

Abstract
Positron Emission Tomography (PET) is playing an increasingly important role in the diagnosis, staging, and monitoring response to treatment in a variety of cancers. Recent efforts have focused on ImmunoPET, which employs antibody-based radiotracers, to image tumors based on expression of tumor-associated antigens. It is postulated that the specificity afforded by antibody targeting should both improve tumor detection and provide phenotypic information related to primary and metastatic lesions that will guide therapy decisions. Advances in antibody-engineering are providing the tools to develop antibody-based molecules with pharmacokinetic properties optimized for use as immunoPET radiotracers. Coupled with technical advances in the design of PET scanners, immunoPET holds promise to improve diagnostic imaging and to guide the use of targeted therapies. An overview of the preclinical immunoPET studies in cancer models is reviewed here.
INTRODUCTION
Medical imaging is a critical tool for the accurate diagnosis and staging of cancer, as well as evaluating response to therapy. Evaluation of therapeutic efficacy through the use of anatomical imaging modalities, such as computed tomography (CT) and magnetic resonance imaging (MRI), is based upon structural changes within tumors that are assessed in relation to standards such as Response Evaluation Criteria In Solid Tumors (RECIST) or WHO criteria (1), (2). By contrast, positron emission tomography (PET) is a nuclear medicine imaging modality that employs radiotracers to image tumors based on a functional readout of biochemical properties, such as metabolism and proliferation rate. Biochemical changes in response to therapies are often manifested much before anatomical changes become apparent by other imaging modalities. The ability of PET to evaluate efficacy at earlier timepoints predicts that it can provide earlier and more sophisticated insight into the efficacy of new agents during both the preclinical and clinical stages of development. It also has the potential to define patient populations that are predicted to have favorable outcomes to both new and currently approved agents. The promise of PET in these types of roles is exemplified by the fact that at the present time there are at least 70 oncology-focused clinical trials in the Clinical Trials.gov database (http://clinicaltrials.gov) that incorporate PET for these purposes. Development of new PET radiotracers has the potential to expand the utility of PET even further.
POSITRON EMISSION TOMOGRAPHY
PET melds the physics of positron decay with the biochemical properties of a tracer compound to map and quantitatively measure specific biochemical processes in vivo. As with all radiological imaging modalities, radiotracers distribute within a subject based upon the biological properties of the individual tracer. As depicted in Figure 1, positrons emitted from the PET isotope used to radiolabel the tracer collide with electrons in nearby tissue, resulting in annihilation and emission of two 511 keV photons oriented at 180 degrees from each other. Detectors, arranged in a ring configuration, allow for coincidence detection of the emitted photons and provide lines of response with which to reconstruct a tomographic image of the radiotracer distribution within the subject. Typical clinical PET scanners have sensitivities that are roughly 10-fold greater than standard SPECT instruments, facilitating detection of radiotracer at levels as low as picomolar concentrations in lesions (3). This sensitivity, combined with the quantitative nature of PET facilitates its use at evaluating the therapeutic response of tumors.
Figure 1.
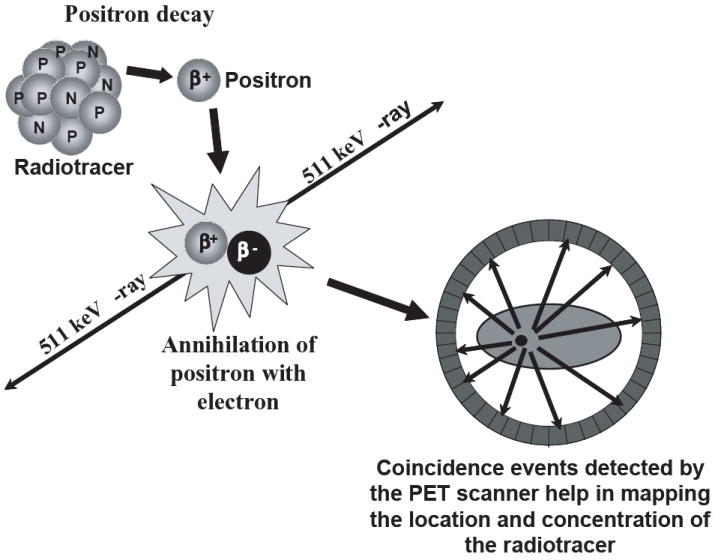
The physical principles underlying PET imaging. A biologically active molecule labeled with a positron emitting radionuclide is administered into the subject. Once injected, the radioisotope emits a positron, which upon traveling a certain distance in the neighboring tissue, annihilates with a nearby electron, emitting two antiparallel 511 keV gamma-ray photons. Pairs of annihilation photons are detected in co-incidence by a multi-ring PET camera, and reconstructed into a whole-body image to map the distribution and concentration of the radiotracer.
In the past decade, small animal imaging has begun to play an ever increasing role in studies designed to both understand the biological underpinnings of cancer as well as in the development of novel therapeutics for the treatment of disease. Imaging modalities such as PET allow for serial evaluation of the tissue distribution and the pharmacokinetics of tracers in individual animals in an unbiased manner. This technology is rapidly replacing “snap-shot” models that rely upon using cohorts of animals to quantify the radioactivity in specific tissues, at specified timepoints, in order to reconstruct the biodistribution of tracer molecules. These efforts have been aided through the development of dedicated small animal PET systems that incorporate CT for anatomical registration. The theoretical maximum spatial resolution is limited in PET by the combination of positron range, a function of the radionuclide being used, and the acolinearity of the annihilation photons. The deleterious effect of acolinearity on resolution is reduced in dedicated animal PET systems, as compared to clinical scanners, as a function of the smaller diameter of the detector ring. Similarly, the reduced cross-sectional area of individual scintillator crystals in small animal PET systems also contributes to the high spatial resolution achievable with these systems. However, this increase in spatial resolution comes at the expense of absolute sensitivity as the reduced area of each crystal covers a smaller solid angle. The limitations imposed by the geometry of detector rings results in commercially available small animal PET systems with 1 – 2 mm full width at half maximum (FWHM) resolution and absolute sensitivities in the range of 2 – 6 % when measured at the center of the field of view (4), (5), (6). The high resolution afforded by these systems, particularly when combined with the anatomical registration available in combined PET/CT systems, facilitates accurate tissue localization of the radiotracers and provides a powerful tool for cancer research.
There are significant numbers of PET radioisotopes that can be employed to radiolabel small molecule and protein-based tracers. Some of these are listed in Table 1 with a subset described in more detail below. Fluorine-18 (18F) represents the most widely used positron emitting radioisotope through its use in 18F-fluoro-deoxy-glucose (FDG). This radioisotope is an almost pure positron emitter (97%, Emax = 635 keV) with a positron range in water of only 0.6 mm that facilitates high resolution imaging in preclinical scanners. Gallium-68 (68Ga) represents another short-lived (t ½ = 68 minutes) radioisotope with high probability of positron emission (89%). It has a higher energy positron emission (Emax = 1.9 MeV, mean=0.89 Mev) and therefore larger positron spread and decreased intrinsic resolution than 18F (Emax =0.63 Mev, mean=0.25 Mev). With the spatial resolution at 5 to 7 mm of current clinical scanners, it is predicted that the imaging quality using 68Ga-based agents can be as good as 18F-based tracers (7), (8). Furthermore, unlike the other PET isotopes relevant to immunoPET imaging, 68Ga is a generator produced radioisotope, providing a potential advantage with regards to radioisotope supply (9). Iodine-124 (124I) and copper-64 (64Cu) are two of the more widely used, longer-lived, PET radioisotopes. In contrast to 18F and 68Ga, positrons emitted by 124I and 64Cu represent a minority of emissions in the decay chains of these two radioisotopes. Iodine-124 decay (t1/2 = 100.3 hours) results in only a 23% probability of positron emission. Positrons emitted by 124I are of relatively high energy (Emax of 1.5 and 2.1 MeV) and limit intrinsic resolution to 2.3 mm (10). In addition, 124I decay results in emission of a number of high energy gamma rays that have been predicted to both decrease resolution through random coincidence detection of the spurious gamma emissions and inhibit quantification of radiotracer kinetics. Methods to address both of these concerns have been described (11), (12), (13). 64Cu,, like 124I, has a low probability (17.4 %) of positron decay. In contrast, it has a positron energy (Emax = 656 keV) closer to that of 18F and an intermediate t1/2 of 12.7 hours making it a versatile PET radioisotope. It has the added benefit of being produced through either reactor or cyclotron-based methods.
Table 1.
Decay characteristics of immunoPET relevant radionuclides
| Isotope | Main β+ energies | Half-life (Hours) | Intrinsic spatial resolution loss (mm) | |
|---|---|---|---|---|
| (keV) | (%) | |||
| Ga-68 | 1899 | 87.9 | 1.13 | 2.3 |
| F-18 | 634 | 97 | 1.83 | 0.66 |
| Cu-64 | 653 | 17.9 | 12.7 | 0.7 |
| Y-86 | 1221 | 12.5 | 14.7 | 1.8 |
| 1545 | 5.6 | |||
| Br-76 | 871 | 5.9 | 16.2 | 5.3 |
| 990 | 5.1 | |||
| 3382 | 27.6 | |||
| 3941 | 6 | |||
| Zr-89 | 897 | 22.7 | 78.4 | 1 |
| I-124 | 1535 | 11.2 | 100.3 | 2.3 |
| 2138 | 11.2 | |||
Emissions greater than 5% as reported by ICRP and NuDat are listed in the table. Intrinsic spatial resolution is calculated based on Pagani et al (10).
PET RADIOTRACERS
Our understanding of the molecular biology of cancer has spurred an immense effort in both the academic and pharmaceutical industry settings to develop targeted therapeutics designed to specifically block cellular events that lead to cancer formation and progression (14), (15). This knowledge has also led, albeit at a reduced rate, to the development of novel PET radiotracers capable of interrogating specific biological processes. Incorporation of PET imaging with these novel radiotracers has the potential to make a dramatic impact on the design and clinical implementation of future targeted therapies.
The use of PET in the preclinical development stage of drug discovery affords the use of FDG, the only PET radiotracer currently approved by the U.S. Food and Drug Administration (FDA) for cancer imaging, as a biomarker for efficacy that can be easily translated into the clinical setting. This fluorinated analog of glucose measures increased glucose uptake in cells as a surrogate for malignant activity. Clinical trials, across all areas of oncology, demonstrate that FDG has an average sensitivity and specificity of 84% and 88%, respectively (16). Other small molecule radiotracers, capable of measuring biological outcomes directly associated with therapeutic mechanism of action can also be employed and are nicely reviewed in Juweid et al (17). Examples include, 3’-deoxy-3’-[18F]fluorothymidine (FLT), a thymidine analog, that has been shown to effectively image proliferating cells in both preclinical and clinical settings (18), (19) and can potentially be used to evaluate the cytostatic effects of novel agents (3). Preliminary studies with 16α-[18F]Fluoro-17β-estradiol (FES), an estrogen analog for targeting hormone-receptor positive tumors, show promise for the use of FES to monitor the effectiveness of hormonal therapy with tamoxifen in breast cancer (20), (21), (22).
The expanded use of monoclonal antibodies (mAbs; e.g. trastuzumab, cetuximab, rituximab, etc) and targeted small molecule inhibitors (e.g. lapatinib, erolitinib, etc.) that are specific for membrane bound tumor-associated antigens, suggests potential roles for immunoPET in guiding therapy decisions. ImmunoPET is well suited to confirm antigen expression in non-biopsied lesions to enable selection of patients who are likely to respond to therapy, or to design informed alternative treatment strategies to improve patient response and/or to avoid unnecessary treatment-induced toxicities. ImmunoPET can be envisioned to inform on the biodistribution and pharmacokinetics of new mAb therapies, both during preclinical and clinical development, to anticipate potential toxicities. Furthermore, immunoPET can be used to provide dosimetry data in the setting of radioimmunotherapy (RAIT) for dose planning. For example, 124I is a relevant surrogate for performing PET-based dosimetry studies related to 131I-radioimmunotherapy (RAIT). In a murine colon cancer xenograft model for studying the biodistribution of a 124I-A33 humanized mAb, Lee et al demonstrated how quantitative PET imaging with a radiolabeled antibody can be used for estimating radiation dose for treatment (23).
ANTIBODIES AS IMMUNOPET RADIOTRACERS FOR CANCER
With the discovery of hybridoma technology for mAb development in 1975, the concept of the “magic bullet” that would be capable of selectively targeting tumors has developed into a plausible option. This is evident from the FDA approval of several mAbs for diagnosis and treatment of cancer. It is postulated that the specificity afforded by antibody targeting should both improve tumor detection as compared to FDG-PET, and provide phenotypic information related to primary and metastatic lesions that will guide therapy decisions. Effective use of antibodies as immunoPET radiotracers requires that: a) the target antigen expression be enriched in the tumor, b) the antibody have sufficient affinity for the antigen to be stably retained in the tumor, c) unbound antibody exhibit rapid systemic clearance to minimize the time necessary to obtain sufficient image contrast. Intact mAbs specific for tumor associated antigens meet the first of these two criteria. To date, five technetium-99m (99mTc) or indium-111 (111In)-labeled murine mAbs have been approved by the FDA for single photon emission computerized tomography (SPECT) imaging (24). Among these, radiolabeled antibodies to tumor associated glycoprotein 72 (TAG-72), prostate-specific membrane antigen (PSMA), carcinoembyonic antigen (CEA), and epithelial cell adhesion molecule have been approved by the FDA for cancer imaging (Table 2). These mAbs are mainly used for staging disease in patients suspected of recurrent or metastatic disease. However, their overall clinical impact has been limited. Advances in protein engineering have facilitated the construction of antibody-based molecules that retain binding characteristics of intact mAbs while exhibiting PK profiles that are optimized for in vivo diagnostic purposes.
Table 2.
FDA-approved antibodies for imaging cancer.
| Generic name | Trade name | Target antigen | Antibody format | Radioiostope | Indication |
|---|---|---|---|---|---|
| Arcitumomab | CEA-Scan™; Immunomedics, Morris Plains, NJ | CEA | Fab | 99mTc | Colorectal cancer |
| Capromab pendetide | ProstaScint™; Cytogen, Princeton, NJ | PSMA | IgG | 111In | Prostate cancer |
| Nofetumomab merpentan | Verluma™; Boehringer Ingelheim, Ingelheim, Germany | EpCAM | Fab | 99mTc | Small cell lung cancer |
| Satumomab pendetide | OncoScint™; Cytogen, Princeton, NJ | TAG-72 | IgG | 111In | Ovarian and colorectal cancer |
The antigen binding specificity of a mAb is dictated by the six complementarity determining regions (CDRs) that are found in its variable domains, three in both the heavy chain and light chain (VH and VL). Molecular engineering techniques have facilitated the construct of single-chain Fv (scFv) molecules that are comprised of the VH and VL domains of an mAb joined together by a short peptide linker. The scFv is approximately 25 kDa and can be readily produced in a wide range of expression systems. Most importantly it recapitulates the binding specificity of the parental mAb. Single-chain Fv molecules represent the basic building blocks of antibody-engineering and can be use to construct a wide range of molecular structures that vary in their size and valency. Representative examples are detailed in Figure 2. Significant effort has led to a detailed understanding of the tumor targeting properties of these engineered antibody fragments (25).
Figure 2.

Schematic representation of intact antibody (IgG) and various genetically engineered [single-chain Fv, scFv; scFv dimers such as (scFv)2 and diabody; and larger fragments such as minibodies and scFv-Fc containing single constant domains and intact Fc regions respectively] and enzymatically derived antibody fragments [Fab and F(ab’)2]. VH and VL represent the heavy and light chain variable regions, CH1, CH2, and CH3 are the heavy chain constant region domains, and CL is the light chain constant region domain.
As stated above, durable tumor retention and rapid systemic clearance are desired characteristics of an immunoPET radiotracer. Physical characteristics of the antibody, such as molecular size, affinity for its target antigen, and valency directly influence the clearance and tumor retention of engineered antibodies. To the first approximation, the PK behavior of a protein correlates with its size relative to the renal threshold; proteins less than approximately 65 kDa can pass through the glomeruli of the kidney and undergo first-pass renal clearance (26). Of the antibody-based molecules described in Figure 2, only the monovalent scFv, the dimeric (scFv’)2 and non-covalent scFv (diabody, ~ 50 kDa) are eliminated in this manner (27), (28), (29). Minibodies (~ 80 kDa) and scFv-Fc (~105 kDa) are larger constructs that contain single constant domains or intact Fc regions, respectively. The lack of first-pass renal clearance of these molecules results in longer serum half-life than diabodies or scFv, allowing higher tumor uptake (30), (31). Although preferable for decreasing scan background, faster clearance kinetics results directly in lower overall tumor uptake (32), and therefore physical characteristics must be evaluated in relationship to each other when designing novel immunoPET radiotracers for the detection of cancer. Of the engineered fragments depicted in Figure 2, the monovalent scFvs have the fastest clearance rate (33), (34), (28), (35), (36), (30). However, monovalent binding fails to result in durable tumor retention as compared to engineered antibody fragments that can bind divalently to the cell surface. Adams et al (37) compared tumor targeting and retention of a homodimeric anti-HER2 (scFv’)2 antibody to that seen with a heterodimeric (scFv’)2 capable of only monovalent binding to HER2. In a xenograft model of ovarian cancer, the homodimeric (scFv’)2 exhibited a 3-fold higher tumor retention than the heterodimeric (scFv’)2. Importantly, the heterodimeric (scFv’)2 behaved identically to its cognate anti-HER2 scFv. Interestingly, avidity may play a larger role in tumor retention than intrinsic affinity for the target antigen. Diabodies spanning a 133-fold range of functional affinity (1.33 × 10-7 to 1 × 10-9) were created from a series of anti-HER2 scFv that bind to the same epitope on HER2 with log variations in affinity, ranging from 10-7 to 10-11 M (38). Although an increase in functional affinity that correlated with intrinsic affinity was observed, this did not translate into better tumor retention. In fact, when radiolabeled with 125I, the diabody with the lowest intrinsic affinity exhibited the highest tumor uptake. Consistent with these findings, five anti-CEA T84.66 antibody variants [IgG, scFv, diabody, minibody, and F(ab’)2] were analyzed in the context of determining their merits as imaging agents (39). A major finding of this study was the importance of divalent binding on tumor retention; all the divalent molecules exhibited similar tumor retention times independent of molecular size. In stark contrast, the scFv exhibited a tumor residence time that was an order of magnitude smaller than the divalent molecules and was predicted to have clearance properties incompatible with an effective imaging tracer.
It is important to note that the vast majority of tumor targeting properties described for engineered antibody fragments, and how they relate to their behavior as immunoPET radiotracers, have been defined in the context of binding to antigens present on the surface of cells in solid tumor models of cancer (25). Antigens found on the surface of tumor vasculature represent an additional set of targets that can be exploited for immunoPET imaging. Access of antibodies to these antigens is not subject to the physical barriers associated with targeting tumor cell antigens. Phosphatidylserine (PS) is normally tightly segregated to the internal leaflet of the plasma membrane. This asymmetry is lost on tumor vascular endothelium, exposing PS on the surface of the endothelial cells. This restricted expression of PS has been exploited for development of the anti-PS mAb bavituximab (40). Bavituximab, when labeled with the positron emitting radioisotope 74As (t½ = 17.8 days, 29% β+,128 keV) was able to effectively localize to tumor vasculature. At 72 hours post-injection, 74As-bavituximab achieved a 22:1 tumor-to-liver ratio in a rat model of prostate cancer. This compared very favorably with the 1.5:1 ratio seen with an isotype-matched control antibody and resulted in effective tumor imaging. PS is exposed on the vasculature of a wide variety of solid tumors, suggesting that bavituximab, or engineered fragments based on its variable domains, have the potential to be effective immunoPET tracers for a broad range of diseases. The potential utility of vasculature imaging is underscored by the increasing use of anti-angiogenic therapies (e.g. bevacizumab). Leukemia and lymphoma also represent a significant percentage of the cancer burden. Using anti-CD20 minibody and scFv-Fc antibodies based on the FDA-approved mAb rituximab, Olafsen et al (41) demonstrated that the 124I-radiolabeled minibody was able to achieve high (7 ± 3-fold) contrast ratios in CD20 positive tumors as compared to CD20 negative tumors. The similarly radiolabeled scFv-Fc failed to obtain this high level of contrast, in large part due to the two-fold lower uptake in CD20 positive tumors. Consistent with the fact that CD20 is not rapidly internalized upon rituximab binding (42), radiolabeling of the minibody with 64Cu-DOTA actually decreased tumor:background ratios by 5-fold. In contrast to the decrease in contrast seen with the scFv-Fc, the decrease in contrast associated with 64Cu-DOTA radiolabeled minibody was due to increased background; a function of the residualizing nature of the radiolabel (see below for more details on residualization). These data suggest that as with solid tumor imaging, biology of the target antigen plays a role in effective immunoPET imaging of hematological malignancies.
CHOICE OF RADIOLABEL FOR IMMUNOPET TRACERS
The physical properties of the radionuclides to be paired with an antibody-based radiotracer must be considered in conjunction with the biological properties of the antibody (described above), to optimize the effectiveness of immunoPET radiotracers for use in either preclinical or clinical oncologic imaging. The physical half-life (t1/2) of the radioisotope should be compatible with the biologic t1/2 of the tracer and the time necessary for the tracer to achieve optimal tumor:background ratios. The chemistry used to conjugate the radioisotope should result in efficient and stable labeling of the immunoPET tracer in a manner that doesn’t negatively impact the antibody’s ability to bind its target antigen. Additionally, data on the cellular trafficking and catabolic pathway of the radiotracer can be used to inform on whether to chose a residualizing (e.g. 64Cu, 68Ga, etc.) or non-residualizing (e.g. 124I, 76Br, etc) radionuclide. Finally, if clinical translation of the radiotracer is a goal, the ease with which the radioisotope is produced and its cost is also of importance.
It is expected that radiometals, or other forms of intracellularly retained (i.e residualizing), radiolabels have an advantage over non-residualizing radiolabels (e.g. iodine) in the diagnosis as well as therapy with mAbs (43). Unlike radiohalogens, radiometals such as 68Ga, 64Cu, 86Y, and 89Zr are retained intracellularly in lysosomes, and may be preferred for antibodies that become internalized after binding antigen. As with all metals, effective use of 64Cu-labeled immunoPET radiotracers requires formation of a stable complex between the antibody and metal ion. Significant effort has been directed toward development of ligands that are amenable to use as immunoPET radiotracers. One such example are the cross-bridged macrocyclics that can stably chelate 64Cu under biological conditions to prevent transchelation. Methods for developing such agents is the topic of a recent review by (44). However, one caveat with radiometal-chelate labeled antibody fragments is high accumulation of radioactive metabolites in liver or kidney following clearance from the blood. Since liver is a major site for metastasis, this may limit the use of radiometal chelate complexes in the detection of metastatic spread to liver. This was illustrated in preclinical imaging studies with an anti-CEA minibody and diabody labeled with 111In. While the size of the minibody resulted in hepatic clearance and significant retention of radiometals in the liver, the diabody showed lower high retention in the kidney due to its renal clearance. Labeling both the fragments with radioiodine decreased the uptake in non target tissues, and resulted in PET images with higher tumor:background ratios. A phase I/II clinical trial with 64Cu-labeled anticolorectal carcinoma 1A3 mAb showed that while the mAb can detect several occult tumor sites (<2cm) that were not visualized by a CT or MRI, but failed to detect lung and liver metastases because of the blood pool activity and accumulation of 64Cu chelate complexes in the liver (45). Similar observations were made in a phase III clinical trial with 99mTc-labeled anti-CEA mAb 88BV59 to image recurrent, metastatic or occult colorectal cancer (46).
Radiohalogens, such as 124I, can be rapidly coupled to tyrosine residues of immunoPET radiotracer using oxidants such as Iodogen or Chloramine-T (47). However, if the radiotracer is internalized upon binding to its antigen target, lysosomal proteolysis of the radiotracer leads to loss of radiohalogen from the target tissue in vivo, and produces a PET image with reduced tumor contrast (48). Direct halogenation is therefore better suited for targets that either do not internalize or do so slowly; in which case, radiolabel will remain high in the target tissue, but decline rapidly in non-target tissues such as kidney and liver due to metabolism and excretion. As described above, 124I-labeled anti-CEA T84.66 diabody and minibody fragments have been applied successfully for imaging tumors in a mouse model of human colon carcinoma. Dehalogenation can be minimized through indirect labeling strategies that incorporate residualizing groups such as non-metabolizable carbohydrate adducts (i.e., tyramine-cellobiose (43), (49) or dilacticol-125I-labeled tyramine (50), (51)), and chelating agents (i.e., diethylenetriaminepentaacetic acid (DTPA) (52)), which enable retention of the radiolabel after internalization, improving target signal:background ratio. Residualizing forms of radioiodine have also been developed and introduced into preclinical animal models (53), (51).
The short t1/2 of 18F and 68Ga make them incompatible for use with larger engineered mAbs. However, Wu and colleagues (54) using Imaging Figures of Merit [IFOM; (55)] predicted that the T84.66 anti-CEA diabody, which exhibits a high apparent affinity for CEA (KA = 8.2 × 1010 M-1) and rapid tumor uptake (13.7% injected dose per gram LS174T tumor at 2 hours post-injection when labeled with 123I) coupled with rapid systemic clearance (tumor:blood ratios exceeding 9 at 6 hours post-injection) could be effectively used as 18F-labeled PET radiotracer in a preclinical mouse model. IFOM predicted that optimal imaging would occur approximately 1.5 hours post-injection. These predictions were validated by Cai et al using N-succinimidyl-4- 18F-fluorobenzoate to label the T84.66 diabody (56). High-contrast images were seen by 1 - 2 hours post-injection in the LS174T xenograft model using a dedicated small animal PET scanner. These data suggest that antibody-based radiotracers, and similar alternative scaffold molecules, that exhibit both rapid systemic clearance and high tumor uptake can be effectively used, at least in the preclinical setting, as 18F-labeled PET radiotracers. Strategies to rapidly radiolabel, purify, and administer the agents will be absolutely necessary for clinical translation of this type of immunoPET radiotracer.
PRE-TARGETED IMAGING
Pre-targeted immunoPET imaging refers to a multi-step process that aims to improve the tumor:background contrast achievable with directly labeled antibody-based radiotracers by uncoupling the PK of antibody targeting from the physical decay characteristics of the PET radioisotope. In a typical strategy, an anti-tumor/anti-hapten bispecific antibody is administered and allowed to both localize in tumor, followed by a clearing step to remove nonbound antibody from circulation (47). Subsequent step includes administration of a radiolabeled hapten, which is captured by tumor-associated antibody. The kinetics of the hapten localization and clearance dictate the choice of radionuclide and as such are optimized for, but not limited to, use of short-lived PET isotopes such as 68Ga and 18F.
Successful pre-targeting strategies that utilize either peptide-based or chelate-based haptens have been described in the literature (57), (58), (53), (59), (47). This is illustrated in work done by Schuhmacher et al (60) to develop a pretargeting strategy capable of effectively imaging MUC-1 positive breast cancer xenografts in a nude mouse model. The preclinical success of this system resulted in its translation into a preliminary clinical PET study conducted in patients with primary breast cancer (61). In this trial, 14 of 17 known lesions (25 mm ± 16 mm) were visualized at 60-90 min after injection of 68Ga-chelate following pretreatment with 10 mg of an anti-MUC1/anti-chelate bispecific antibody. Successful detection of the lesions was despite relatively low overall levels of 68Ga-chelate retained in tumor (0.003% ID/g tumor) and only a 3-fold ratio in uptake when comparing tumor to normal breast. Methods to address the significant levels of MUC1 antigen shed into circulation and the relatively low affinity of the antibody for MUC-1 represent potential areas for improving this system.
The benefit of uncoupling the PK of the bispecific antibody from the PK of the radiolabeled hapten is balanced, in part, by decreased tumor retention observed when antibodies bind monovalently to the tumor. When a bispecific antibody-based pretargeting system is considered in the context of the findings by Adams et al (37) and Williams et al (39) that are discussed above, it suggests that, in general, pre-targeting strategies that depend upon monovalent association with the tumor cell surface may be sub-optimal. Strategies to easily generate multivalent antibodies to address this shortcoming have been developed (57). When considering the design of pre-targeting strategies it is also critical that the biology of the antigen target be considered. Rapid internalization of the antibody/antigen complex will decrease functional antibody on the cell surface required to trap radiolabeled hapten, potentially limiting the breadth of antigens amenable to pre-targeting strategies.
SELECTION OF APPROPRIATE ANIMAL MODELS FOR IMMUNOPET
By definition, animal models are necessary for the preclinical testing of new radiotracers for in vivo imaging of cancer. Just as the selection of appropriate animal models is critical for effective evaluation of novel therapeutics the same is true for evaluation of new imaging radiotracers. Besides predicting dose-limiting toxicity, tissue distribution and metabolism of drug, animal models are increasingly being used to guide dose escalation studies in phase I clinical trials, and to provide tumor microenvironments that closely mimic the clinical setting. While no animal model is perfect, the main objective of preclinical animal studies should be to choose an animal model system based on its validity, specificity, predictability, and reproducibility for the specific human cancer being studied. In selecting the best model system, consideration should be given to the heterogeneity of the transplanted cell line, immunogenicity within the host species, and the appropriate biological end point (62).
In general, animal model systems can be broadly classified into either spontaneous or artificially transplanted systems. While idiopathic spontaneous tumors mimic human cancer most closely, practical issues, such as identification of a suitable surrogate for the human disease of interest or variability in the natural course of tumor development, can preclude the use of such models. Therefore xenograft models, either ectopic or orthotopic, represent the most widely used form of animal models for the evaluation of novel cancer therapies and diagnostics. Although orthotopic mouse models are hypothesized to better reflect human tumor biology their use in drug development has been limited by technical challenges of monitoring tumor growth. A potential pitfall that could benefit from improved immunoPET radiotracers. Ectopic xenograft studies using human cancer cell lines are: 1) easy to conduct, 2) result in predictable tumor growth rates, and 3) recognized by the FDA for preclinical evaluation. However, the tumors formed through use of established cell lines often lack the biochemical and morphological hallmarks of the tumors from which they originated. This can often be overcome by using primary tumor isolates that are maintained by serially passage in immunodeficient animal (62) (63).
To date, development of antibody-based radiotracers for PET has focused almost exclusively on determining the appropriate size and molecular format of the antibody molecule necessary to provide optimal tumor uptake and retention combined with rapid systemic clearance. While very important, this work has been performed in the absence of an understanding of how variables such as the density of antigen molecules on the surface of tumor cells or the levels of shed antigen in circulation will affect radiotracer performance. Additionally, one drawback with the human tumor xenografts models used for preclinical evaluation is that they provide an unrealistic tumor-restricted expression of the target antigen due to fact that the tumor xenografts are the sole source of antigen expression. A goal of future research should be to evaluate how each of these variables impact on the performance of antibody-based tracers. Transgenic mouse models that express distinct, clinically relevant, human antigens on normal tissues and shed those antigens into the blood stream similarly to patients would be one way to address this. Such studies would allow evaluation of antibody-based imaging probes in a more realistic setting of normal tissue expression and develop protocols to overcome the hypothesized deleterious effects.
FUTURE PERSPECTIVES
Major challenges of new targeted therapy approaches include: 1) selection of patients that are likely to respond to treatment, 2) identification of the biologically active concentration and appropriate dosing schedule, and 3) assessment of the tumor response to therapy. Future work should focus on the integration of immunoPET into the process of drug development and to address key questions in the preclinical and clinical evaluation of novel targeted agents with special regard to the imaging of expression and inhibition of drug targets, pharmacokinetics of new drugs, and early assessment of the tumor response to treatment. In order to adequately assess clinical endpoints, markers that allow precise measurement of tumor targets on a whole-body image upon administration of a functional agent are required. Such agents are expected to provide image-guided therapy that may allow discontinuation of ineffective treatments early on in the course of treatment, and guide alternate more efficient treatment strategies that would be beneficial to patients.
Acknowledgments
This work was supported by the Health Resources and Services Administration through a grant to the American-Russian Cancer Alliance (MKR) and by funding from Postdoctoral Training Grant NIH-CA09035 (SR). The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the NCI or HRSA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Smitha Reddy, Email: Smitha.Reddy@fccc.edu.
Matthew Robinson, Email: Matthew.Robinson@fccc.edu.
Reference List
- 1.Fass L. Imaging and cancer: a review. Mol Oncol. 2008;2(2):115–152. doi: 10.1016/j.molonc.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tumeh PC, Radu CG, Ribas A. PET imaging of cancer immunotherapy. J Nucl Med. 2008;49(6):865–868. doi: 10.2967/jnumed.108.051342. [DOI] [PubMed] [Google Scholar]
- 3.Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nat Rev Cancer. 2002;2(9):683–693. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- 4.Bao Q, Newport D, Chen M, Stout DB, Chatziioannou AF. Performance evaluation of the inveon dedicated PET preclinical tomograph based on the NEMA NU-4 standards. J Nucl Med. 2009;50(3):401–408. doi: 10.2967/jnumed.108.056374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tai YC, Ruangma A, Rowland D, et al. Performance evaluation of the microPET focus: a third-generation microPET scanner dedicated to animal imaging. J Nucl Med. 2005;46(3):455–463. [PubMed] [Google Scholar]
- 6.Wang Y, Seidel J, Tsui BM, Vaquero JJ, Pomper MG. Performance evaluation of the GE healthcare eXplore VISTA dual-ring small-animal PET scanner. J Nucl Med. 2006;47(11):1891–1900. [PubMed] [Google Scholar]
- 7.Reichert DE, Hancock RD, Welch MJ. Molecular Mechanics Investigation of Gadolinium(III) Complexes. Inorg Chem. 1996;35(24):7013–7020. doi: 10.1021/ic960495m. [DOI] [PubMed] [Google Scholar]
- 8.Mathias CJ, Lewis MR, Reichert DE, et al. Preparation of 66Ga- and 68Ga-labeled Ga(III)-deferoxamine-folate as potential folate-receptor-targeted PET radiopharmaceuticals. Nucl Med Biol. 2003;30(7):725–731. doi: 10.1016/s0969-8051(03)00080-5. [DOI] [PubMed] [Google Scholar]
- 9.Yang DJ, Kim EE, Inoue T. Targeted molecular imaging in oncology. Ann Nucl Med. 2006;20(1):1–11. doi: 10.1007/BF02985584. [DOI] [PubMed] [Google Scholar]
- 10.Pagani M, Stone-Elander S, Larsson SA. Alternative positron emission tomography with non-conventional positron emitters: effects of their physical properties on image quality and potential clinical applications. Eur J Nucl Med. 1997;24(10):1301–1327. doi: 10.1007/s002590050156. [DOI] [PubMed] [Google Scholar]
- 11.Daghighian F, Pentlow KS, Larson SM, et al. Development of a method to measure kinetics of radiolabelled monoclonal antibody in human tumour with applications to microdosimetry: positron emission tomography studies of iodine-124 labelled 3F8 monoclonal antibody in glioma. Eur J Nucl Med. 1993;20(5):402–409. doi: 10.1007/BF00208998. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez Trotter DE, Manjeshwar RM, Doss M, et al. Quantitation of small-animal (124)I activity distributions using a clinical PET/CT scanner. J Nucl Med. 2004;45(7):1237–1244. [PubMed] [Google Scholar]
- 13.Pentlow KS, Graham MC, Lambrecht RM, et al. Quantitative imaging of iodine-124 with PET. J Nucl Med. 1996;37(9):1557–1562. [PubMed] [Google Scholar]
- 14.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 15.Lawrence S. Billion dollar babies--biotech drugs as blockbusters. Nat Biotechnol. 2007;25(4):380–382. doi: 10.1038/nbt0407-380. [DOI] [PubMed] [Google Scholar]
- 16.Gambhir SS, Czernin J, Schwimmer J, Silverman DH, Coleman RE, Phelps ME. A tabulated summary of the FDG PET literature. J Nucl Med. 2001;42(5 Suppl):1S–93S. [PubMed] [Google Scholar]
- 17.Juweid ME, Cheson BD. Positron-emission tomography and assessment of cancer therapy. N Engl J Med. 2006;354(5):496–507. doi: 10.1056/NEJMra050276. [DOI] [PubMed] [Google Scholar]
- 18.Krohn KA, Mankoff DA, Eary JF. Imaging cellular proliferation as a measure of response to therapy. J Clin Pharmacol. 2001;Suppl:96S–103S. [PubMed] [Google Scholar]
- 19.Shields AF, Grierson JR, Dohmen BM, et al. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat Med. 1998;4(11):1334–1336. doi: 10.1038/3337. [DOI] [PubMed] [Google Scholar]
- 20.Dehdashti F, Mortimer JE, Siegel BA, et al. Positron tomographic assessment of estrogen receptors in breast cancer: comparison with FDG-PET and in vitro receptor assays. J Nucl Med. 1995;36(10):1766–1774. [PubMed] [Google Scholar]
- 21.Dehdashti F, Flanagan FL, Mortimer JE, Katzenellenbogen JA, Welch MJ, Siegel BA. Positron emission tomographic assessment of “metabolic flare” to predict response of metastatic breast cancer to antiestrogen therapy. Eur J Nucl Med. 1999;26(1):51–56. doi: 10.1007/s002590050359. [DOI] [PubMed] [Google Scholar]
- 22.Mortimer JE, Dehdashti F, Siegel BA, Katzenellenbogen JA, Fracasso P, Welch MJ. Positron emission tomography with 2-[18F]Fluoro-2-deoxy-D-glucose and 16alpha-[18F]fluoro-17beta-estradiol in breast cancer: correlation with estrogen receptor status and response to systemic therapy. Clin Cancer Res. 1996;2(6):933–939. [PubMed] [Google Scholar]
- 23.Lee FT, Hall C, Rigopoulos A, et al. Immuno-PET of human colon xenograft-bearing BALB/c nude mice using 124I-CDR-grafted humanized A33 monoclonal antibody. J Nucl Med. 2001;42(5):764–769. [PubMed] [Google Scholar]
- 24.Zuckier LS, DeNardo GL. Trials and tribulations: oncological antibody imaging comes to the fore. Semin Nucl Med. 1997;27(1):10–29. doi: 10.1016/s0001-2998(97)80033-5. [DOI] [PubMed] [Google Scholar]
- 25.Hudson PJ, Souriau C. Engineered antibodies. Nat Med. 2003;9(1):129–134. doi: 10.1038/nm0103-129. [DOI] [PubMed] [Google Scholar]
- 26.Wochner RD, Strober W, Waldmann TA. The role of the kidney in the catabolism of Bence Jones proteins and immunoglobulin fragments. J Exp Med. 1967;126(2):207–221. doi: 10.1084/jem.126.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams GP, McCartney JE, Tai MS, et al. Highly specific in vivo tumor targeting by monovalent and divalent forms of 741F8 anti-c-erbB-2 single-chain Fv. Cancer Res. 1993;53(17):4026–4034. [PubMed] [Google Scholar]
- 28.Adams GP, Schier R, McCall AM, et al. Prolonged in vivo tumour retention of a human diabody targeting the extracellular domain of human HER2/neu. Br J Cancer. 1998;77(9):1405–1412. doi: 10.1038/bjc.1998.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yokota T, Milenic DE, Whitlow M, Wood JF, Hubert SL, Schlom J. Microautoradiographic analysis of the normal organ distribution of radioiodinated single-chain Fv and other immunoglobulin forms. Cancer Res. 1993;53(16):3776–3783. [PubMed] [Google Scholar]
- 30.Yazaki PJ, Wu AM. Construction and characterization of minibodies for imaging and therapy of colorectal carcinomas. Methods Mol Biol. 2003;207:351–364. doi: 10.1385/1-59259-334-8:351. [DOI] [PubMed] [Google Scholar]
- 31.Wu AM, Yazaki PJ. Designer genes: recombinant antibody fragments for biological imaging. Q J Nucl Med. 2000;44(3):268–283. [PubMed] [Google Scholar]
- 32.Adams GP, Schier R, McCall AM, et al. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001;61(12):4750–4755. [PubMed] [Google Scholar]
- 33.Adams GP, McCartney JE, Wolf EJ, et al. Optimization of in vivo tumor targeting in SCID mice with divalent forms of 741F8 anti-c-erbB-2 single-chain Fv: effects of dose escalation and repeated i.v. administration. Cancer Immunol Immunother. 1995;40(5):299–306. doi: 10.1007/BF01519629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adams GP. Improving the tumor specificity and retention of antibody-based molecules. In Vivo. 1998;12(1):11–21. [PubMed] [Google Scholar]
- 35.Olafsen T, Tan GJ, Cheung CW, et al. Characterization of engineered anti-p185HER-2 (scFv-CH3)2 antibody fragments (minibodies) for tumor targeting. Protein Eng Des Sel. 2004;17(4):315–323. doi: 10.1093/protein/gzh040. [DOI] [PubMed] [Google Scholar]
- 36.Olafsen T, Kenanova VE, Sundaresan G, et al. Optimizing radiolabeled engineered anti-p185HER2 antibody fragments for in vivo imaging. Cancer Res. 2005;65(13):5907–5916. doi: 10.1158/0008-5472.CAN-04-4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adams GP, Tai MS, McCartney JE, et al. Avidity-mediated enhancement of in vivo tumor targeting by single-chain Fv dimers. Clin Cancer Res. 2006;12(5):1599–1605. doi: 10.1158/1078-0432.CCR-05-2217. [DOI] [PubMed] [Google Scholar]
- 38.Nielsen UB, Adams GP, Weiner LM, Marks JD. Targeting of bivalent anti-ErbB2 diabody antibody fragments to tumor cells is independent of the intrinsic antibody affinity. Cancer Res. 2000;60(22):6434–6440. [PubMed] [Google Scholar]
- 39.Williams LE, Wu AM, Yazaki PJ, et al. Numerical selection of optimal tumor imaging agents with application to engineered antibodies. Cancer Biother Radiopharm. 2001;16(1):25–35. doi: 10.1089/108497801750095989. [DOI] [PubMed] [Google Scholar]
- 40.Jennewein M, Lewis MA, Zhao D, et al. Vascular imaging of solid tumors in rats with a radioactive arsenic-labeled antibody that binds exposed phosphatidylserine. Clin Cancer Res. 2008;14(5):1377–1385. doi: 10.1158/1078-0432.CCR-07-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olafsen T, Betting D, Kenanova VE, et al. Recombinant anti-CD20 antibody fragments for small-animal PET imaging of B-cell lymphomas. J Nucl Med. 2009;50(9):1500–1508. doi: 10.2967/jnumed.108.060426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jilani I, O’Brien S, Manshuri T, et al. Transient down-modulation of CD20 by rituximab in patients with chronic lymphocytic leukemia. Blood. 2003;102(10):3514–3520. doi: 10.1182/blood-2003-01-0055. [DOI] [PubMed] [Google Scholar]
- 43.Sharkey RM, Behr TM, Mattes MJ, et al. Advantage of residualizing radiolabels for an internalizing antibody against the B-cell lymphoma antigen, CD22. Cancer Immunol Immunother. 1997;44(3):179–188. doi: 10.1007/s002620050371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderson CJ, Wadas TJ, Wong EH, Weisman GR. Cross-bridged macrocyclic chelators for stable complexation of copper radionuclides for PET imaging. Q J Nucl Med Mol Imaging. 2008;52(2):185–192. [PMC free article] [PubMed] [Google Scholar]
- 45.Philpott GW, Schwarz SW, Anderson CJ, et al. RadioimmunoPET: detection of colorectal carcinoma with positron-emitting copper-64-labeled monoclonal antibody. J Nucl Med. 1995;36(10):1818–1824. [PubMed] [Google Scholar]
- 46.Serafini AN, Klein JL, Wolff BG, et al. Radioimmunoscintigraphy of recurrent, metastatic, or occult colorectal cancer with technetium 99m-labeled totally human monoclonal antibody 88BV59: results of pivotal, phase III multicenter studies. J Clin Oncol. 1998;16(5):1777–1787. doi: 10.1200/JCO.1998.16.5.1777. [DOI] [PubMed] [Google Scholar]
- 47.Wu AM, Olafsen T. Antibodies for molecular imaging of cancer. Cancer J. 2008;14(3):191–197. doi: 10.1097/PPO.0b013e31817b07ae. [DOI] [PubMed] [Google Scholar]
- 48.Zalutsky MR. Radiohalogens for radioimmunotherapy. In: Abrams PG, Frizberg AR, editors. Radioimmunotherapy of cancer. 2000. pp. 81–106. [Google Scholar]
- 49.Pittman RC, Carew TE, Glass CK, Green SR, Taylor CA, Jr, Attie AD. A radioiodinated, intracellularly trapped ligand for determining the sites of plasma protein degradation in vivo. Biochem J. 1983;212(3):791–800. doi: 10.1042/bj2120791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shih LB, Thorpe SR, Griffiths GL, et al. The processing and fate of antibodies and their radiolabels bound to the surface of tumor cells in vitro: a comparison of nine radiolabels. J Nucl Med. 1994;35(5):899–908. [PubMed] [Google Scholar]
- 51.Stein R, Goldenberg DM, Thorpe SR, Basu A, Mattes MJ. Effects of radiolabeling monoclonal antibodies with a residualizing iodine radiolabel on the accretion of radioisotope in tumors. Cancer Res. 1995;55(14):3132–3139. [PubMed] [Google Scholar]
- 52.Stein R, Govindan SV, Mattes MJ, et al. Improved iodine radiolabels for monoclonal antibody therapy. Cancer Res. 2003;63(1):111–118. [PubMed] [Google Scholar]
- 53.Reist CJ, Archer GE, Kurpad SN, et al. Tumor-specific anti-epidermal growth factor receptor variant III monoclonal antibodies: use of the tyramine-cellobiose radioiodination method enhances cellular retention and uptake in tumor xenografts. Cancer Res. 1995;55(19):4375–4382. [PubMed] [Google Scholar]
- 54.Wu AM, Williams LE. Anti-carcinoembryonic antigen (CEA) diabody for rapid tumor targeting and imaging. Tumor Targeting. 1999;4:1–12. [Google Scholar]
- 55.Williams LE, Liu A, Wu AM, et al. Figures of merit (FOMs) for imaging and therapy using monoclonal antibodies. Med Phys. 1995;22(12):2025–2027. doi: 10.1118/1.597646. [DOI] [PubMed] [Google Scholar]
- 56.Cai W, Olafsen T, Zhang X, et al. PET imaging of colorectal cancer in xenograft-bearing mice by use of an 18F-labeled T84.66 anti-carcinoembryonic antigen diabody. J Nucl Med. 2007;48(2):304–310. [PubMed] [Google Scholar]
- 57.Goldenberg DM, Rossi EA, Sharkey RM, McBride WJ, Chang CH. Multifunctional antibodies by the Dock-and-Lock method for improved cancer imaging and therapy by pretargeting. J Nucl Med. 2008;49(1):158–163. doi: 10.2967/jnumed.107.046185. [DOI] [PubMed] [Google Scholar]
- 58.McBride WJ, Zanzonico P, Sharkey RM, et al. Bispecific antibody pretargeting PET (immunoPET) with an 124I-labeled hapten-peptide. J Nucl Med. 2006;47(10):1678–1688. [PubMed] [Google Scholar]
- 59.Sharkey RM, Karacay H, Vallabhajosula S, et al. Metastatic human colonic carcinoma: molecular imaging with pretargeted SPECT and PET in a mouse model. Radiology. 2008;246(2):497–507. doi: 10.1148/radiol.2462070229. [DOI] [PubMed] [Google Scholar]
- 60.Schuhmacher J, Klivenyi G, Kaul S, et al. Pretargeting of human mammary carcinoma xenografts with bispecific anti-MUC1/anti-Ga chelate antibodies and immunoscintigraphy with PET. Nucl Med Biol. 2001;28(7):821–828. doi: 10.1016/s0969-8051(01)00246-3. [DOI] [PubMed] [Google Scholar]
- 61.Schuhmacher J, Kaul S, Klivenyi G, et al. Immunoscintigraphy with positron emission tomography: gallium-68 chelate imaging of breast cancer pretargeted with bispecific anti-MUC1/anti-Ga chelate antibodies. Cancer Res. 2001;61(9):3712–3717. [PubMed] [Google Scholar]
- 62.Khleif SN, Curt GA. Animal models in developmental therapeutics. In: Holland JF, Frei E III, editors. Cancer Medicine. 2000. [Google Scholar]
- 63.Marks CL. Animal models for human diseases:Is there a future without them? The journal of nuclear medicine. 2006;47(12):50N–51N. [Google Scholar]