

SUMMARY
Synthetic peptides encoding protective pathogen-derived epitopes represent – in principle – an ideal approach to T cell vaccination. Empirically, however, these strategies have not been successful. In the current study, we profiled the early activation of CD8+ T cells by MHC class I-restricted peptide immunization to better understand the biology of this response. We found that CD8+ T cells proliferated robustly in response to low doses of short synthetic peptides in PBS, but failed to acquire effector function or form memory populations in the absence of the TLR ligand CpG. CpG was unique among TLR ligands in its ability to enhance the response to peptide and its adjuvant effects had strict temporal requirements. Interestingly, CpG treatment modulated T cell expression of the surface receptors PD-1 and CD25, providing insight into its possible adjuvant mechanism. The effects of CpG on peptide immunization were dramatically enhanced in the absence of B cells, demonstrating a unique system of regulation of T cell responses by these lymphocytes. The results reported here provide insight into the complex response to a simple vaccination regimen, as well as provide a framework for a rational peptide-based vaccine design to both exploit and overcome targeted aspects of the immune response.
Keywords: T cell, peptide, CpG, B cell, PD-1
INTRODUCTION
CD8+ T cells specific for the SYVPSAEQI epitope of the Plasmodium yoelii circumsporozoite protein (CSP) are induced by immunization with radiation-attenuated sporozoites and strongly inhibit the development of liver stage parasites [1-5]. In view of their efficiency at inducing protective immunity, attenuated parasites have been proposed as a vaccine for humans. Obtaining these parasites is, however, a laborious and costly process, as they need to be isolated aseptically from the salivary glands of infected mosquitoes and maintained in a viable state until immediately before vaccination. As an alternative approach, the development of subunit vaccines containing parasite-derived antigenic moieties has been the focus of research in many laboratories in the last two decades. While encouraging results have been obtained on the induction of protective humoral responses, only modest success has been achieved on the induction of protective parasite-specific T cell mediated immune responses.
Immunization with short synthetic peptides encompassing MHC class I-restricted epitopes could be – in principle – the simplest subunit vaccine that targets the adaptive immune system. Peptide-based vaccination strategies would have many advantages, including low cost, safety, stability and ease of synthesis and modification. However, peptide vaccine approaches have not been successful. The reasons for the poor outcomes of peptide vaccinations are not well understood, but some studies in mice have demonstrated that, instead of activating T cells, soluble peptides tolerize and/or delete antigen-specific T cells [6-9]. Immunization with peptides together with adjuvants such as CFA, LPS, or CpG, is able to induce small populations of memory CD8+ T cells. Unfortunately, these populations accumulate primarily in the local draining lymph nodes and are largely undetectable by direct ex vivo assays, requiring in vitro secondary expansion for detection [10-13]. Recent studies have reported some success at improving these apparent limitations and describe the induction of memory T cell populations using synthetic peptide antigens [14-19]. However, these studies have employed repeated immunizations, high doses of antigen, large quantities of recombinant cytokines, and/or potent agonistic antibodies to T cell costimulatory machinery – strategies that may not be feasible in a mass vaccination setting.
Here we describe studies aimed to characterize the basic features of the CD8+ T cell responses induced by immunization with short synthetic peptides. We tracked the response of TCR-transgenic T cells to a vaccination of peptide alone and in combination with different TLR agonists and found that soluble peptides alone are highly immunogenic in vivo, but fail to induce mechanisms promoting the survival of activated T cells. Indeed, peptide-primed CD8+ T cells display unique phenotypic features indicative of poor survival and inability to expand. Further, we identify the TLR-9 agonist, CpG, and B cells as major factors that can positively and negatively affect, respectively, the establishment of long-term memory CD8+ T cell populations in response to peptide immunization.
RESULTS
CD8+ T cells induced by soluble peptide proliferate but fail to survive
To study the CD8+ T cell responses to soluble peptide immunization, we used an experimental system based on the adoptive transfer of naive CD8+ T cells expressing a transgenic T-cell receptor (TCR-Tg) specific for the epitope SYVPSAEQI from the CS protein of P. yoelii malaria parasites. Given that primary T cell responses to peptide-based immunization have been difficult to detect directly ex vivo or upon transfer of small numbers (2×103) TCR-Tg cells (Supplementary Figure 1), we began our studies by transferring 5×105 CFSE-labeled TCR-Tg T cells so that early priming events could be readily visualized by the dilution profile of the labeled T cells. We established that as little as 2.5 μg of peptide in PBS induced a strong proliferative response, detectable as early as three days after immunization in the spleen and in the lymph nodes draining the site of immunization (Fig 1a). In fact, as little as 0.25 μg of peptide was able to induce measurable T cell proliferation in the lymph nodes draining the site of immunization, though a systemic response was not observed. Increasing the amount of peptide to 25 μg resulted in an unphysiological T cell proliferation profile. Thus, we carried out further experiments with a peptide dose range of 2.5-5 μg.
Figure 1. CD8+ T cells proliferate in response to peptide, but do not survive.

TCR-Tg cells (5 × 105) were labeled with CFSE and transferred into naïve BALB/c mice. The following day, the mice were immunized with either indicated amounts of SYVPSAEQI peptide (A), 2.5 μg peptide (B and C), or 5 × 104 γ-irradiated sporozoites (γ-SPZ) (D) subcutaneously at the base of the tail. Control mice received PBS alone. (A) Three days later, spleens and draining lymph nodes were removed and single cell suspensions were stained and analyzed by FACS. CFSE profiles of CD8+tetramer+ cells from pooled draining lymph nodes of three mice and a representative spleen are shown. (B) Pooled lymph node and (C) individual spleen samples were analyzed as in (A) at three and five days after immunization. Numbers represent percent CD8+tetramer+ cells among total CD8+ cells. Values for lymph nodes samples are from a pool of three mice and spleen samples are the mean ± s.d. of three mice. (D) Spleen samples from 2-3 γ-SPZ-immunized mice were pooled and analyzed by flow cytometry. (E) Mice were immunized with peptide either the day of adoptive transfer of CFSE-labeled T cells or four days prior to transfer. Three days after adoptive transfer, the percent of proliferating T cells was determined by CFSE dilution. Data show mean + SD of three independent experiments. *p<0.05, ***p< 0.001, compared to ‘No Peptide’ control; Student’s t-test. Data are representative of two (A), three (D) and four (B and C) independent experiments.
Despite the robust proliferation observed in the T cells recovered from the lymph nodes and spleen at three days post-immunization, we observed only three-fold and two-fold increases, respectively, in the population size of antigen specific T cells compared to non-immunized mice (Figure 1b, c). Five days after peptide immunization, the frequency of tetramer+ CD8+ T cells in the spleen and lymph nodes of immunized mice had contracted >90% from their frequency at day 3 (Figure 1b,c). However, given the modest expansion observed at day 3, this contraction resulted in significantly fewer CD8+ tetramer+ T cells in the lymph nodes and spleens of immunized mice compared to non-immunized mice. Interestingly, among those cells that remained at day 5, CFSE levels were moderate to high, with most cells in the spleen having divided only once or not at all. In sharp contrast, the response of CD8+ T cells activated after immunization with radiation-attenuated P. yoelii sporozoites display robust proliferation at day 3 that results in accumulation of large numbers of CFSElo T cells at day 5 (Fig.1d).
The lack of accumulation of CFSE− cells among the tetramer+ population demonstrates that unlabeled endogenous T cells (non-TCR-Tg) are not recruited into the response to soluble peptide immunization at a detectable level. Increasing the dose of peptide induced more intense proliferation at day 3 (Figure 1a), but did not result in an increased population size at day 5 (not shown). Moreover, emulsifying the peptide in IFA to create an antigen depot and extend antigen presentation did not improve T cell survival, regardless of peptide dose (not shown). It is noteworthy that aborted T cell responses may not be due to a premature clearance of peptide, as we determined that TCR-Tg cells are activated even if transferred four days after immunization with peptide (Figure 1e), indicating that this epitope is presented for at least four days post-immunization. This indicates that premature loss of antigen due to clearance from circulation or degradation was not likely the reason for the development of poor T cell responses to peptide. Restriction of peptide presentation due to killing of professional APCs by large numbers of activated CD8+ T cells could introduce self-regulatory mechanisms that limit T cell expansion, though we do not believe this is the root of the peptide immunization failure. When transferring low numbers of TCR-Tg CD8+ T cells (Supplementary Figure 1) or when measuring endogenous responses in the absence of transgenic cells (not shown), we still fail to detect T cell expansion, suggesting that the elimination of peptide-presenting APCs by a small number of T cells is not likely a limiting factor.
Clonal expansion and IFN-γ production after immunization with TLR agonists
Given the prominent and critical role of innate signaling to support T cell priming in vivo [20], we evaluated the impact of TLR signaling on the survival of CD8+ T cells activated by soluble peptide in vivo. For these purposes, we immunized mice with peptide and different TLR agonists and evaluated the CD8+ T cell responses three and five days post-immunization. Administration of CpG with peptide greatly enhanced the number of tetramer+ T cells recovered from the lymph nodes and spleens of immunized mice after three days (Figure 2a). However, the number of antigen-specific cells recovered at day 5 was not different between CpG-treated and control mice. Co-injection of poly(I:C), LPS, or imiquimod did not modify the number of tetramer+ cells recovered from the spleens or lymph nodes at these early time points compared to mice immunized with peptide alone (data not shown), demonstrating a selective potency of CpG to enhance the early expansion of CD8+ T cells in response to soluble peptide in vivo.
Figure 2. CpG delivered with peptide enhances clonal expansion and IFN-γ production.
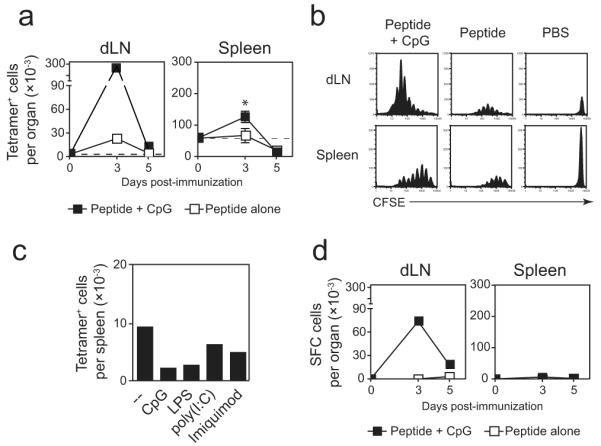
TCR-Tg cells (5 × 105) were transferred into naïve BALB/c mice. The following day, the mice were immunized with 2.5 μg SYVPSAEQI subcutaneously at the base of the tail in PBS alone or with the indicated TLR ligand. (A) Three and five days after immunization, the number of antigen-specific CD8+ T cells in the spleen and draining lymph nodes was analyzed by flow cytometry. Lymph nodes were pooled from three mice and the data from the spleens are plotted as the mean ± SD (n=3). *p<0.05, Student’s t-test. (B) TCR-Tg cells were labeled with CFSE prior to transfer and were harvested from the spleen and lymph nodes three days after immunization. CFSE profiles of CD8+tetramer+ cells are shown from pooled lymph nodes of three mice and a representative individual spleen sample. Vertical scales were maintained for all lymph node or spleen samples to demonstrate relative differences in population size among a constant total bulk population of cells. (C) Ten days after immunization, the number of CD8+tetramer+ in the spleen and lymph nodes (not shown) was determined by flow cytometry. Bars represent pooled spleens of three mice per group. (D) Mice were treated as in (A) and assayed for IFN-γ production by ELISPOT. Data are representative of two (C), three (A, D), and four (B) independent experiments.
Consistent with the increased clonal population size at day 3 post-immunization, tetramer+ cells recovered from mice treated with CpG displayed a more robust proliferation profile compared to control mice that received peptide alone, as indicated by CFSE dilution (Figure 2b). The effects of CpG were not as striking in the spleen, though similar trends were observed. By day 5, however, there was no accumulation of CFSElo cells regardless of CpG treatment, with proliferation profiles similar to those observed previously at day 5 in all groups (not shown). Further, the numbers of tetramer+ cells recovered from the spleens of immunized mice ten days post-immunization were not changed by treatment with any TLR, including CpG (Figure 2c). Thus, in spite of inducing more robust early proliferative activity, CpG treatment could not modify the widespread cell death observed after peptide immunization. Addition of MHC class II-restricted peptides to the inoculum to elicit help from CD4+ T cells did not enhance the survival of the peptide-stimulated CD8+ T cells, even in the presence of CpG (Supplementary Figure 2).
In mice that were immunized with peptide alone, we could not detect antigen-specific T cells by ELISPOT, suggesting that they were unable to produce IFN-γ (Figure 2d). However, antigen-specific cells from the dLN of mice treated with CpG and peptide were readily detected by IFN-γ ELISPOT. These differences were not merely due to differences in frequency, as there was a 10-fold increase in tetramer+ cells measured by FACS, but there were greater than 300-fold differences in the number of IFN-γ-producing cells. Curiously, antigen-specific IFN-γ secreting T cells were not detected in the spleen when immunizing mice with either peptide alone or CpG with peptide.
CpG administered two days before peptide immunization can rescue T cells from death
CpG clearly modulates the CD8+ T cell response to soluble peptide by promoting cell division and clonal expansion, as well as supporting IFN-γ production. However, CpG could not induce T cell survival, as there was no significant increase in the final magnitude of the CD8+ T cell after the contraction phase. Since CpG has been shown to have many effects on the immune system [21] that may change over time, we modified the timing of the CpG administration relative to the peptide to investigate whether there were temporal effects of the CpG that could enhance T cell survival. We found that mice given CpG two days before peptide had significantly more antigen-specific CD8+ T cells in the spleens and lymph nodes compared to mice that were given CpG at the same time as peptide, as seen both by FACS (Figure 3a) and IFN-γ ELISPOT (Figure 3b). Pre-treatment of mice with CpG four days prior to peptide resulted in an increase in the number of peptide-stimulated T cells recovered from the spleen, which was significant compared to mice that received peptide alone (p < 0.01). Importantly, these results were obtained ten days post-immunization with peptide, demonstrating survival of large numbers of activated T cells past the contraction phase measured previously at day 5. Thus, there are time-dependent effects of CpG that can affect the survival of peptide-stimulated CD8+ T cells. Other TLR ligands (LPS, poly(I:C), imiquimod) were ineffective at promoting enhanced T cell survival when administered two days before prior to peptide (Figure 3c), demonstrating a selective potency of CpG to modify synthetic peptide-induced CD8+ T cell responses.
Figure 3. CpG delivered two days prior to peptide uniquely enhances cell survival.

Thy-1.1+ TCR-Tg cells (1 × 105) were transferred into naïve BALB/c mice. Mice were then immunized with 30 μg CpG 1826 and peptide simultaneously, or with CpG two or four days before peptide. Control mice received peptide alone. Ten days after peptide immunization, spleens and draining lymph nodes were removed and the number of cells in the spleen and lymph nodes was evaluated by (A) flow cytometry and (B) ELISPOT. (A) The total number of CD8+Thy-1.1+ cells in the spleens of immunized mice that received CpG at the indicated times or (4, 2, or 0 days before peptide) or peptide alone (—). Data show mean + SD from three independent experiments with 3-10 mice for each group. (B) IFN-γ ELISPOT assay of pools of spleen (black bars) and lymph node (grey bars) samples. Data are representative of two independent experiments. (C) Mice were pre-treated with the indicated TLR ligand two days prior to peptide and the number of CD8+Thy-1.1+ T cells in the spleens was evaluated ten days post-peptide immunization. Data show mean + SD of three mice. *p<0.05, **p<0.01, ***p< 0.001, compared to peptide-only treated mice (--); Student’s t-test.
CpG administered before peptide modifies surface marker expression in activated T cells
Pre-treatment with CpG resulted in an enhanced survival of the peptide-stimulated T cells. While the mechanisms underlying these time-dependent effects are not immediately clear, analysis of surface activation marker expression of the stimulated T cells provided some insights into possible reasons. We compared the surface marker phenotype of T cells obtained from mice immunized with peptide after CpG treatment with those from mice receiving peptide alone (Figure 4 and Supplementary Figure 3). While many of these markers of not differentially-regulated between treatments (eg. CD44, CD11a, CD69, CD62L, CD27), we found some notable differences in surface expression of PD-1 and CD25. On CD8+ T cells stimulated by peptide, PD-1 expression was greatly increased 3 days after immunization, regardless of CpG pre-treatment (Figure 4a). Over the next three days, PD-1 expression levels decreased on CD8+ T cells from mice that were pre-treated with CpG. This rapid increase in PD-1 expression and gradual down-regulation on activated T cells has been previously reported by others in the context of a viral infection [22]. In mice that received peptide alone, PD-1 expression levels remained high and unchanged through day 6 post-peptide immunization. In other systems, sustained expression of PD-1 has been considered indicative of “exhausted” T cells, suggesting that perhaps peptide immunization in the absence of CpG results in repeated TCR engagement that leads to cell exhaustion or death.
Figure 4. CpG given two days before peptide modifies surface marker expression.

Naïve BALB/c mice received TCR-Tg cells (2×104) and were immunized with peptide three days later. Mice also received CpG two days before peptide or no CpG at all. (A) Mice were sacrificed at the indicated time points after peptide immunization and PD-1 expression of CD8+Thy-1.1+ cells in the lymph nodes is shown from non-immunized (shaded) or peptide-immunized (solid line) from either CpG-pre-treated (left column) or non-CpG-treated mice (right column). The vertical dashed line on the plots indicates the median PD-1 expression level on day 3 from mice treated with CpG + peptide. (B) Mice were treated as in (A) and sacrificed at three days post-peptide immunization and stained for PD-1 and CD25 expression on lymph node-resident CD8+Thy-1.1+ cells. Data are from individual lymph node samples representative of three mice per group. Similar observations were made on cells from the spleen. Data are representative of two independent experiments.
In addition to inducing down-regulation of PD-1 in peptide-activated T cells, CpG also induced expression of the high affinity IL-2 receptor (CD25). Robust expression of CD25 was seen at day 3 after peptide in cells pre-immunized with CpG, but not in cells that received peptide alone (Figure 4b). The lack of CD25 expression by CD8+ T cells exposed to peptide alone would suggest that these cells may not be receiving IL-2 signals [23], providing an additional possible mechanism of peptide-induced cell death. However, administration of exogenous IL-2 did not recreate the effects of CpG (not shown), indicating that IL-2 alone is not sufficient to ensure T cell survival.
B cells inhibit CD8+ T cell responses to peptide
While CpG pre-treatment resulted in enhanced CD8+ T cell expansion and survival compared to peptide immunization alone, the population size of the resultant surviving T cell pool was still much lower than the T cell response to radiation-attenuated parasites [2]. Since this lower response is not due to lack of recruitment of antigen-specific T cells into the effector phase (based on percentage of CFSEbright cells at day 3, Figure 2b), an exaggerated amount of cell death still appeared to be occurring with CpG treatment. Others have demonstrated that soluble peptide antigen can be found systemically on the surface of non-professional APC following peptide immunization [10, 11], suggesting that naïve and recently-primed T cells may repeatedly engage their antigen in an inappropriate context on the “wrong” kind of cells. Given that 40-60% of resting lymph node cells are B cells, it is possible, if not likely, that T cells engage their cognate antigen on the surface of B cells following peptide immunization. Since previous studies have shown that B cells could have detrimental effects on the development of CD8+ T cell responses [24-28], we examined the effects of these cells on the response to soluble peptide immunization. For this purpose, we immunized wild type and B cell-deficient (JHT) BALB/c mice with peptide following adoptive transfer of TCR-Tg cells and pre-immunization with CpG. Ten days after peptide immunization, the frequencies of TCR-Tg cells recovered from the spleens of B cell-deficient mice were much greater than those observed in wild type mice (Figure 5a,b). This striking phenotype was dependent upon pre-immunizing with CpG, as peptide immunization alone in B cell-deficient mice did not result in increased T cell survival (Figure 5c), indicating that homeostatic mechanisms cannot account for the phenotype observed with the mutant host. Importantly, these experiments were done with low numbers of TCR-Tg T cells (2×103 per mouse), minimizing possible artifacts that may be introduced with a high precursor frequency of naïve T cells. To confirm that the B cells played an inhibitory role in T cell priming with CpG and peptide, B cell-deficient mice were reconstituted with 3×106 sortpurified B cells from normal BALB/c mice prior to immunization. This reconstitution resulted in near complete reversal of phenotype, with an 85% reduction in the frequency of antigen-specific CD8+ T cells recovered from these mice compared to non-reconstituted B cell-deficient mice (Figure 5d). Similar results were obtained by adoptive transfer of non-sorted spleen cells containing 3×106 B cells from normal BALB/c mice as a source of unmanipulated B cells (Supplementary Figure 5). Thus, in the context of immunization with soluble peptide and CpG, B cells are detrimental to the survival of CD8+ T cells.
Figure 5. B cell-deficient mice support peptide-stimulated CD8+ T cell priming.

(A and B) Naïve BALB/c and B cell-deficient (JHT) mice received Thy-1.1+ TCR-Tg cells (2×103) and were immunized with CpG the following day and peptide two days after CpG. All mice were sacrificed ten days after peptide immunization and the frequency of TCR-Tg cells in the spleen and draining lymph nodes was determined by flow cytometry. (A) Representative FACS plots of the frequency of CD8+Thy-1.1+ cells among total CD8+ spleen cells. (B) Data summary of frequencies from part (A) with four mice per group. (C) Mice were immunized as in (A, B), in addition to a group of B cell-deficient mice that received peptide alone without CpG. FACS plots are representative spleen samples from each group. Values represent the mean percent of CD8+Thy-1.1+ cells among total CD8+ cells ± SD, n=3 per group. (D) Mice were immunized as in (A,B), in addition to a group of B cell-deficient mice that received 3×106 purified B cells from normal BALB/c mice prior to CpG treatment. Ten days later, the percent of TCR-Tg cells in the spleens was evaluated by FACS. Each symbol if representative of one mouse and data are pooled from two independent experiments; vertical lines represent the mean values for each group. *p<0.05, **p<0.01, ***p<0.001; Student’s t-test. Data are representative of two (C, D) and three (B) independent experiments
DISCUSSION
Despite the fact that, in principle, peptide-based immunization would serve as an ideal vaccination regimen, this strategy has found limited success. It has been assumed that the failure of synthetic peptides to induce robust T cell responses is related to an inherent lack of immunogenicity, even when delivered amidst intense inflammatory agents. However, data presented here indicates that synthetic peptides are strong immunogens capable of inducing robust responses of antigen-specific CD8+ T cells in the absence of any other immunologic cues. These antigen-specific T cells, perhaps coerced into proliferation by high number and density of MHC complexes bearing cognate antigen, fail to reach optimal clonal expansion or form a memory population. The stimulation of innate immune signaling by CpG co-administration is able to rescue a small percentage of activated effectors from death, but only when given 2-4 days before peptide immunization.
While the mechanisms mediating the survival effects of CpG are not clear, the phenotype of the responding CD8+ T cells can provide clues – of particular interest is the marker PD-1. Under conditions of productive priming, T cells express PD-1 during acute expansion and down-regulate its expression following contraction and sustained PD-1 expression has been associated with chronic exposure to antigen and states of T cell dysfunction [22, 29]. In our model, sustained expression of PD-1 could be an indicator of an aberrant T cell response due to peptide-MHC abundance or possibly a mechanism by which T cells are eliminated, though blocking PDL1 in vivo as described in previous studies [30] did not rescue T cell survival (data not shown), suggesting that in our system, PD-1/PDL1 interaction is not the sole regulatory mechanism. In addition to the down regulation of PD-1, CpG also induces expression of CD25, which may also allow these activated cells to benefit from IL-2-induced signaling. Remarkably, in mice that received peptide and CpG simultaneously – which resulted in enhanced peak expansion, but not survival – no expression of CD25 was observed at day 3 (Supplementary Figure 4A). Further, this population contained cells with an expression pattern of PD-1 that overlapped both cells from mice treated with peptide alone and those treated with CpG two days prior to peptide (Supplementary Figure 4B).
A remarkable feature of the CpG treatment was the induced ability of the peptide-stimulated T cells to produce IFN-γ. In response to peptide immunization without CpG, T cells failed to produce IFN-γ, even though proliferation was observed. However, when mice were treated with CpG, the responding T cells were able to produce IFN-γ at day 3 (Figure 2). Perhaps the increased proliferation under CpG treatment may have allowed for further differentiation of the responding T cells compared to T cells that were primed by peptide alone.
A striking finding of our studies is the potent regulation of the CD8+ T cell response by B cells to a basic vaccination regimen. In such a simplified system with only two elements, i.e. short synthetic peptide and CpG, there are limited means by which the B cells could be impacting the CD8+ T cell responses. Previous studies have demonstrated that B cell presentation of antigen directly to CD8+ T cells could lead to aberrant T cell responses or deletion of antigen-specific T cells altogether [24, 25, 28]. It has been shown that direct antigen presentation to CD4+ helper T cells by antigen-specific B cells is important to optimal antibody responses [31]. However, their role in priming CD8+ T cells is unclear. Thus, while B cells are considered professional APCs because of their expression of MHC class II and other T cell costimulatory machinery, they may be unable to properly prime cytotoxic CD8+ T cells. In our experiments, reconstitution of B cell-deficient mice with only 3×106 B cells largely restored the phenotype of wildtype mice. The ability of this relatively low number of cells suggests that direct antigen presentation of peptide to T cells by B cells may not be the mechanism of B cell regulation, though this possibility cannot be ruled out entirely. Despite significantly enhanced survival of CD8+ T cells in the absence of B cells, the T cells were unable to provide protection against live P. yoelii parasite challenge (data not shown). Studies are currently underway to determine if there are defects in T cell effector function in the absence of B cells or if there are limitations of this immunization protocol in generating large enough numbers of T cells required for protection in this assay.
B cells could regulate CD8+ T cell responses to peptide by responding to CpG in a manner that is detrimental to effector T cell survival [32]. Indeed, B cells have been shown to proliferate [33-36] and upregulate costimulatory molecules [35, 36] in response to LPS or CpG, but they also potently produce IL-10 and TGF-β [26, 37-40]. Thus, while CpG pre-treatment could induce factors that promote T cell survival such as production of IFN-α [41, 42] and increased numbers of DCs in the lymph node [33], it may also induce suppressive factors from B cells that drive T cell death. There is likely a delicate balance of these factors that allows for the survival of a small number of T cells in normal mice that receive CpG and peptide. Differential kinetics of the production of enhancing and detrimental soluble factors could help to explain the positive effects of delaying antigen delivery after CpG pre-treatment. It has been proposed that B cell responses to innate stimuli, such as CpG, contribute to immune suppression through promotion of regulatory T cell activity [43]. However, depletion of CD4+ cells did not alleviate the suppression of the CD8+ T cell response to CpG and peptide in intact mice, suggesting that regulatory T cells were not playing a direct role (data not shown). Future experiments using adoptive transfer of mutant B cells will be important to gain insight into the exact mechanisms of regulation by the B cells to control the peptide-stimulated CD8+ T cell responses. From a vaccination standpoint, regulation of T cell responses by B cells must be better understood to better design effective vaccines. In our hands, the use of CpG as an adjuvant for peptide immunizations is superior to other TLR ligands for reasons that are not clear. Strategies for avoiding stimulation of B cells with CpG in peptide-based vaccinations could make these approaches more effective.
MATERIALS AND METHODS
Mice
Female BALB/c mice 5-8 weeks of age were purchased from Taconic Farms and housed in microisolater cages. T cell receptor (TCR)-transgenic mice expressing a TCR specific for H2Kd-SYVPSAEQI have been previously described [5]. B cell-deficient mice (JHT) were purchased from Taconic Farms. For adoptive transfer, indicated numbers of TCR-transgenic CD8+ T cells (TCR-Tg) from whole splenocytes were injected intravenously into naïve BALB/c mice. Experiments involving mice were approved by the Institutional Care and Use Committee of the Johns Hopkins University.
CFSE labeling
Vybrant CFDA-SE Cell Tracer Kit (Molecular Probes) was used to label cells to track proliferation according to the manufacturer’s instructions. Briefly, spleen cell suspensions were suspended in CFSE solution (5 μM in PBS) at 107 cells per mL for six minutes at room temperature. The reaction was then quenched by five-fold dilution of suspension with media containing 10% serum. Cells were then washed in cold media and transferred into mice.
Immunizations
Synthetic peptide representing the immunodominant epitope of P. yoelii circumsporozoite (CS) protein and cognate antigen of the TCR-Tg cells (SYVPSAEQI) was diluted in PBS and injected subcutaneously at the base of the tail in 100 μL. When peptide was emulsified in incomplete Freund’s adjuvant (IFA), peptide stock is diluted to in sterile PBS and emulsified 1:1 with IFA.
CpG oligodinucleotide 1826 was synthesized by Integrated DNA Technologies and solubilized in sterile PBS (5′-TCC-ATG-ACG-TTC-CTG-ACG-TT-3′). Intranucleotide bonds were phosphorothioated to enhance stability in vivo. CpG stock solution was diluted to 0.3 mg/ml in sterile PBS just prior to immunization and mice were injected subcutaneously at the base of the tail with 30 μg CpG.
FACS Analysis
Spleens and draining lymph nodes were removed following euthanasia and placed in cold media on ice. Single-cell suspensions of lymphocytes were obtained by grinding organs between the frosted ends of two microscope slides and filtering twice through 100-μm pore size nylon mesh. Cells were washed and resuspended in fresh media containing 10% serum. Lymph node cells were pooled among mice of the same group and spleens were analyzed individually for statistical analyses. All antibodies for flow cytometry were purchased from eBioscience unless otherwise noted. Frequency of parasite-specific TCR-Tg T cells was determined by staining of single cell suspensions with anti-CD8-APC and either anti-Thy-1.1-PE (BD Biosciences) or PE-conjugated H2Kd-CS260 tetramer, as previously described [5]. Cells were analyzed on a BD FACSCalibur (BD Biosciences) and data was analyzed in FlowJo (TreeStar).
ELISPOT
ELISPOT analysis of antigen-specific IFN-γ production by CD8+ T cells has been previously described [44].
Purification and transfer of B cells
Lymphocytes were harvested from the spleens of wildtype BALB/c mice and sorted for B220+Thy-1.2−120G8− cells on a FACSAria. 3×106 purified (>98% purity by FACS) B cells were adoptively transferred by intravenous injection into naïve BALB/c mice prior to adoptive transfer of TCR-Tg cells and immunization.
Statistical Analysis
Data analysis and presentation was performed using Prism (GraphPad Software).
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIH grant AI44375. M.G.O. was supported by a fellowship from the Malaria Research Institute. The authors are grateful for the support of the Bloomberg Family Foundation. PDL-1 blocking antibodies were kindly provided by Lieping Chen.
Footnotes
CONFLICT OF INTEREST The authors declare no financial or commercial conflict of interest.
Current address: Michael G. Overstreet’ David H. Smith Center for Vaccine Biology and Immunology, University of Rochester Medical Center, 601 Elmwood Avenue, Rochester, NY 14642, USA.
References
- 1.Chakravarty S, Baldeviano GC, Overstreet MG, Zavala F. Effector CD8+ T lymphocytes against liver stages of Plasmodium yoelii do not require gamma interferon for antiparasite activity. Infect Immun. 2008;76:3628–3631. doi: 10.1128/IAI.00471-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chakravarty S, Cockburn IA, Kuk S, Overstreet MG, Sacci JB, Zavala F. CD8+ T lymphocytes protective against malaria liver stages are primed in skin-draining lymph nodes. Nat Med. 2007;13:1035–1041. doi: 10.1038/nm1628. [DOI] [PubMed] [Google Scholar]
- 3.Rodrigues M, Nussenzweig RS, Romero P, Zavala F. The in vivo cytotoxic activity of CD8+ T cell clones correlates with their levels of expression of adhesion molecules. J Exp Med. 1992;175:895–905. doi: 10.1084/jem.175.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodrigues MM, Cordey AS, Arreaza G, Corradin G, Romero P, Maryanski JL, Nussenzweig RS, Zavala F. CD8+ cytolytic T cell clones derived against the Plasmodium yoelii circumsporozoite protein protect against malaria. Int Immunol. 1991;3:579–585. doi: 10.1093/intimm/3.6.579. [DOI] [PubMed] [Google Scholar]
- 5.Sano G, Hafalla JC, Morrot A, Abe R, Lafaille JJ, Zavala F. Swift development of protective effector functions in naive CD8(+) T cells against malaria liver stages. J Exp Med. 2001;194:173–180. doi: 10.1084/jem.194.2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aichele P, Brduscha-Riem K, Zinkernagel RM, Hengartner H, Pircher H. T cell priming versus T cell tolerance induced by synthetic peptides. J Exp Med. 1995;182:261–266. doi: 10.1084/jem.182.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kyburz D, Aichele P, Speiser DE, Hengartner H, Zinkernagel RM, Pircher H. T cell immunity after a viral infection versus T cell tolerance induced by soluble viral peptides. Eur J Immunol. 1993;23:1956–1962. doi: 10.1002/eji.1830230834. [DOI] [PubMed] [Google Scholar]
- 8.Toes RE, Blom RJ, Offringa R, Kast WM, Melief CJ. Enhanced tumor outgrowth after peptide vaccination. Functional deletion of tumor-specific CTL induced by peptide vaccination can lead to the inability to reject tumors. J Immunol. 1996;156:3911–3918. [PubMed] [Google Scholar]
- 9.Toes RE, Offringa R, Blom RJ, Melief CJ, Kast WM. Peptide vaccination can lead to enhanced tumor growth through specific T-cell tolerance induction. Proc Natl Acad Sci U S A. 1996;93:7855–7860. doi: 10.1073/pnas.93.15.7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, van der Burg SH, Offringa R. Superior induction of anti-tumor CTL immunity by extended peptide vaccines involves prolonged, DC-focused antigen presentation. Eur J Immunol. 2008;38:1033–1042. doi: 10.1002/eji.200737995. [DOI] [PubMed] [Google Scholar]
- 11.Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, Offringa R, van der Burg SH. CD8+ CTL priming by exact peptide epitopes in incomplete Freund’s adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J Immunol. 2007;179:5033–5040. doi: 10.4049/jimmunol.179.8.5033. [DOI] [PubMed] [Google Scholar]
- 12.Valmori D, Romero JF, Men Y, Maryanski JL, Romero P, Corradin G. Induction of a cytotoxic T cell response by co-injection of a T helper peptide and a cytotoxic T lymphocyte peptide in incomplete Freund’s adjuvant (IFA): further enhancement by pre-injection of IFA alone. Eur J Immunol. 1994;24:1458–1462. doi: 10.1002/eji.1830240633. [DOI] [PubMed] [Google Scholar]
- 13.Widmann C, Romero P, Maryanski JL, Corradin G, Valmori D. T helper epitopes enhance the cytotoxic response of mice immunized with MHC class I-restricted malaria peptides. J Immunol Methods. 1992;155:95–99. doi: 10.1016/0022-1759(92)90275-x. [DOI] [PubMed] [Google Scholar]
- 14.Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF, Barth RJ, Vasilakos JP, Noelle RJ, Kedl RM. Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J Exp Med. 2004;199:775–784. doi: 10.1084/jem.20031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Assudani D, Cho HI, Devito N, Bradley N, Celis E. In vivo Expansion, Persistence, and Function of Peptide Vaccine-Induced CD8 T Cells Occur Independently of CD4 T Cells. Cancer Res. 2008;68:9892–9899. doi: 10.1158/0008-5472.CAN-08-3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kochenderfer JN, Chien CD, Simpson JL, Gress RE. Synergism between CpG-containing oligodeoxynucleotides and IL-2 causes dramatic enhancement of vaccine-elicited CD8+ T cell responses. J Immunol. 2006;177:8860–8873. doi: 10.4049/jimmunol.177.12.8860. [DOI] [PubMed] [Google Scholar]
- 17.Kochenderfer JN, Chien CD, Simpson JL, Gress RE. Maximizing CD8+ T cell responses elicited by peptide vaccines containing CpG oligodeoxynucleotides. Clin Immunol. 2007;124:119–130. doi: 10.1016/j.clim.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toka FN, Gierynska M, Suvas S, Schoenberger SP, Rouse BT. Rescue of memory CD8+ T cell reactivity in peptide/TLR9 ligand immunization by codelivery of cytokines or CD40 ligation. Virology. 2005;331:151–158. doi: 10.1016/j.virol.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 19.Toubaji A, Hill S, Terabe M, Qian J, Floyd T, Simpson RM, Berzofsky JA, Khleif SN. The combination of GM-CSF and IL-2 as local adjuvant shows synergy in enhancing peptide vaccines and provides long term tumor protection. Vaccine. 2007;25:5882–5891. doi: 10.1016/j.vaccine.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 20.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 21.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 22.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 23.Minami Y, Kono T, Miyazaki T, Taniguchi T. The IL-2 receptor complex: its structure, function, and target genes. Annu Rev Immunol. 1993;11:245–268. doi: 10.1146/annurev.iy.11.040193.001333. [DOI] [PubMed] [Google Scholar]
- 24.Bennett SR, Carbone FR, Toy T, Miller JF, Heath WR. B cells directly tolerize CD8(+) T cells. J Exp Med. 1998;188:1977–1983. doi: 10.1084/jem.188.11.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuchs EJ, Matzinger P. B cells turn off virgin but not memory T cells. Science. 1992;258:1156–1159. doi: 10.1126/science.1439825. [DOI] [PubMed] [Google Scholar]
- 26.Tian J, Zekzer D, Hanssen L, Lu Y, Olcott A, Kaufman DL. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J Immunol. 2001;167:1081–1089. doi: 10.4049/jimmunol.167.2.1081. [DOI] [PubMed] [Google Scholar]
- 27.Watt V, Ronchese F, Ritchie D. Resting B cells suppress tumor immunity via an MHC class-II dependent mechanism. J Immunother. 2007;30:323–332. doi: 10.1097/CJI.0b013e31802bd9c8. [DOI] [PubMed] [Google Scholar]
- 28.Werner-Klein M, Dresch C, Marconi P, Brocker T. Transcriptional Targeting of B Cells for Induction of Peripheral CD8 T Cell Tolerance. The Journal of Immunology. 2007;178:7738. doi: 10.4049/jimmunol.178.12.7738. [DOI] [PubMed] [Google Scholar]
- 29.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–245. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 30.Tsushima F, Yao S, Shin T, Flies A, Flies S, Xu H, Tamada K, Pardoll DM, Chen L. Interaction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood. 2007;110:180–185. doi: 10.1182/blood-2006-11-060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lanzavecchia A. Antigen-specific interaction between T and B cells. Nature. 1985;314:537–539. doi: 10.1038/314537a0. [DOI] [PubMed] [Google Scholar]
- 32.Gray D, Gray M, Barr T. Innate responses of B cells. Eur J Immunol. 2007;37:3304–3310. doi: 10.1002/eji.200737728. [DOI] [PubMed] [Google Scholar]
- 33.Lipford GB, Sparwasser T, Zimmermann S, Heeg K, Wagner H. CpG-DNA-mediated transient lymphadenopathy is associated with a state of Th1 predisposition to antigen-driven responses. J Immunol. 2000;165:1228–1235. doi: 10.4049/jimmunol.165.3.1228. [DOI] [PubMed] [Google Scholar]
- 34.Yi AK, Chang M, Peckham DW, Krieg AM, Ashman RF. CpG oligodeoxyribonucleotides rescue mature spleen B cells from spontaneous apoptosis and promote cell cycle entry. J Immunol. 1998;160:5898–5906. [PubMed] [Google Scholar]
- 35.Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 36.Heit A, Huster KM, Schmitz F, Schiemann M, Busch DH, Wagner H. CpG-DNA Aided Cross-Priming by Cross-Presenting B Cells. The Journal of Immunology. 2004;172:1501. doi: 10.4049/jimmunol.172.3.1501. [DOI] [PubMed] [Google Scholar]
- 37.Barr TA, Brown S, Ryan G, Zhao J, Gray D. TLR-mediated stimulation of APC: Distinct cytokine responses of B cells and dendritic cells. Eur J Immunol. 2007;37:3040–3053. doi: 10.1002/eji.200636483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lenert P, Brummel R, Field EH, Ashman RF. TLR-9 activation of marginal zone B cells in lupus mice regulates immunity through increased IL-10 production. J Clin Immunol. 2005;25:29–40. doi: 10.1007/s10875-005-0355-6. [DOI] [PubMed] [Google Scholar]
- 39.Parekh VV, Prasad DV, Banerjee PP, Joshi BN, Kumar A, Mishra GC. B cells activated by lipopolysaccharide, but not by anti-Ig and anti-CD40 antibody, induce anergy in CD8+ T cells: role of TGF-beta 1. J Immunol. 2003;170:5897–5911. doi: 10.4049/jimmunol.170.12.5897. [DOI] [PubMed] [Google Scholar]
- 40.Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28:639–650. doi: 10.1016/j.immuni.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 41.Eppihimer MJ, Gunn J, Freeman GJ, Greenfield EA, Chernova T, Erickson J, Leonard JP. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation. 2002;9:133–145. doi: 10.1038/sj/mn/7800123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schreiner B, Mitsdoerffer M, Kieseier BC, Chen L, Hartung HP, Weller M, Wiendl H. Interferon-beta enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: relevance for the immune modulatory effect in multiple sclerosis. J Neuroimmunol. 2004;155:172–182. doi: 10.1016/j.jneuroim.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 43.Fillatreau S, Gray D, Anderton SM. Not always the bad guys: B cells as regulators of autoimmune pathology. Nat Rev Immunol. 2008;8:391–397. doi: 10.1038/nri2315. [DOI] [PubMed] [Google Scholar]
- 44.Carvalho LH, Hafalla JC, Zavala F. ELISPOT assay to measure antigen-specific murine CD8(+) T cell responses. J Immunol Methods. 2001;252:207–218. doi: 10.1016/s0022-1759(01)00331-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.