

Abstract
AIMS
Pharmacokinetic (PK) and pharmacodynamic (PD) monitoring strategies and clinical outcome were evaluated in enteric-coated mycophenolate sodium (EC-MPS)-treated renal allograft recipients.
METHODS
PK [mycophenolic acid (MPA)] and PD [inosine monophosphate dehydrogenase (IMPDH) activity] data were analysed in 66 EC-MPS and ciclosporin A (CsA)-treated renal allograft recipients. Adverse events were considered in a follow-up period of 12 weeks.
RESULTS
Analyses confirmed a limited sampling strategy (LSS) consisting of PK and PD data at predose, 1, 2, 3 and 4 h after oral intake as an appropriate sampling method (MPA r2= 0.812; IMPDH r2= 0.833). MPA AUC0–12 of patients with early biopsy-proven acute rejection was significantly lower compared with patients without a rejection (median MPA AUC0–12 28 µg*h ml−1 (7–45) vs. 40 µg*h ml−1 (16–130), P < 0.01), MPA AUC0–12 of patients with recurrent infections was significantly higher compared with patients without infections (median MPA AUC0–12 65 µg*h ml−1 (range 37–130) vs. 37 µg*h ml−1 (range 7–120), P < 0.005). Low 12-h IMPDH enzyme activity curve (AEC0–12) was associated with an increased frequency of gastrointestinal side-effects (median IMPDH AEC0–12 43 nmol*h mg−1 protein h−1[range 12–67) vs. 75 nmol*h mg−1 protein h−1 (range 15–371), P < 0.01].
CONCLUSIONS
Despite highly variable absorption data, an appropriate LSS might be estimated by MPA AUC0–4 and IMPDH AEC0–4 in renal transplant patients treated with EC-MPS and CsA. Regarding adverse events, the suggested MPA-target AUC0–12 from 30 to 60 µg*h ml−1 seems to be appropriate in renal allograft recipients.
Keywords: EC-MPS, IMPDH, limited sampling strategy, renal transplantation, therapeutic drug monitoring
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Therapeutic drug monitoring of mycophenolate mofetil is a promising tool for reducing acute rejection episodes after renal transplantation.
Limited sampling algorithms of mycophenolic acid in mycophenolate mofetil-treated renal transplant patients have been established.
Recently published study results indicate that the intensity of early drug exposure might determine the risk of acute rejection episodes.
WHAT THIS STUDY ADDS
This study provides pharmacokinetic and pharmacodynamic data of mycophenolic acid in enteric-coated mycophenolate sodium-treated renal transplant patients.
Limited sampling algorithms are evaluated and a practical sampling strategy with five sampling time points within the first 4 h after oral drug intake is provided in renal transplant patients with combined immunosuppression consisting of enteric-coated mycophenolate sodium and ciclosporin A.
The association between pharmacokinetic and pharmacodynamic parameters and the risk of adverse events are evaluated.
Both pharmacokinetic and pharmacodynamic parameters contribute to optimized individual immunosuppression.
Introduction
Mycophenolic acid (MPA) is a potent immunosuppressant that in combination with calcineurin inhibitors and steroids decreases the frequency of acute rejection episodes and chronic allograft failure in renal transplant patients [1–4]. However, data on pharmacokinetics (PK) and pharmacodynamics (PD) of MPA are still incomplete, especially for the enteric-coated mycophenolate sodium (EC-MPS) formulation. MPA is generally administered in a fixed dose, with a standard dose of 1000 mg mycophenolate mofetil (MMF) or the equal dose of 720 mg EC-MPS twice daily. Reduction of the standard dose results in fewer side-effects, although increases the risk of acute rejection. It has been shown that the area under the concentration–time curves (AUCs) vary widely in patients following the same MPA dosage [5]. Therapeutic drug monitoring (TDM) might be useful for optimizing individual immunosuppressive therapy and improving allograft function [6–10]. In the French APOMYGRE study, titration of MMF dose according to plasma MPA concentrations resulted in an increased MPA exposure and a reduced incidence of acute rejection episodes without an increased risk of infections or haematological disorders [11]. In addition, the fixed-dose concentration-controlled (FDCC) trial and earlier studies showed that in the early post-transplant period most patients did not reach the intended target AUC0–12 of 30–60 µg*h ml−1[12, 13]. A panel consensus report monitoring MPA stated that evaluation of MPA AUC0–12 might be valuable for establishing adequate MPA concentrations early after transplant surgery and providing a basis for flexibility for the practitioner to reduce MMF dose to avoid adverse reactions [14]. However, PK and PD data on EC-MPS are sparse and no formal target AUC has been defined, although most practitioners consider both formulations as equivalent.
In addition to the confounding discussion about TDM in mycophenolic therapy, there is another debate about the best PK monitoring strategy [15–17]. Few data regarding PK and PD analysis of plasma MPA concentration of EC-MPS are available. Consistent with the enteric-coating of the formulation, EC-MPS provides delayed release of MPA in the small intestine instead of the stomach; maximum plasma concentrations occur later compared with MMF [18]. EC-MPS is anticipated to have high interindividual variability in absorption because of a varying release of EC-MPS from the stomach to the small intestine. This observed variation in PK profiles makes TDM for EC-MPS even more challenging. Only one study has evaluated whether a limited sampling strategy (LSS) is appropriate in EC-MPS-treated renal transplant patients, and did not find a suitable algorithm [19].
MPA is a selective, reversible, noncompetitive inhibitor of inosine monophosphate dehydrogenase (IMPDH), the rate-limiting enzyme of lymphocyte proliferation. IMPDH activity is proposed as a potential PD parameter for optimizing MPA therapy [20]. In this study, PK and PD data of this new formulation EC-MPS are presented in renal allograft recipients. This is the first study aiming to verify the current MPA-target AUC from 30 to 60 µg*h ml−1 in EC-MPS-treated patients. In addition, a LSS is evaluated for introduction in daily clinical practice.
Methods
Patients
Renal transplant patients from the Department of Nephrology, University of Heidelberg, Germany, who met the following inclusion criteria, were eligible to take part in the present PK and PD analysis: adequate renal transplant function [glomerular filtration rate (GFR) ≥ 20 ml min−1], treatment with at least 720 mg EC-MPS daily (from 360 to 1440 mg twice daily), ciclosporin A (CsA) therapy, aged 18–70 years, 8–56 days after transplantation and written informed consent. All patients were on triple immunosuppressive treatment consisting of EC-MPS (Myfortic; Novartis Pharma GmbH, Nuremberg, Germany), CsA (Sandimmun Optoral; Novartis Pharma GmbH, Nuremberg, Germany) and methylprednisolone and all patients had an induction therapy with interleukin-2 antibody (Basiliximab, Simulect; Novartis Pharma GmbH) on days 0 and 4 after transplantation. Exclusion criteria were suspected noncompliance, a history of surgery of the stomach or small intestine, co-medication with an immunosuppressive agent other than CsA, EC-MPS and methylprednisolone and co-medication with cholestyramine, magnesium- or aluminium-containing antacids and rifampicin. The study was approved by the local ethics committee and conducted in accordance with the Helsinki Declaration.
Study design
Blood samples were collected for routine analysis and to determine trough (C0) plasma CsA and MPA concentrations. Subsequently, each patient was given the morning dose of CsA and EC-MPS and underwent evaluation for PK and PD profiles. Blood samples were taken at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10 and 12 h after the morning dose of EC-MPS. The blood samples were immediately placed at 4°C after drawing and transferred directly for measurement of MPA concentration and IMPDH activity.
The follow-up period was 12 weeks after transplantation. Clinical data and information about the adverse events, e.g. acute rejection episodes, recurrent infections and gastrointestinal side-effects, were obtained during monthly visits to the outpatient clinic. GFR was calculated by the modification of diet in renal disease (MDRD) formula [21]. Renal allograft biopsies were performed depending on clinical decision. Biopsies were classified in accordance with the BANFF 2005 classification [22]. Early antigen of cytomegalovirus (CMV pp65) was assessed routinely. CMV prophylaxis with valganciclovir was given to patients with CMV IgG+ donors and all patients had a Pneumocystis jirovecii prophylaxis with cotrimoxazole.
Pharmacokinetic assessment
All CsA blood levels were measured using the enzyme-multiplied immunoassay technique (EMIT; Dimension XL, Dade Behring, Marburg, Germany). The within-run and between-run precision of this assay was 8% and 10%, respectively. The limit of detection was <25 µg l−1.
MPA concentrations were measured using the EMIT. The within- and between-run precision of this assay was <9% and <11%, respectively. The limit of detection was <0.5 µg ml−1.
Noncompartmental PK parameters were derived from each individual plasma MPA concentration–time profile using WinNonlin Professional (v.5.2) software (Pharsight Corporation, Montreal, Canada). The AUC for the 12-h MPA exposure (AUC0–12) was calculated by the linear trapezoidal rule and 12-h MPA exposure was estimated by abbreviated sampling strategies and calculated by the developed equations (AUC0–3, AUC0–4). Cmax and Cmin were defined as the maximum and minimum daytime MPA concentration after dosing with MPA within the dosing interval. Tmax was defined as the time to reach the maximum daytime MPA concentration.
Pharmacodynamic assessment
PD analysis was performed by assessing IMPDH activity, as published previously [20, 23, 24]. Peripheral blood mononuclear cells (PBMC) were isolated using Ficoll–Paque gradient centrifugation, and lysed PBMC were incubated with inosine 5′-monophosphate. The xanthosine 5′-monophosphate produced was determined by reversed-phase high-performance liquid chromatography using UV detection (Jasco Corporation, Tokyo, Japan). This method showed intra-assay variation coefficients of <15% across a concentration range of 10–150 nmol mg−1 protein h−1, and a lower quantification limit of 10 nmol mg−1 protein h−1. The within- and between-run precision of this assay was 11% and 18%, respectively.
The PD parameters derived from each enzyme activity–time profile were calculated using WinNonlin Professional (v.5.2) software. The area under the 12-h IMPDH enzyme activity curve (AEC0–12) was calculated by the linear trapezoidal rule and 12-h IMPDH enzyme activity was estimated by abbreviated sampling strategies and calculated by the developed equations (AEC0–3, AEC0–4). Amax and Amin were defined as the maximum and minimum daytime enzyme activity after dosing with MPA within the dosing interval. Maximum inhibition of IMPDH activity (Imax) was calculated with the following formula (1 −Amin/Amax) × 100. Tmin was defined as the time to reach the minimum daytime enzyme activity.
Statistical analysis
All statistical analyses were performed using SPSS for Windows, Version 11.5 (SPSS Inc., Chicago, IL, USA). Data for continuous variables are expressed as median and ranges. Categorical variables are expressed as absolute numbers and percentages. PK and PD parameters were not normally distributed in the study population and the following nonparametric statistical tests were performed: the Mann–Whitney U-test (for comparison of sample medians) and the nonparametric Spearman rank correlation coefficient (for correlation between variables). Multivariate logistic regression analyses were calculated to determine the influence of several variables. Correlations and differences between groups were considered significant with an α error of 0.05 and a β error of 0.2 (power of 80%) in a two-sided statistical test.
LSS were established by the following procedure. MPA concentration and IMPDH activity at each sampling time were correlated by linear regression analysis with the total measured MPA AUC0–12 and IMPDH AEC0–12 in all patients. Multiple stepwise linear regression analysis was performed to give improved correlations with total measured MPA AUC0–12 and IMPDH AEC0–12. Equations were in the form of MPA AUC0–12=K+K0×C0+K1×C1 … K n×C n, where K, K0, K n are fitted constants associated with each timed MPA concentration, C0, C1 … C n are MPA concentrations at 0, 1 … nth h post dose. Prediction bias of these LSS-derived estimates was assessed by calculating the percentage of prediction error (PE%) from the formula PE% = 100% × (LSS AUC − total measured AUC)/total measured AUC. Corresponding analyses were performed on IMPDH data.
Results
Patient demographics
A total of 66 full 12-h MPA profiles (AUC0–12) in renal transplant patients [32 male, 34 female; median age 41 years (19–68); median time after transplantation 14 days (10–56)] on a broad range of EC-MPS therapy and stable renal allograft function [median estimated GFR 46 ml min−1 (30–76)] were obtained. Daily EC-MPS dosage was 1440 mg in 57 patients, 2880 mg in eight patients and 720 mg in one patient. Median methylprednisolone dosage was 20 mg (12–20) once daily. Median CsA dosage was 150 mg (100–300) twice daily with a median CsA C0 level of 178 µg l−1 (156–281). Patient characteristics are shown in Table 1.
Table 1.
Demographic data of 66 renal allograft recipients
| Patient characteristic | |
|---|---|
| Male gender (%)* | 48.5 (32/66) |
| Age (years)† | 41 (19–68) |
| Body mass index (kg m−2)† | 27 (17–38) |
| Time after transplantation (days)† | 14 (10–56) |
| Living donation (%)* | 42.4 (28/66) |
| First transplantation (%)* | 95.5 (63/66) |
| Second transplantation (%)* | 4.5 (3/66) |
| Cold ischaemia time (h)† | 9.6 (2–24) |
| HLA mismatches† | 3 (0–6) |
| Panel reactive antibodies > 25%* | 0 (0/66) |
| eGFR (ml min−1)† | 46 (30–76) |
| Methylprednisolone dosage (mg day−1)† | 20 (12–20) |
| Ciclosporin A dosage (mg day−1)† | 150 (100–300) |
| Ciclosporin C0 level (µg l−1)† | 178 (156–281) |
| Mycophenolate sodium dosage (mg day−1)† | 1440 (720–2880) |
| Mycophenolate sodium dosage per body weight (mg kg−1)† | 20 (10–40) |
Number and percentages.
Median and ranges are shown. C0, predose concentration; eGFR, estimated glomerular filtration rate; HLA, human leucocyte antigen.
Pharmacokinetic and pharmacodynamic analysis
An overview of the PK and PD data is given in Table 2 demonstrating a broad range of MPA exposure in the patient population. MPA AUC0–12 correlated poorly with MPA C0 concentration (r2= 0.239; P < 0.01) and MPA C12 concentration (r2= 0.361; P < 0.01). Individual MPA C0 and MPA C12 differed significantly (P < 0.05).
Table 2.
Pharmacokinetic and pharmacodynamic parameters of enteric-coated mycophenolate sodium (EC-MPS) in 66 renal transplant patients (median and range)
| All patients | EC-MPS 1440 mg day−1 | EC-MPS 2880 mg day−1 | Significance | |
|---|---|---|---|---|
| n= 66 | n= 57 | n= 8 | P | |
| MPA AUC0–12 (µg*h ml−1) | 38 (7–130) | 38 (7–130) | 40 (20–65) | NS |
| MPA C0 (µg ml−1) | 1.8 (0.5–23) | 1.8 (0.5–23) | 1.8 (0.6–16) | NS |
| MPA C12 (µg ml−1) | 1.5 (0.3–19.4) | 1.5 (0.3–19.4) | 1.5 (0.5–9.9) | NS |
| MPA Cmin (µg ml−1) | 1.0 (0.5–3.8) | 0.9 (0.5–3.8) | 1.2 (0.5–1.5) | NS |
| MPA Cmax (µg ml−1) | 11.6 (2.2–45.7) | 12 (2.2–45.7) | 11.8 (4.7–30.8) | NS |
| MPA Tmax (h) | 3.0 (0.5–10) | 3.0 (0.5–10) | 3.0 (2.0–6.0) | NS |
| Pharmacodynamic data | ||||
| IMPDH AEC0–12 (nmol*h mg−1 protein h−1) | 74 (12–371) | 79 (12–371) | 64 (15–110) | <0.05 |
| IMPDH A0 (nmol mg−1 protein h−1) | 8.4 (1.0–28.5) | 8.5 (1.0–28.5) | 5.6 (1.1–13.3) | NS |
| IMPDH A12 (nmol mg−1 protein h−1) | 8.0 (0.5–33.5) | 8.6 (0.5–33.5) | 4.9 (0.7–15.4) | NS |
| IMPDH Amax (nmol mg−1 protein h−1) | 15 (3–48) | 15 (5–48) | 11 (3–19) | <0.05 |
| IMPDH Amin (nmol mg−1 protein h−1) | 1.7 (0–20.7) | 1.7 (0–20.7) | 1.0 (0–2.3) | <0.05 |
| IMPDH Tmin (h) | 3 (0.5–10) | 3 (0.5–10) | 3 (2–5) | NS |
| IMPDH Imax (%) | 87 (57–100) | 87 (57–100) | 93 (71–100) | NS |
Fifty-seven patients were on 1440 mg day−1 and eight patients on 2880 mg day−1. One patient was on EC-MPS 720 mg day−1 (data not shown). Significance P is given for difference between the patient groups with EC-MPS 1440 mg day−1 and with EC-MPS 2880 mg day−1 (Mann–Whitney U-test). A, enzyme activity; AEC, area under the enzyme activity curve; AUC, area under the concentration–time curve; C, concentration; EC-MPS, enteric-coated mycophenolate sodium; I, inhibition; IMPDH, inosine monophosphate dehydrogenase; MPA, mycophenolic acid; T, time.
IMPDH AEC0–12 correlated with IMPDH A0 (r2= 0.412; P < 0.001) and with IMPDH A12 (r2= 0.450, P < 0.05). Individual IMPDH A0 and IMPDH A12 were not significantly different (P= 0.07).
MPA AUC0–12 and IMPDH AEC0–12 did not correlate significantly (Figure 1). The overall PD response is demonstrated by plotting IMPDH activity against MPA plasma concentration (Figure 2). The median MPA concentrations and median IMPDH activities observed over the 12-h sampling period are shown in Figure 3.
Figure 1.
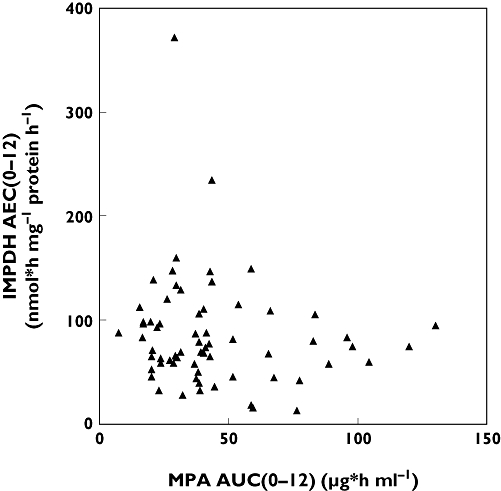
Association of MPA AUC0–12 and IMPDH AEC0–12 is demonstrated. AUC, area under the concentration–time curve; AEC, area under the enzyme activity curve; IMPDH, inosine monophosphate dehydrogenase; MPA, mycophenolic acid
Figure 2.
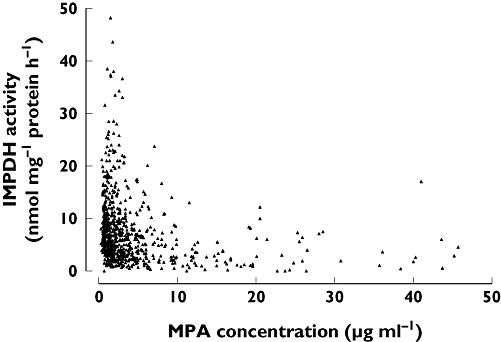
Assessment of pharmacodynamic response by plotting inosine monophosphate dehydrogenase (IMPDH) activity against plasma mycophenolic acid (MPA) concentration
Figure 3.
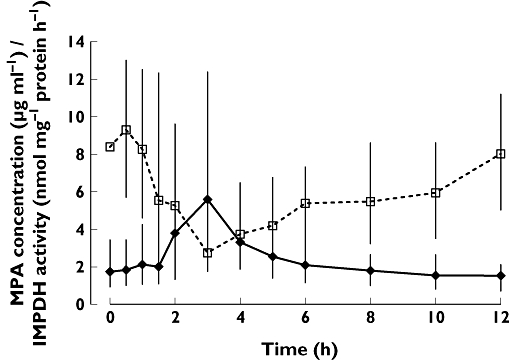
Median time profiles of plasma mycophenolic acid (MPA) concentration and inosine monophosphate dehydrogenase (IMPDH) activity in peripheral mononuclear cells of 66 renal transplant patients on enteric-coated mycophenolate sodium (EC-MPS) therapy are presented (median, 25 and 75 percentile; solid line, MPA concentration; dashed line, IMPDH activity). MPA concentration [µg ml−1] ( ); IMPDH activity [nmol/mg protein/h] (
); IMPDH activity [nmol/mg protein/h] ( )
)
Assessment of an abbreviated area under the concentration–time curve
Each MPA concentration at a single time point correlated poorly with AUC0–12. MPA AUC0–12 correlated best with MPA C1 (r2= 0.321, P < 0.001), MPA C3 (r2= 0.320, P < 0.001), MPA C8 (r2= 0.335, P < 0.001) and MPA C12 (r2= 0.424, P < 0.001). Using stepwise linear regression analysis, the best sampling algorithm with a maximum of three sampling time points included plasma MPA concentration at 1, 3 and 8 h after oral EC-MPS intake (C1, C3, C8; r2= 0.836, P < 0.001). Sampling points and corresponding equations are given in Table 3. Two highly predictive formulae with sampling points within the first 4 h were selected: MPA AUC0–3 including C0, C1 and C3 and MPA AUC0–4 including C0, C1, C2, C3 and C4. There was a significant correlation between estimated abbreviated MPA AUC0–3 as well as MPA AUC0–4 and AUC0–12 (r2= 0.702 and r2= 0.812). Inclusion of C0.5 and C1.5 did not rigorously improve accuracy of the LSS. Median PE% were −1% (−73–54) with MPA AUC0–3 and −8% (−45–47) with MPA AUC0–4.
Table 3.
Multiple stepwise linear regression analysis was made to identify a limited sampling strategy to estimate a 12-h exposure of mycophenolate acid in enteric-coated mycophenolate sodium (EC-MPS)-treated renal transplant patients (n= 66)
| Sampling time (h) | Equation for estimation of AUC0–12 | Coefficient of determination (r2) |
|---|---|---|
| Best limited sampling strategies with regard to number of sampling points | ||
| 1, 3 | 21.2 + 2.13 C1+ 1.55 C3 | 0.671 |
| 1, 3, 8 | 12.7 + 2.05 C1+ 1.51 C3+ 3.99 C8 | 0.836 |
| 1, 2, 3, 8 | 8.36 + 1.61 C1+ 0.75 C2+ 1.63 C3+ 4.13 C8 | 0.886 |
| 1, 2, 3, 4, 8 | 3.31 + 1.56 C1+ 0.89 C2+ 1.07 C3+ 1.95 C4+ 4.09 C8 | 0.950 |
| Best limited sampling strategies within the first 4 h (r2 > 0.7) | ||
| 0, 1, 3 | 19.7 + 1.22 C0+ 1.93 C1+ 1.39 C3 | 0.702 |
| 1, 3, 4 | 16.5 + 2.16 C1+ 1.06 C3+ 1.92 C4 | 0.738 |
| 1, 2, 3, 4 | 12.4 + 1.70 C1+ 0.78 C2+ 0.99 C3+ 2.15 C4 | 0.792 |
| 0, 1, 2, 3, 4 | 11.5 + 0.99 C0+ 1.56 C1+ 0.74 C2+ 0.87 C3+ 2.09 C4 | 0.812 |
| 0, 0.5, 1, 1.5, 2, 3, 4 | 2.56 + 0.57 C0+ 0.87 C0.5+ 0.48 C1+ 0.61 C1.5+ 0.55 C2+ 0.99 C3+ 2.08 C4 | 0.842 |
| Best limited sampling strategies within the first 6 h (r2 > 0.7) | ||
| 1, 3, 6 | 11.5 + 1.79 C1+ 1.59 C3+ 4.76 C6 | 0.797 |
| 1, 2, 3, 6 | 5.72 + 1.17 C1+ 0.92 C2+ 1.52 C3+ 5.70 C6 | 0.867 |
| 1, 2, 3, 4, 6 | 3.21 + 1.21 C1+ 0.97 C2+ 1.14 C3+ 1.50 C4+ 4.99 C6 | 0.904 |
Assessment of an abbreviated area under the enzyme activity curve
Correlation between IMPDH activity at a single time point and AEC0–12 ranged between r2= 0.239 and r2= 0.689. The best correlations between AEC0–12 and IMPDH activity at single time points were found at 6 h (C6, r2= 0.684, P < 0.001) and 8 h (C8, r2= 0.689, P < 0.001). Corresponding to the abbreviated PK sampling strategies, an abbreviated sampling model including A0 to A4 to predict AEC0–12 was evaluated (Table 4). As shown in Table 3, there was an acceptable correlation between estimated abbreviated IMPDH AEC and AEC0–12 using IMPDH AEC0–3 including A0, A1 and A3 and IMPDH AEC0–4 including A0, A1, A2, A3 and A4 (r2= 0.819 and r2= 0.833). Median PE% were 8% (−46–139) with IMPDH AEC0–3 and 1% (−43–83) with IMPDH AEC0–4.
Table 4.
Limited sampling algorithms to estimate a 12-h inhibition of inosine monophosphate dehydrogenase activity in enteric-coated mycophenolate sodium (EC-MPS)-treated renal transplant patients (n= 66). Algorithms were calculated by linear stepwise regression analysis
| Sampling time (h) | Equation for estimation of AEC0–12 | Coefficient of determination (r2) |
|---|---|---|
| 1, 3 | 30.8 + 3.03 A1+ 4.27 A3 | 0.791 |
| 0, 1, 3 | 24.2 + 1.81 A0+ 2.26 A1+ 4.0 A3 | 0.819 |
| 0, 1, 2, 3, 4 | 19.9 + 1.53 A0+ 1.78 A1+ 1.02 A2+ 2.81 A3+ 1.61 A4 | 0.833 |
Clinical data – univariate analysis
Patient and graft survival was 100% in the follow-up period of 12 weeks after transplantation. Median MDRD GFR was 52 ml/min (28–96) and neither MPA AUC0–12 nor MPA AUC0–3 or MPA AUC0–4 determined renal allograft function assessed by MDRD GFR at week 12. In the follow-up period, 13 biopsy proven acute rejection episodes were documented (nine borderline rejection, three Banff IA and one Banff IB). MPA AUC0–12 and MPA AUC0–4 of patients with early acute rejection episodes were significantly lower compared with patients without acute rejection episodes (MPA AUC0–12: median 28 µg*h ml−1 (7–45) vs. 40 µg*h ml−1 (16–130), P < 0.01; MPA AUC0–4: median 32 µg*h ml−1 (17–45) vs. 40 µg*h ml−1 (20–121), P < 0.05; MPA AUC0–3: median 35 µg*h ml−1 (22–56) vs. 38 µg*h ml−1 (23–125), n.s.). Infections were noted in nine of 66 (13.6%) patients and gastrointestinal side-effects in six of 66 (9.1%). MPA AUC0–12, MPA AUC0–4 and MPA AUC0–3 of patients with infections were significantly higher compared with patients without infections (MPA AUC0–12: median 65 µg*h ml−1 (37–130) vs. 37 µg*h ml−1 (7–120), P < 0.005; MPA AUC0–4: median 62 µg*h ml−1 (31–121) vs. 36 µg*h ml−1 (17–115), P < 0.05; MPA AUC0–3: median 53 µg*h ml−1 (27–125) vs. 35 µg*h ml−1 (22–115), P < 0.05). Neither MPA AUC0–12 nor MPA AUC0–4 or MPA AUC0–3 affected gastrointestinal side-effects (MPA AUC0–12: median 51 µg*h ml−1 (29–76) vs. 38 µg*h ml−1 (7–130), n.s.; MPA AUC0–4: median 39 µg*h ml−1 (30–68) vs. 36 µg*h ml−1 (17–121), n.s.; MPA AUC0–3: median 36 µg*h ml−1 (22–125) vs. 34 µg*h ml−1 (25–56), n.s.). The evaluated LSS MPA AUC0–4 confirmed significant MPA AUC0–12 differences between patients with and without acute rejections or infections. Association between MPA AUC0–4 and clinical outcome is given in Figure 4. Nevertheless, it was impossible to detect patients at risk of acute rejection, infection or gastrointestinal side-effects with the LSS MPA AUC0–3.
Figure 4.
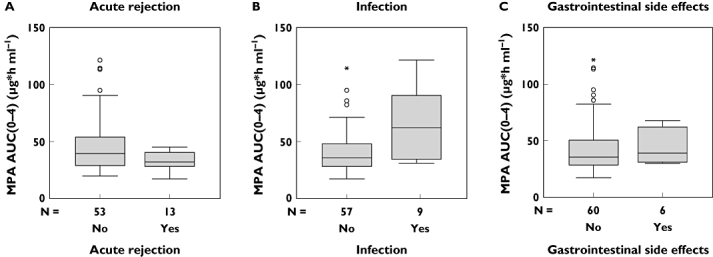
Box and whisker plot shows the median, interquartile range, outliers (°) and extreme values (*) of the evaluated abbreviated sampling strategy MPA AUC0–4 in patients (a) with and without acute rejection episode (P < 0.05), (b) with and without recurrent infections (P < 0.05), and with and without gastrointestinal side-effects (NS). AUC, area under the concentration–time curve; MPA, mycophenolic acid
Neither IMPDH AEC0–12 nor IMPDH AEC0–4 or IMPDH AEC0–3 determined renal allograft function assessed by MDRD GFR at week 12. There was no significant difference between IMPDH AEC0–12, IMPDH AEC0–4 and IMPDH AEC0–3 of patients with and without acute rejections [IMPDH AEC0–12: median 64 nmol*h mg−1 protein h−1 (28–110) vs. 74 nmol*h mg−1 protein h−1 (12–371), NS; IMPDH AEC0–4: median 64 nmol*h mg−1 protein h−1 (40–97) vs. 73 nmol*h mg−1 protein h−1 (30–315), NS; IMPDH AEC0–3: median 61 nmol*h mg−1 protein h−1 (44–100) vs. 78 nmol*h mg−1 protein h−1 (36–321), NS]. Two patients with an acute rejection episode despite a MPA AUC0–12 > 38 µg*h ml−1 showed an IMPDH AEC0–12 > 100 nmol*h mg−1 protein h−1. IMPDH AEC0–12, IMPDH AEC0–4 and IMPDH AEC0–3 of patients with and without infections did not differ significantly [IMPDH AEC0–12: median 68 nmol*h mg−1 protein h−1 (12–94) vs. 74 nmol*h mg−1 protein h−1 (15–371), NS; IMPDH AEC0–4: median 71 nmol*h mg−1 protein h−1 (35–124) vs. 72 nmol*h mg−1 protein h−1 (30–315), NS; IMPDH AEC0–3: median 72 nmol*h mg−1 protein h−1 (41–117) vs. 76 nmol*h mg−1 protein h−1 (36–321), NS]. IMPDH AEC0–12 and IMPDH AEC0–4 of patients with gastrointestinal side-effects were significantly lower compared with patients without gastrointestinal symptoms [IMPDH AEC0–12: median 43 nmol*h mg−1 protein h−1 (12–67) vs. 75 nmol*h mg−1 protein h−1 (15–371), P < 0.01; IMPDH AEC0–4: median 55 nmol*h mg−1 protein h−1 (35–73) vs. 75 nmol*h mg−1 protein h−1 (30–315), P < 0.05; IMPDH AEC0–3: median 59 nmol*h mg−1 protein h−1 (41–78) vs. 76 nmol*h mg−1 protein h−1 (36–321), NS].
The evaluated LSS IMPDH AEC0–4 confirmed an increased risk of low IMPDH AEC0–12 concerning gastrointestinal side-effects. Association of IMPDH AEC0–4 and clinical outcome is given in Figure 5. Nevertheless, it was impossible to detect patients at risk of acute rejection, infection or gastrointestinal side-effects with the LSS IMPDH AEC0–3.
Figure 5.
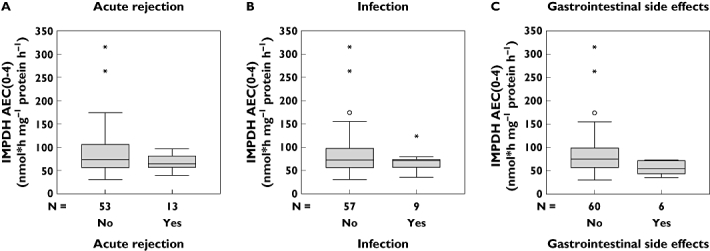
Box and whisker plot shows the median, interquartile range, outliers (°) and extreme values (*) of the evaluated abbreviated sampling strategy IMPDH AEC0–4 in patients (a) with and without acute rejection episode (NS), (b) with and without recurrent infections (NS), and (c) with and without gastrointestinal side-effects (P < 0.05). AEC, area under the enzyme activity curve; IMPDH, inosine monophosphate dehydrogenase
Clinical data – multivariate analysis
Multiple regression analyses were performed including the following variables: MPA dosage (mg kg−1), MPA Cmax, MPA AUC0–12, IMPDH Amin, IMPDH AEC0–12 and IMPDH Imax. MPA AUC0–12, IMPDH Imax and IMPDH Amin determined the risk of an acute rejection episode. MPA AUC0–12 predicted the risk of recurrent infections and IMPDH AEC0–12 independently revealed the risk of gastrointestinal side-effects. Data of multivariate logistic regression analyses are given in Table 5.
Table 5.
Results of multivariate logistic regression analyses concerning the association between pharmacokinetic and pharmacodynamic data and adverse events are given. Regression coefficient β, significance value P and 95% confidence interval are shown
| Coefficient β | P-value | Exp [b] (95% CI) | |
|---|---|---|---|
| Acute rejection (R= 0.50, SE = 0.36, P < 0.005) | |||
| MPA AUC0–12 | −0.49 | 0.02 | 0.90 (0.83, 0.98) |
| MPA Cmax | 0.32 | 0.13 | 1.12 (0.98, 1.27) |
| IMPDH AEC0–12 | −0.10 | 0.42 | 1.02 (0.98, 1.06) |
| IMPDH Amin | −0.43 | 0.01 | 0.23 (0.05, 1.0) |
| IMPDH Imax | −0.50 | 0.005 | 0.78 (0.65, 0.95) |
| MPA dosage* | −0.01 | 0.94 | 1.03 (0.92, 1.15) |
| Infections (R= 0.44, SE = 0.32, P < 0.05) | |||
| MPA AUC0–12 | 0.43 | 0.05 | 1.04 (1.00, 1.09) |
| MPA Cmax | −0.04 | 0.87 | 1.00 (0.88, 1.13) |
| IMPDH AEC0–12 | −0.10 | 0.41 | 0.99 (0.96, 1.02) |
| IMPDH Amin | −0.05 | 0.76 | 0.83 (0.31, 2.23) |
| IMPDH Imax | −0.06 | 0.76 | 0.98 (0.84, 1.14) |
| MPA dosage* | 0.12 | 0.32 | 1.05 (0.94, 1.18) |
| Gastrointestinal side effects (R= 0.36, SE = 0.28, P= 0.05) | |||
| MPA AUC0–12 | 0.11 | 0.62 | 0.99 (0.93, 1.06) |
| MPA Cmax | −0.07 | 0.76 | 1.05 (0.89, 1.24) |
| IMPDH AEC0–12 | −0.30 | 0.02 | 0.95 (0.90, 1.00) |
| IMPDH Amin | −0.06 | 0.73 | 0.87 (0.55, 1.39) |
| IMPDH Imax | −0.07 | 0.70 | 0.94 (0.81, 1.10) |
| MPA dosage* | 0.13 | 0.30 | 1.05 (0.92, 1.19) |
MPA dosage given in mg kg−1. A, activity; AUC, area under the concentration–time curve; C, concentration; CI, confidence interval; I, inhibition; IMPDH, inosine monophosphate dehydrogenase; MPA, mycophenolic acid; R, multiple correlation coefficient; SE, standard error of estimation.
Discussion
EC-MPS as an enteric-coated formulation of MPA has approved equal clinical efficacy and safety compared with MMF in de novo renal transplant patients [25]. However, some PK and PD data differ significantly in both drug formulations [18, 26]. This is the first study assessing PK and PD parameters of EC-MPS in de novo renal transplant patients with stable allograft function. Results are similar to published studies [18, 25]. In an individual patient's IMPDH activity, the pharmacological targets of EC-MPS are inversely related to plasma MPA concentration. Evaluation of an appropriate limited PK and PD sampling strategy suggested formulae consisting of five blood samples from 0 to 4 h after oral EC-MPS intake as appropriate tools for estimating MPA 12-h exposure in patients treated with CsA. Clinical associations of MPA exposure were revealed with respect to acute rejection, infection and gastrointestinal side-effects.
Steady-state PK data of MPA (Cmin, Cmax, Tmax and AUC) after oral intake of EC-MPS were consistent with previous results [18, 25]. Consistent with the enteric-coated formulation and delayed release, EC-MPS demonstrated a postponed MPA peak level at 3 h (0.5–10) after oral intake. This study confirmed that predose MPA levels are highly variable in EC-MPS-treated renal transplant patients and differ significantly from MPA C12. In addition, MPA AUC0–12 correlated poorly with MPA C0 and MPA C12. It is of note that MPA exposure varied widely despite corresponding daily MPA dosages and even body-weight-normalized MPA dosages did not determine MPA exposure. These findings support the request for individual drug dosing.
TDM is one of the main aspects of optimizing and personalizing immunosuppressive therapy. As mentioned above, the use of MPA C0 for monitoring of MPA in EC-MPS treatment is not appropriate because of an absent correlation between MPA C0 and MPA AUC0–12. Individual MPA exposure should be assessed by MPA AUC0–12 profiles. However, these strategies are time consuming, costly and inconvenient for the patients, and therefore impractical in daily clinical work. In the APOMYGRE study, the new dosage was calculated by the Bayesian formula and the results of the LSS correlated significantly with AUC0–12[11]. FDCC investigators used a three-point abbreviated sampling strategy including C0, C0.5 and C2[12]. However, limited data on TDM in EC-MPS-treated patients are available. Budde et al. [18] and Cattaneo et al. [27] demonstrated delayed MPA peak concentration compared with MMF. MPA 12-h exposure was comparable in 18 stable renal transplant patients on MMF therapy converted to EC-MPS [18]. Until now, only de Winter et al. [19] have evaluated most recently a LSS for TDM of EC-MPS in renal transplant recipients. However, the authors analysed only the estimation of MPA AUC0–12 with LSS for EC-MPS drawn within 2 or 3 h post dose in the maintenance period after transplantation. TDM using an easy abbreviated sampling strategy is challenging with respect to the observed variation in PK profiles of EC-MPS [26].
In this study, stepwise logistic analysis revealed a five-point sampling strategy including blood samples at predose, 1, 2, 3 and 4 h post-EC-MPS intake as a promising tool for adequate and convenient assessment of drug exposure in renal allograft recipients treated with CsA. Such a LSS might be appropriately performed in outpatient clinics. Additional sampling time points at 0.5 and 1.5 h after oral intake did not improve accuracy when a LSS of 4 h was applied. It is of note that in this study CsA was the concomitant immunosuppressive agent in all patients. CsA is known to inhibit the multidrug resistance 2 transporters followed by reduction of enterohepatic MPA recycling [28]. A relevant second MPA peak level is not expected in combined EC-MPS and CsA treatment and therefore the establishment of an abbreviated sampling strategy might be possible [29]. The alternative widely used calcineurin inhibitor tacrolimus presents a recirculation issue of MPA affecting the 12-h MPA profiles [30, 31]. This is likely to influence possible LSS and equations in MPA- and tacrolimus-treated patients. However, in this study no tacrolimus-treated patients were evaluated.
Randomized, concentration-controlled trials in renal transplant patients on MMF, CsA and corticosteroids provide the basis of the currently recommended therapeutic range of MPA AUC0–12 of 30 to 60 µg*h ml−1 to achieve efficacy and avoid toxicity in the early post-transplantation period [6, 16, 32]. Most patients treated with the ester prodrug MMF did not reach the intended target MPA AUC0–12 of 30 to 60 µg*h ml−1 in the early post-transplant period and TDM might be especially useful in this early time period [12, 13, 32, 33]. In the present study, median MPA AUC0–12 was 38 µg*h ml−1, suggesting that a considerable number of the patients exhibited a MPA exposure below MPA AUC0–12 of 30 µg*h ml−1. Even patients with increased EC-MPS dosages did not reach sufficient drug exposure. These results are comparable with published data that reveal a low EC-MPS exposure in the first weeks after renal transplantation with a time-dependent PK of MPA despite standard dosing. It is reported that the mean AUC0–12 of MPA in the early post-transplantation period is approximately 30–50% lower for the same dose than in the late post-transplantation period [34]. This is especially important because clinical data confirmed an association between MPA exposure and efficacy. The AUC values <30 µg*h ml−1 were associated with an increased risk for development of acute rejection, whereas no further reduction in acute rejection was observed in patients with AUC values > 60 µg*h ml−1 in MMF-treated renal allograft recipients [32]. However, clinical experience indicates that escalation of MMF dosages is limited by gastrointestinal intolerance.
Several clinical trials have been conducted to investigate whether MPA TDM and concentration-controlled MMF dosing is feasible in clinical practice and whether it is superior to using a fixed MMF dose [33, 35]. Recently, three prospective randomized multicentre studies – the French APOMYGRE study, the FDCC trial and the OPTICEPT trial – have confirmed a clinical benefit of TDM in patients treated with the ester prodrug MMF [9, 10, 36]. In the APOMYGRE trial, the incidence of treatment failure (acute rejection, graft loss, death, discontinuation of MMF) was significantly lower in the concentration-controlled group [11]. In particular, the incidence of acute rejection was significantly lower during the first year (12.3% vs. 30.7%) and there was no difference in the incidence of adverse events. In the FDCC trial, immunosuppression consisted of MMF in combination with CsA or tacrolimus [12]. Despite of a similar MPA exposure in patients on fixed-dose and concentration-controlled MPA regimens, in all patients a low MPA exposure at day 3 after transplantation was followed by an increased risk of biopsy-proven acute rejections within the first year after transplantation. In the recently published OPTICEPT trial, MMF dose adjustments were performed based on the MPA trough levels and MPA trough concentrations ≥ 1.6 µg ml−1 were associated with a longer time to first biopsy-proven acute rejection episode in tacrolimus-treated patients from all three groups [36]. Despite a relatively clear relationship between MPA exposure and clinical efficacy, these prospective randomized multicentre studies have shown that it might be difficult to establish a similarly strong association between the PK parameters of MPA and drug-related adverse events [37]. Our present study has confirmed a relationship between MPA exposure in EC-MPS- and CsA-treated renal transplant patients and acute rejection episodes. In contrast to the mentioned multicentre studies, the present study also suggested a relationship between recurrent infections and MPA exposure.
In this study, all patients diagnosed with biopsy-proven acute rejections displayed a MPA AUC0–12 < 45 µg*h ml−1 and the median MPA AUC0–12 was approximately 30 µg*h ml−1– demonstrating for the first time a exposure response relationship for EC-MPS-treated patients, similar to MMF-treated allograft recipients. In contrast, patients with high MPA exposure were at an increased risk of adverse events. MPA AUC0–12 of nearly all patients with recurrent infections was >45 µg ml−1 with a median MPA AUC0–12 of 65 µg*h ml−1. Only three patients with recurrent infections had a MPA AUC0–12 between 39 and 42 µg*h ml−1. PK monitoring of plasma MPA concentrations in renal transplant patients on EC-MPS therapy might be a useful tool for achieving sufficient drug exposure and optimizing individual immunosuppressive treatment. Results of this study support the established MPA AUC0–12 target level of 30–60 µg*h ml−1 in patients on EC-MPS and CsA immunosuppression.
A PD monitoring strategy by IMPDH activity in lymphocytes as the biological target of MPA was proposed recently in renal transplant patients and in patients with IgA nephritis treated with MMF [23, 38–40]. Assessment of IMPDH activity serves as a surrogate marker of MPD-induced immunosuppression and, in comparison with conventional TDM, might better predict drug efficacy and toxicity. IMPDH activity exhibited high interindividual variability in patients with low intraindividual variability [23]. After oral administration of MMF, close temporal associations between MPA plasma levels and IMPDH inhibition have been reported [18, 40]. This study confirms that individual MPA levels and IMPDH activities run inversely in a daytime observation in EC-MPS treatment. However, the correlation between MPA trough levels and predose IMPDH activity is weak. In most patients, the highest inhibition of IMPDH activity was observed within the first 4 h after oral EC-MPS intake. This is in accordance with earlier studies, which demonstrated the lowest IMPDH activity within the first 3 h in renal transplant patients treated with MMF and CsA [18]. Therefore, a LSS including at least the first 4 h might cover the highly variable absorption period.
There are only limited data on a clinical benefit of IMPDH monitoring in MPA-treated patients. In earlier studies, it has been suggested that assessment of individual IMPDH activity provides clinical benefits. A significant association between IMPDH activity before transplantation and the necessity to reduce MMF dose after transplantation because of MPA-induced complications has been shown. Conversely, dose reduction due to side-effects in patients with low pre-transplant IMPDH activity was followed by an increased risk of acute rejection [23]. This study showed that the incidence of infections was increased in patients with low IMPDH activity despite adequate MPA exposure. In addition, independent of MPA exposure, patients with low IMPDH activity were at an increased risk of gastrointestinal side-effects. These results confirmed an augmented risk of side-effects in patients on EC-MPS treatment with low IMPDH activity assessed by IMPDH AEC0–12. Multivariate regression analysis proved that both PK and PD monitoring contribute to an optimized individual immunosuppression. These preliminary data are promising and indicate that IMPDH activity might be a useful surrogate marker of MPA-induced immunosuppression.
There are some study limitations to be considered. First, EC-MPS is combined with CsA, which is known to interfere with the enterohepatic recirculation of MPA. In this study, LSS of EC-MPS are only evaluated in combination with the calcineurin inhibitor CsA in renal allograft recipients. The study population size was relatively small because of the single-centre design. Prognostic impact of PK and PD EC-MPS monitoring should be prospectively assessed in further clinical trials with long-term follow-up.
Conclusions
This study has confirmed a high interindividual variability of PK and PD data in EC-MPS-treated renal transplant patients. On the basis of the presented results, it is possible to use a limited sampling algorithm for practical monitoring of MPA in renal transplant patients on EC-MPS and CsA therapy. Including PK and practical aspects, a LSS with blood samples drawn within the first 4 h after intake of EC-MPS is recommended. Both PK and PD parameters determine the risk of adverse events. Further data are necessary to confirm clinical benefits of drug monitoring by PK and PD data in EC-MPS-treated patients. In particular, the optimal time point of MPA monitoring has to be evaluated with respect to short- and long-term clinical benefits.
Competing interests
None to declare.
The authors thank Friederike Hug and Sabine Bönisch-Schmidt (Department of Nephrology, University Hospital Heidelberg) for excellent technical assistance.
REFERENCES
- 1.Grinyo J, Groth C, Pichlmayr R, Sadek SA, Vanrenterghem Y the European Mycophenolate Mofetil Cooperative Study Group. Placebo-controlled study of mycophenolate mofetil combined with ciclosporin and corticosteroids for prevention of acute rejection. Lancet. 1995;345:1321–5. [PubMed] [Google Scholar]
- 2.Keown P, Häyry P, Mathew T, Morris P, Stiller C, Barker C, Carr L the Tricontinental Mycophenolate Mofetil Renal Transplantation Study Group. A blinded, randomised clinical trial of mycophenolate mofetil for the prevention of acute rejection in cadaveric renal transplantation. Transplantation. 1996;61:1029–37. [PubMed] [Google Scholar]
- 3.Sollinger HW. Mycophenolate mofetil for the prevention of acute rejection in primary cadaveric renal allograft recipients. U.S. Renal Transplant Mycophenolate Mofetil Study Group. Transplantation. 1995;60:225–32. doi: 10.1097/00007890-199508000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Meier-Kriesche HU, Steffen BJ, Hochberg AM, Gordon RD, Liebman MN, Morris JA, Kaplan B. Mycophenolate mofetil versus azathioprine therapy is associated with a significant protection against long-term renal allograft function deterioration. Transplantation. 2003;75:1341–6. doi: 10.1097/01.TP.0000062833.14843.4B. [DOI] [PubMed] [Google Scholar]
- 5.Bullingham R, Monroe S, Nicholls A, Hale M. Pharmacokinetics and bioavailability of mycophenolate mofetil in healthy subjects after single-dose oral and intravenous administration. J Clin Pharmacol. 1996;36:315–24. doi: 10.1002/j.1552-4604.1996.tb04207.x. [DOI] [PubMed] [Google Scholar]
- 6.Van Gelder T, Hilbrands LB, Vanrenterghem Y, Weimar W, de Fijter JW, Squifflet JP, Hené RJ, Verpooten GA, Navarro MT, Hale MD, Nicholls AJ. A randomised double-blind, multicenter plasma concentration controlled study of the safety and efficacy of oral mycophenolate mofetil for the prevention of acute rejection after kidney transplantation. Transplantation. 1999;68:261–6. doi: 10.1097/00007890-199907270-00018. [DOI] [PubMed] [Google Scholar]
- 7.Mourad M, Malaise J, Eddour DC, De Meyer M, König J, Schepers R, Squifflet JP, Wallemacq P. Correlation of mycophenolic acid pharmacokinetic parameters with side effects in kidney transplant patients treated with mycophenolate mofetil. Clin Chem. 2001;47:88–94. [PubMed] [Google Scholar]
- 8.Pascual M, Theruvath T, Kawai T, Tolkoff-Rubin N, Cosimi AB. Strategies to improve long-term outcomes after renal transplantation. N Engl J Med. 2002;346:580–90. doi: 10.1056/NEJMra011295. [DOI] [PubMed] [Google Scholar]
- 9.Hyunyoung J, Kaplan B. Therapeutic monitoring of mycophenolate mofetil. Clin J Am Soc Nephrol. 2007;2:184–91. doi: 10.2215/CJN.02860806. [DOI] [PubMed] [Google Scholar]
- 10.Knight SR, Morris PJ. Does the evidence support the use of mycophenolate mofetil therapeutic drug monitoring in clinical practice? A systematic review. Transplantation. 2008;85:1675–85. doi: 10.1097/TP.0b013e3181744199. [DOI] [PubMed] [Google Scholar]
- 11.Le Meur Y, Buchler M, Thierry A, Caillard S, Villemain F, Lavaud S, Etienne I, Westeel PF, de Ligny BH, Rostaing L, Thervet E, Szelag JC, Rérolle JP, Rousseau A, Touchard G, Marquet P. Individualised mycophenolate mofetil dosing based on drug exposure significantly improves patient outcomes after renal transplantation. Am J Transplant. 2007;7:2496–503. doi: 10.1111/j.1600-6143.2007.01983.x. [DOI] [PubMed] [Google Scholar]
- 12.van Gelder T, Tedesco-Silva H, de Fijter JW, Budde K, Kypers D, Tyden G, Lohmus A, Sommerer C, Hartmann A, Le Meur Y, Oellerich M, Holt DW, Tönshoff B, Keown P, Campbell S, Mamelok RD. Comparing mycophenolate mofetil regimens for de novo renal transplant recipients: the fixed-dose concentration-controlled (FDCC) trial. Transplantation. 2008;86:1043–51. doi: 10.1097/TP.0b013e318186f98a. [DOI] [PubMed] [Google Scholar]
- 13.Van Hest RM, Mathot RA, Pescovitz MD, Gordon R, Mamelok RD, van Gelder T. Explaining variability in mycophenolic acid exposure to optimise mycophenolate mofetil dosing: a population pharmacokinetic meta-analysis of mycophenolic acid in renal transplant recipients. J Am Soc Nephrol. 2006;17:871–80. doi: 10.1681/ASN.2005101070. [DOI] [PubMed] [Google Scholar]
- 14.Shaw LM, Nicholls A, Hale M, Armstrong VW, Oellerich M, Yatscoff R, Morris RE, Holt DW, Venkataramanan R, Haley J, Halloran P, Ettenger R, Keown P, Morris RG. Therapeutic monitoring of mycophenolic acid. A consensus panel report. Clin Biochem. 1998;31:317–22. doi: 10.1016/s0009-9120(98)00040-x. [DOI] [PubMed] [Google Scholar]
- 15.Shaw LM, Korecka M, Venkataramanan R, Goldberg L, Bloom R, Brayman KL. Mycophenolic acid pharmacodynamics and pharmacokinetics provide a basis for rational monitoring strategies. Am J Transplant. 2003;3:534–42. doi: 10.1034/j.1600-6143.2003.00079.x. [DOI] [PubMed] [Google Scholar]
- 16.van Gelder T, Le Meur Y, Shaw LM, Oellerich M, DeNofrio D, Holt C, Holt DW, Kaplan B, Kuypers D, Meiser B, Toenshoff B, Mamelok RD. Therapeutic drug monitoring of mycophenolate mofetil in transplantation. Ther Drug Monit. 2006;28:145–54. doi: 10.1097/01.ftd.0000199358.80013.bd. [DOI] [PubMed] [Google Scholar]
- 17.Arns W, Cibrik DM, Walker RG, Mourad G, Budde K, Mueller EA, Vincenti F. Therapeutic drug monitoring of mycophenolic acid in solid organ transplant patients treated with mycophenolate mofetil: review of the literature. Transplantation. 2006;82:1004–12. doi: 10.1097/01.tp.0000232697.38021.9a. [DOI] [PubMed] [Google Scholar]
- 18.Budde K, Bauer S, Hambach P, Hahn U, Röblitz H, Mai I, Diekmann F, Neumayer HH, Glander P. Pharmacokinetic and pharmacodynamic comparison of enteric-coated mycophenolate sodium and mycophenolate mofetil in maintenance renal transplant patients. Am J Transplant. 2007;7:888–98. doi: 10.1111/j.1600-6143.2006.01693.x. [DOI] [PubMed] [Google Scholar]
- 19.de Winter BC, van Gelder T, Mathot RA, Glander P, Tedesco-Silva H, Hilbrands L, Budde K, van Hest RM. Limited sampling strategies drawn within 3 hours postdose poorly predict mycophenolic acid area-under-the-curve after enteric-coated mycophenolate sodium. Ther Drug Monit. 2009;31:585–91. doi: 10.1097/FTD.0b013e3181b8679a. [DOI] [PubMed] [Google Scholar]
- 20.Glander P, Braun KP, Hambach P, Bauer S, Mai I, Roots I, Waiser J, Fritsche L, Neumayer HH, Budde K. Non-radioactive determination of inosine 5’-monophosphate dehydrogenase (IMPDH) in peripheral mononuclear cells. Clin Biochem. 2001;34:543–9. doi: 10.1016/s0009-9120(01)00267-3. [DOI] [PubMed] [Google Scholar]
- 21.Levey AS, Greene T, Schluchter MD, Cleary PA, Teschan PE, Lorenz RA, Molitch ME, Mitch WE, Siebert C, Hall PM. Glomerular filtration rate measurements in clinical trials. Modification of Diet in Renal Disease Study Group and the Diabetes Control and Complications Trial Research Group. J Am Soc Nephrol. 1993;4:1159–71. doi: 10.1681/asn.v451159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solez K, Colvin RB, Racusen LC, Sis B, Halloran PF, Birk PE, Campbell PM, Cascalho M. Banff ‘05 Meeting Report: differential diagnosis of chronic allograft injury and elimination of chronic allograft nephropathy (‘CAN’) Am J Transplant. 2007;7:3518–26. doi: 10.1111/j.1600-6143.2006.01688.x. [DOI] [PubMed] [Google Scholar]
- 23.Glander P, Hambach P, Braun KP, Fritsche L, Giessing M, Mai I, Einecke G, Waiser J, Neumayer HH, Budde K. Pre-transplant inosine monophosphate dehydrogenase activity is associated with clinical outcome after renal transplantation. Am J Transplant. 2004;4:237–43. doi: 10.1111/j.1600-6143.2004.00617.x. [DOI] [PubMed] [Google Scholar]
- 24.Montero C, Duley JA, Fairbanks LD, McBride MB, Micheli V, Cant AJ, Morgan G. Demonstration of induction of erythrocyte inosine monophosphate dehydrogenase activity in Ribavirin-treated patients using a high-performance liquid chromatography linked method. Clin Chem Acta. 1995;238:169–78. doi: 10.1016/0009-8981(95)06088-u. [DOI] [PubMed] [Google Scholar]
- 25.Salvadori M, Holzer H, de Mattos A, Sollinger H, Arns W, Oppenheimer F, Maca J, Hall M The ERL B301 Study Groups. Enteric-coated mycophenolate sodium is therapeutically equivalent to mycophenolate mofetil in de novo renal transplant patients. Am J Transplant. 2004;4:231–6. doi: 10.1046/j.1600-6143.2003.00337.x. [DOI] [PubMed] [Google Scholar]
- 26.Budde K, Glander P, Kramer BK, Fischer W, Hoffmann U, Bauer S, Grohmann J, Neumayer HH, Arns W. Conversion from mycophenolate mofetil to enteric-coated mycophenolate sodium in maintenance renal transplant recipients receiving tacrolimus: clinical, pharmacokinetic and pharmacodynamic outcomes. Transplantation. 2007;83:417–24. doi: 10.1097/01.tp.0000251969.72691.ea. [DOI] [PubMed] [Google Scholar]
- 27.Cattaneo D, Cortinovis M, Baldelli S, Bitto A, Gotti E, Remuzzi G, Perico N. Pharmacokinetics of mycophenolate sodium and comparison with the mofetil formulation in stable kidney transplant recipients. Clin J Am Soc Nephrol. 2007;2:1147–55. doi: 10.2215/CJN.02820707. [DOI] [PubMed] [Google Scholar]
- 28.Hesselink DA, van Hest RM, Mathot RA, Bonthuis F, Weimar W, de Bruin RW, van Gelder T. Ciclosporin interacts with mycophenolic acid by inhibiting the multidrug resistance-associated protein 2. Am J Transplant. 2005;5:987–94. doi: 10.1046/j.1600-6143.2005.00779.x. [DOI] [PubMed] [Google Scholar]
- 29.Shipkova M, Armstrong VW, Kuypers D, Perner F, Fabrizi V, Holzer H, Wieland E, Oellerich M for the investigators of the MMF Creeping Creatinine Study Group. Effect of ciclosporin withdrawal on mycophenolic acid pharmacokinetics in kidney transplant recipients with deteriorating renal function: preliminary report. Ther Drug Monit. 2001;23:717–21. doi: 10.1097/00007691-200112000-00020. [DOI] [PubMed] [Google Scholar]
- 30.van Gelder T, Klupp J, Barten MJ, Christians U, Morris RE. Comparison of the effects of tacrolimus and ciclosporin on the pharmacokinetics of mycophenolic acid. Ther Drug Monit. 2001;23:119–28. doi: 10.1097/00007691-200104000-00005. [DOI] [PubMed] [Google Scholar]
- 31.Kuypers D, Ekberg H, Grinyo J, Nashan B, Vincenti S, Snell P, Mamelok RD, Bouw RM. Mycophenolic acid exposure after administration of mycophenolate mofetil in the presence and absence of ciclosporin in renal transplant recipients. Clin Pharmacokinet. 2009;48:329–41. doi: 10.2165/00003088-200948050-00005. [DOI] [PubMed] [Google Scholar]
- 32.Hale MD, Nicholls AJ, Bullingham RE, Hené R, Hoitsma A, Squifflet JP, Weimar W, Vanrenterghem Y, Van de Woude FJ, Verpooten GA. The pharmacokinetic–pharmacodynamic relationship for mycophenolate mofetil in renal transplantation. Clin Pharmacol Ther. 1998;64:672–83. doi: 10.1016/S0009-9236(98)90058-3. [DOI] [PubMed] [Google Scholar]
- 33.Kiberd BA, Lawen J, Fraser AD, Keough-Ryan T, Belitsky P. Early adequate mycophenolic acid exposure is associated with less rejection in kidney transplantation. Am J Transplant. 2004;4:1079–83. doi: 10.1111/j.1600-6143.2004.00455.x. [DOI] [PubMed] [Google Scholar]
- 34.Weber LT, Shipkova M, Armstrong VW, Wagner N, Schütz E, Mehls O, Zimmerhackl LB, Oellerich M, Tönshoff B. Comparison of the EMIT immunoassay with HPLC for therapeutic drug monitoring of mycophenolic acid in paediatric renal transplant recipients on mycophenolate mofetil therapy. Clin Chem. 2002;48:517–25. [PubMed] [Google Scholar]
- 35.Borrows R, Chusney G, Loucaidou M, James A, Lee J, Tromp JV, Owen J, Cairns T, Griffith M, Hakim N, McLean A, Palmer A, Papalois V, Taube D. Mycophenolic acid 12-h trough level monitoring in renal transplantation: association with acute rejection and toxicity. Am J Transplant. 2006;6:121–8. doi: 10.1111/j.1600-6143.2005.01151.x. [DOI] [PubMed] [Google Scholar]
- 36.Gaston RS, Kaplan B, Shah T, Cibrik D, Shaw LM, Angelis M, Mulgaonkar S, Meier-Kriesche HU, Patel D, Bloom RD. Fixed- or controlled-dose mycophenolate mofetil with standard- or reduced-dose calcineurin inhibitors: the OPTICEPT trial. Am J Transplant. 2009;9:1607–19. doi: 10.1111/j.1600-6143.2009.02668.x. [DOI] [PubMed] [Google Scholar]
- 37.Kuypers DRJ, de Jonge H, Naesens M, de Loor H, Halewick E, Dekens M, Vanrenterghem Y. Current target ranges of mycophenolic acid exposure and drug-related adverse events: a five-year, open-label, prospective, clinical follow-up study in renal allograft recipients. Clin Ther. 2008;30:673–83. doi: 10.1016/j.clinthera.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 38.Czock D, Rasche FM, Carius A, Glander P, Budde K, Bauer S, Keller F, von Müller L. Pharmacokinetics and pharmacodynamics of mycophenolic acid after enteric-coated mycophenolate versus mycophenolate mofetil in patients with progressive IgA nephritis. J Clin Pharmacol. 2007;47:850–9. doi: 10.1177/0091270007301624. [DOI] [PubMed] [Google Scholar]
- 39.Weimert NA, Derotte M, Alloway RR, Woodle ES, Vinks AA. Monitoring of inosine monophosphate dehydrogenase activity as a biomarker for mycophenolic acid effect: potential clinical implications. Ther Drug Monit. 2007;29:141–9. doi: 10.1097/FTD.0b013e31803d37b6. [DOI] [PubMed] [Google Scholar]
- 40.Langman LJ, LeGatt DF, Halloran PF, Yatscoff RW. Pharmacodynamic assessment of mycophenolic acid-induced immunosuppression in renal transplant recipients. Transplantation. 1996;62:666–72. doi: 10.1097/00007890-199609150-00022. [DOI] [PubMed] [Google Scholar]