to pathogenesis of AD [25]. In mild cognitive impairment patients, alterations of Csp3 and Bcl2 levels are associated with disease progression [26]. Therefore, Csp3 and Bcl2 might be involved in amyloid toxicity and could be regulated by protective agents, such as CE.
Taken together, the data from this study confirm that CE is protective against Aβ toxicity in cell cultures, which makes it a promising candidate for amyloid-based AD prevention or therapy.
Acknowledgments
The authors thank the supports from 111 Project (B08044) to X.Q., Minzu University 985 fund (MUC985) to X.Q., the National Program of Basic Research sponsored by the Ministry of Science and Technology of China (2009CB941301) to Y.Z., Peking University President Research Grant and Ministry of Education Recruiting Research Grant to Y.Z. Also, the authors thank technical assistance of Yong Cheng (College of Life Sciences, Peking University) and inspiring advice from Dr. Long-Chuan Yu (College of Life Sciences, Peking University).
Glossary
List of Abreviations
- Ab
amyloid β
- AD
Alzheimer's disease
- ANOVA
analysis of variance
- APP
amyloid precursor protein
- Csp3
caspase-3
- DMEM
Dulbecco's modified Eagle's medium
- FAD
familial AD
- FBS
fetal bovine serum
- GABA
g-aminobutyric acid
- HRP
horseradish peroxidase
- MTT
methyl thiazolyl tetrazolium
- PBS
phosphate buffered saline
- PMSF
phenylmethanesulfonyl fluoride
- PVDF
polyvinylidene fluioride
- TBST
tris-buffered saline Tween-20
- SP
senile plaque
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling.
References
- 1.Bondareff W, Mountjoy CQ, Roth M. Loss of neurons of origin of the adrenergic projection to cerebral cortex (nucleus locus ceruleus) in senile dementia. Neurology. 1982;32:164–168. doi: 10.1212/wnl.32.2.164. [DOI] [PubMed] [Google Scholar]
- 2.Colle MA, Duyckaerts C, Laquerriere A, Pradier L, Czech C, Checler F, Hauw JJ. Laminar specific loss of isocortical presenilin 1 immuno-reactivity in Alzheimer's disease. Correlations with the amyloid load and the density of taupositive neurofibrillary tangles. Neuropathol Appl Neurobiol. 2000;26:117–123. doi: 10.1046/j.1365-2990.2000.026002117.x. [DOI] [PubMed] [Google Scholar]
- 3.Lippa CF, Hamos JE, Pulaski-Salo D, DeGennaro LJ, Drachman DA. Alzheimer's disease and aging: effects on perforant pathway perikarya and synapses. Neurobiol Aging. 1992;13:405–411. doi: 10.1016/0197-4580(92)90115-e. [DOI] [PubMed] [Google Scholar]
- 4.Gomez-Isla T, Growdon WB, McNamara M, Newell K, Gomez-Tortosa E, Hedley-Whyte ET, Hyman BT. Clinicopathologic correlates in temporal cortex in dementia with Lewy bodies. Neurology. 1999;53:2003–2009. doi: 10.1212/wnl.53.9.2003. [DOI] [PubMed] [Google Scholar]
- 5.Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 6.Gomez-Isla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hohmann GF, Wenk GL, Lowenstein P, Brown ME, Coyle JT. Age-related recurrence of basal forebrain lesion-induced cholinergic deficits. Neurosci Lett. 1987;82:253–259. doi: 10.1016/0304-3940(87)90265-5. [DOI] [PubMed] [Google Scholar]
- 8.Pearson RC, Powell TP. Anterograde vs. retrograde degeneration of the nucleus basalis medialis in Alzheimer's disease. J Neural Transm Suppl. 1987;24:139–146. [PubMed] [Google Scholar]
- 9.Mesulam MM. Alzheimer plaques and cortical cholinergic innervation. Neuroscience. 1986;17:275–276. doi: 10.1016/0306-4522(86)90242-3. [DOI] [PubMed] [Google Scholar]
- 10.Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 11.Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol. 1981;10:122–126. doi: 10.1002/ana.410100203. [DOI] [PubMed] [Google Scholar]
- 12.Weinstock M. Possible role of the cholinergic system and disease models. J Neural Transm Suppl. 1997;49:93–102. doi: 10.1007/978-3-7091-6844-8_10. [DOI] [PubMed] [Google Scholar]
- 13.Francis PT, Webster MT, Chessell IP, Holmes C, Stratmann GC, Procter AW, Cross AJ, Green AR, Bowen DM. Neurotransmitters and second messengers in aging and Alzheimer's disease. Ann N Y Acad Sci. 1993;695:19–26. doi: 10.1111/j.1749-6632.1993.tb23021.x. [DOI] [PubMed] [Google Scholar]
- 14.Lorenzo A, Yankner BA. Amyloid fibril toxicity in Alzheimer's disease and diabetes. Ann N Y Acad Sci. 1996;777:89–95. doi: 10.1111/j.1749-6632.1996.tb34406.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhang D, Liu G, Shi J, Zhang J. Coeloglossum viride var. bracteatum extract attenuates D-galactose and NaNO2 induced memory impairment in mice. J Ethnopharmacol. 2006;104:250–256. doi: 10.1016/j.jep.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 16.Huang SY, Li GQ, Shi JG, Mo SY. Chemical constituents of the rhizomes of Coeloglossum viride var. bracteatum. J Asian Nat Prod Res. 2004;6:49–61. doi: 10.1080/1028602031000119826. [DOI] [PubMed] [Google Scholar]
- 17.Zhang D, Zhang Y, Liu G, Zhang J. Dactylorhin B reduces toxic effects of beta-amyloid fragment (25-35) on neuron cells and isolated rat brain mitochondria. Naunyn Schmiedebergs Arch Pharmacol. 2006;374:117–125. doi: 10.1007/s00210-006-0095-9. [DOI] [PubMed] [Google Scholar]
- 18.Yamatsuji T, Okamoto T, Takeda S, Murayama Y, Tanaka N, Nishinoto I. Expression of V642 APP mutant causes cellular apoptosis as Alzheimer trait-linked phenotype. EMBO J. 1996;15:498–509. [PMC free article] [PubMed] [Google Scholar]
- 19.Games D, Adams D, Alesandrini R, Barbour R, Berthelette P. Alzheimer-type neuropathology in transgenic mice overexpression V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 20.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S. Accelerate Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 21.Nijhawan D, Honarpour N, Wang X. Apoptosis in neural development and disease. Annu Rev Neurosci. 2000;23:73–87. doi: 10.1146/annurev.neuro.23.1.73. [DOI] [PubMed] [Google Scholar]
- 22.Bursztajn S, De Souza R, McPhie DL. Overexpression in neurons of human presenilin-1 familial Alzheimer disease mutant does not enhance apoptosis. J Neurosci. 1998;18:9790–9799. doi: 10.1523/JNEUROSCI.18-23-09790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shoji M, Iwakami N, Takeuchi S, Waragai M, Suzuki M, Kanazawa I, Lippa CF, Ono S, Okazawa H. JNK activation is associated with intracellular beta-amyloid accumulation. Brain Res Mol Brain Res. 2000;85:221–233. doi: 10.1016/s0169-328x(00)00245-x. [DOI] [PubMed] [Google Scholar]
- 24.Qin XY, Cheng Y, Cui J, Zhang Y, Yu LC. Potential protection of curcumin against amyloid beta-induced toxicity on cultured rat prefrontal cortical neurons. Neurosci Lett. 2009;463:158–161. doi: 10.1016/j.neulet.2009.07.047. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Liu P, Zhu H, Xu Y, Ma C, Dai X, Huang L, Liu Y, Zhang L, Qin C. miR-34a, a microRNA up-regulated in a double transgenic mouse model of Alzheimer's disease, inhibits bcl2 translation. Brain Res Bull. 2009;80:268–273. doi: 10.1016/j.brainresbull.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Sultana R, Banks WA, Butterfield DA. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer's disease. J Neurosci Res. 88:469–477. doi: 10.1002/jnr.22227. [DOI] [PMC free article] [PubMed] [Google Scholar]