

Abstract
Background:
Acute and chronic systemic inflammation are characterized by the systemic production of the proinflammatory cytokine tumor necrosis factor α (TNF-α) that plays a role in immune to brain communication. Previous preclinical research shows that acute systemic inflammation contributes to an exacerbation of neurodegeneration by activation of primed microglial cells.
Objective:
To determine whether acute episodes of systemic inflammation associated with increased TNF-α would be associated with long-term cognitive decline in a prospective cohort study of subjects with Alzheimer disease.
Methods:
Three hundred community-dwelling subjects with mild to severe Alzheimer disease were cognitively assessed, and a blood sample was taken for systemic inflammatory markers. Each subject’s main caregiver was interviewed to assess the presence of incident systemic inflammatory events. Assessments of both patient and caregiver were repeated at 2, 4, and 6 months.
Results:
Acute systemic inflammatory events, found in around half of all subjects, were associated with an increase in the serum levels of proinflammatory cytokine TNF-α and a 2-fold increase in the rate of cognitive decline over a 6-month period. High baseline levels of TNF-α were associated with a 4-fold increase in the rate of cognitive decline. Subjects who had low levels of serum TNF-α throughout the study showed no cognitive decline over the 6-month period.
Conclusions:
Both acute and chronic systemic inflammation, associated with increases in serum tumor necrosis factor α, is associated with an increase in cognitive decline in Alzheimer disease.
GLOSSARY
- AD
= Alzheimer disease;
- ADAS-COG
= Alzheimer’s Disease Assessment Scale–Cognitive subscale;
- ANOVA
= analysis of variance;
- CheI
= cholinesterase inhibitor;
- CI
= confidence interval;
- CRP
= C-reactive protein;
- IQR
= interquartile range;
- MSD
= Meso Scale Discovery;
- NINCDS-ADRDA
= National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association;
- SIE
= systemic inflammatory event;
- TNF-α
= tumor necrosis factor α.
Studies in subjects without dementia have suggested that low-grade peripheral systemic inflammation is associated with increased cognitive decline, including reduced hippocampal volume.1,2 Some studies,3–6 but not all,7 have also suggested that increased systemic inflammation may be associated with increased risk of developing Alzheimer disease (AD). However, although some pilot studies exist,8,9 no prospective longitudinal studies have examined the relationship between acute or chronic inflammation on disease progression in large groups of subjects with established AD. Both acute and chronic systemic inflammation is characterized by the systemic production of C-reactive protein (CRP) from the liver and the proinflammatory cytokine tumor necrosis factor α (TNF-α) from macrophages. TNF-α then plays a role in immune to brain communication by activating the central innate immune response, including microglial cells.10 In animal models of neurodegeneration, where the microglia are already activated by the presence of chronic neurodegenerative changes, we have shown that experimentally induced acute systemic inflammation results in an exaggerated central innate immune response leading to the release of cytotoxic inflammatory mediators that exacerbate neurodegeneration.11 These effects, over time, result in accelerated disease progression.11,12 There is abundant morphologic evidence for different states of microglial activation in subjects with AD.13 We therefore hypothesized that acute systemic inflammatory events (SIEs) associated with an increased production of the peripheral proinflammatory cytokine TNF-α would be associated with long-term (i.e., delirium-independent) cognitive decline.
METHODS
Study design.
Three hundred community-dwelling subjects (and their caregivers) with mild to severe dementia were recruited during the period November 2003 to May 2006 from clinical referrals to memory assessment services in Southampton, UK. After consent procedures, all subjects fulfilling National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria14 for probable or possible AD were cognitively tested using the Alzheimer’s Disease Assessment Scale (ADAS-COG), in which positive changes imply worsening cognition.15 Immediately after cognitive assessment at baseline, a blood sample for CRP and the proinflammatory cytokine TNF-α was taken. The subjects’ main caregiver was then interviewed within 2 days by a separate registered general medical nurse, who was blind to the cognitive assessment of the subject, using a standardized questionnaire to assess the presence of any acute incident SIEs at interview or in the previous 2 months. An SIE was defined as a short-lived (less than 2 months’ duration) infection or trauma not directly involving the CNS with a minimum serum CRP level of 1 μg/mL after the event. In addition, long-term medical history and medication use over the previous 2 months was also documented. At the end of the interview, the main caregiver was given a once-weekly checklist diary, as an aide memoir, in which to enter the occurrence of any subsequent SIEs based on a systems-based checklist and medication changes before the next interview. The subject and main caregiver were revisited at 2, 4, and 6 months and reassessed in an identical manner but also including an assessment of the presence of delirium in the subject with AD using the Confusion Assessment Method.16
Standard protocol approvals, registrations, and patient consents.
This study received approval from the South and West Hampshire Local Research Ethics Committee (ref 237/03/w). Written informed consent was obtained from all patients (or guardians of patients) participating in the study.
Systemic inflammation assays.
All blood sera were immediately placed on ice and stored within 2 hours at −80°C. CRP was assayed using ELISA and had a detection limit of 1 μg/mL. TNF-α was assayed using sandwich immunoassay multiplex cytokine assay (Meso Scale Discovery [MSD]). A protocol provided by MSD for custom assays was used, with no major modifications. The lowest detectable limit was 1.1 pg/mL for TNF-α.
Statistical analysis.
Assessment of normality of continuous variables was determined by quantile–quantile plots of the residuals. Baseline ADAS-COG, change in ADAS-COG, and change in serum TNF-α levels were normally distributed. Serum CRP and TNF-α levels were not normally distributed. Serum TNF-α levels were simplified by the use of quartile ranges, based on subject numbers found at baseline assessment. Low serum TNF-α was defined as the range found in the lowest quartile for TNF-α (<2.4pg/mL); high serum levels were defined as all serum ranges above 2.4pg/mL. Following previous guidelines,17 low serum CRP was defined as <1.0 μg/mL; moderate/high levels were defined as all serum ranges above 1.0 μg/mL. Allowing for a 10% dropout rate, 300 subjects gave 90% power to detect a significant difference (α = 0.05) increase of 3.5 points (½ SD) on the ADAS-COG in subjects with an SIE (minimum expected from previous data 20%) compared with subjects without an SIE. Data interaction was assessed using a mixture of linear regression analysis and analysis of variance (ANOVA).
RESULTS
Twenty-five subjects were clinically unresponsive at baseline with severe end-stage dementia, i.e., ADAS-COG scores greater than 60 points, and were excluded from further analysis to reduce floor effects.
Baseline data.
Of the 275 subjects, 161 (59%) fulfilled NINCDS-ADRDA criteria for probable AD and 114 (41%) fulfilled criteria for possible AD. The mean age of the cohort at baseline was 82.7 (SD 7.4) years. Ninety-nine subjects (36%) were men. One hundred seventeen (43%) of the 275 subjects at baseline were taking a cholinesterase inhibitor (CheI), of whom 97 (83%) had been taking the drug for more than 2 months. Eighty-eight subjects (32%) had a history of hypertension, 45 (16%) had a history of hypercholesterolemia, and 19 (7%) had a history of type 2 diabetes. Eighteen subjects (6.5%) had delirium at baseline. Table 1 shows the type and period prevalence of SIEs at, or retrospectively, in the 2 months preceding, baseline. Thirty-nine subjects (14.2% of all subjects) had a total of 50 SIEs identified at or during the 2 months preceding baseline. Twenty-nine of these 39 subjects had 1 SIE, 9 subjects had 2 SIEs, and 1 subject had 3 SIEs at or preceding baseline assessment. Blood sampling was obtained in 269 of 275 subjects (98%) at baseline. Serum CRP was moderate/high (serum level ≥1 μg/mL) in 192 of all subjects (70%). Serum TNF-α was detectable in all subjects (TNF-α median 3.3 [interquartile range (IQR) 2.4–4.2] pg/mL).
Table 1 Period prevalence of systemic inflammatory events at baseline and during the 6-month follow-up period and effect size on cognitive decline

Baseline cognitive score and systemic inflammation.
At baseline, subjects had a mean ADAS-COG score of 29.6 (SD 13.0) points.
No relationship was found between the presence of low serum CRP and baseline ADAS-COG score (low CRP 28.0 [SE 1.5] pts, c.f. moderate/high CRP 30.7 [SE 0.9] pts; mean difference 2.7 [95% confidence interval (CI) −6.1 to 0.7] pts, t test p = 0.1). Low serum CRP was associated with a younger age (low CRP 81.0 [SE 0.8] years, c.f. moderate/high CRP 83.5 [SE 0.6] years; mean difference 2.5 [95% CI 0.6 to 4.5] years, t test p = 0.01) and with the prescription of a cholinesterase inhibitor (χ2 13.2, p < 0.001) but not sex (χ2 0.7, p = 0.4).
Low baseline serum TNF-α was associated with younger age, lower ADAS-COG scores, and CheI use, but there was no relationship with sex or SIEs preceding baseline (table 2).
Table 2 Baseline serum TNF-α systemic inflammatory markers and demographics
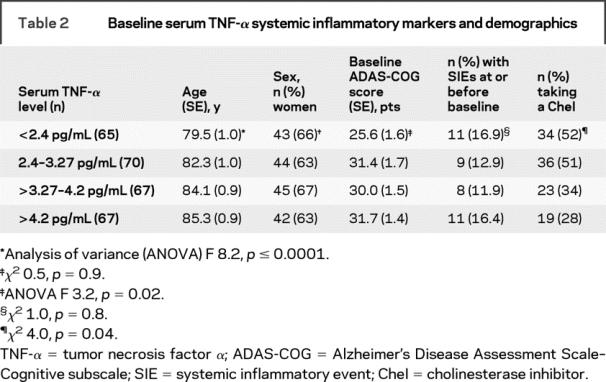
Linear regression analysis with baseline ADAS-COG score as the dependent variable showed that the relationship between baseline ADAS-COG score and low or high serum baseline TNF-α levels remained unchanged (mean difference 4.1 [95% CI 0.5–7.7] pts, t test p = 0.02) after correction for age, CheI use, history of hypertension, type 2 diabetes, hypercholesterolemia, and the presence of delirium at baseline as possible confounders.
Six-month follow-up period.
Two hundred twenty-two (81%) of all subjects had complete clinical and systemic inflammatory marker follow-up at 2, 4, and 6 months. Of the 53 subjects who did not complete the study, 15 subjects died before study completion and 38 subjects declined phlebotomy or further cognitive assessment at some point during the study. A trend relationship was found between study completion and baseline serum levels of TNF-α (completers median level 3.1 [IQR 2.3–4.3] pg/mL, c.f. noncompleters 3.7 [IQR 3.0–4.2] pg/mL, Mann–Whitney U p = 0.07).
Systemic inflammatory events.
One hundred ten (49.5%) of the 222 subjects followed up had a combined total of 150 SIEs (table 1). Eighty-one subjects (36.5%) had clinical evidence of 1 acute SIE, 22 (9.9%) subjects had evidence of 2 SIEs, and 7 (3.2%) had evidence of 3 or more SIEs in the 6 months after baseline assessment.
Subjects with 1 or more SIEs during the 6-month follow-up period had an increase in TNF-α levels compared with subjects with no SIEs over the next 6 months (SIEs absent: TNF-α increased by 0.03 [SE 0.07] pg/mL, c.f. SIEs present: TNF-α increased by 0.32 [SE 0.07] pg/mL; mean difference 0.29 [95% CI 0.08–0.50] pg/mL, t test p = 0.005).
Cognitive decline and systemic inflammation.
The mean change in ADAS-COG score over the 6-month follow-up period was 2.6 (SD 7.0) points.
Change in ADAS-COG score over the 6-month follow-up period showed a low correlation with baseline ADAS-COG score (Pearson correlation 0.14, p = 0.03).
There was no difference in change in ADAS-COG score over the 6-month follow-up period in those subjects with low CRP levels at baseline compared with subjects with moderate/high CRP levels at baseline (low CRP 1.9 [SE 0.7] pts, c.f. moderate/high CRP 3.0 [SE 0.6] pts; mean difference 1.1 [95% CI −3.1 to 1.0] pts, t test p = 0.3). However, change in ADAS-COG score over the 6-month follow-up period was greater in those subjects with high TNF-α levels at baseline compared with subjects with low TNF-α levels at baseline (low TNF-α 0.8 [SE 0.8] pts, c.f. high TNF-α 3.2 [SE 0.6] pts; mean difference 2.4 [95% CI 0.3–4.5] pts, t test p = 0.02).
Subjects with 1 or more SIEs during the 6-month follow-up period had a faster rate of cognitive decline from baseline to 6 months compared with those with no recorded SIEs (SIE absent ADAS-COG change 1.6 [SE 0.6] pts, c.f. SIE present 3.5 [SE 0.8] pts; mean difference 2.0 [95% CI 0.1–3.8] pts, t test p = 0.04) (figure 1). There was a trend for patients with more than 1 SIE to have an increased rate of cognitive decline (SIEs absent ADAS-COG change 1.6 [SE 0.6] pts, 1 SIE ADAS-COG change 3.3 [SE 0.9] pts, 2 or more SIEs ADAS-COG change 4.3 [SE 1.6] pts; ANOVA F 2.4, p = 0.09). Effect sizes for individual SIEs varied (table 1).

Figure 1 Presence or absence of systemic inflammatory events and mean change in cognitive score from baseline
ADAS-COG = Alzheimer’s Disease Assessment Scale–Cognitive subscale; SIE = systemic inflammatory events.
Change in ADAS-COG score had a low correlation with the change in serum TNF-α levels over the 6-month follow-up period (Pearson correlation 0.20, p = 0.004).
Linear regression analysis was performed with change in ADAS-COG as the dependent variable and age, baseline ADAS-COG, the presence or absence of an SIE through the 6-month follow-up period, presence of low or high TNF-α level at baseline, change in TNF-α, and CheI use as independent variables. This analysis showed a change in the relationship between change in ADAS-COG and baseline ADAS-COG score (adjusted mean difference 0.06 [95% CI −0.02 to 0.14] pts, p = 0.1) and the presence or absence of SIEs (adjusted mean difference 1.2 [95% CI −0.7 to 3.2] pts, p = 0.2). However, there was little change in the relationship between change in ADAS-COG and either baseline TNF-α level (adjusted mean difference 2.5 [95% CI 0.3–4.7] pts, p = 0.03) or change in serum TNF-α (adjusted mean difference 1.8 [95% CI 0.6–3.0] pts, p = 0.003). Correction for the presence of a history of hypertension, hypercholesterolemia, and type 2 diabetes did not alter these relationships.
Delirium was identified in 25 subjects during the 6-month follow-up period and was more frequent in subjects with increased serum TNF-α at baseline (χ2 7.4, p = 0.007) but was not related to the presence of SIEs (χ2 0.7, p = 0.2). Correcting for the presence of delirium altered the relationship between change in ADAS-COG and baseline TNF-α level (adjusted mean difference 1.6 [95% CI −0.6 to 3.7] pts, p = 0.1), but no difference was found between change in ADAS-COG score and change in serum TNF-α (adjusted mean difference 1.8 [95% CI 0.6–2.9] pts, p = 0.002).
Allocating subjects to 1 of 4 groups based on low or high levels of TNF-α at baseline and the presence or absence of SIEs in the 6-month follow-up period showed a difference in rates of cognitive decline at 6 months between groups (ANOVA F 3.3, p = 0.02) (figure 2). There was no interaction effect between low or high levels of TNF-α at baseline and the presence or absence of SIEs in the 6-month follow-up period (ANOVA F 0.5, p = 0.5).

Figure 2 Rate of cognitive decline at 6 months by presence of incident systemic inflammatory events and baseline serum TNF-α levels
ADAS-COG = Alzheimer’s Disease Assessment Scale–Cognitive subscale; TNF-α = tumor necrosis factor α; SIE = systemic inflammatory events.
In total, 27 of the 83 subjects at baseline with low serum CRP levels had low levels throughout the 6-month follow-up period. Examination of those 27 subjects showed no differences in their rate of cognitive decline compared with those with moderate/high CRP levels (low CRP 1.9 [SE 1.3] pts, c.f. moderate/high CRP 2.6 [SE 0.5] pts; mean difference 0.7 [95% CI −3.7 to 2.1] pts, p = 0.6).
In total, 34 of the 59 subjects at baseline with low serum TNF-α levels had serum TNF-α levels that remained at less than 2.4 pg/mL during the 6-month follow-up. Examination of those 34 subjects with low TNF-α throughout the course of the study showed them to have a markedly lower rate of cognitive decline compared with those with high TNF-α (low TNF-α −0.3 [SE 1.2] pts, c.f. high TNF-α 3.1 [SE 0.5] pts; mean difference 3.4 [95% CI 0.7–5.9] pts, p = 0.01) (figure 3).

Figure 3 Rate of cognitive decline by mean serum TNF-α levels during the follow-up period
ADAS-COG = Alzheimer’s Disease Assessment Scale–Cognitive subscale; TNF-α = tumor necrosis factor α.
DISCUSSION
Serum TNF-α levels found in this study were comparable to those found in other studies of subjects with AD, with the lower quartile being comparable to that found in age-matched control populations.18,19
In this prospective study, increased baseline levels of serum TNF-α were associated with a 4-fold increase in the rate of cognitive decline over a 6-month follow-up period. At baseline, high levels of serum TNF-α were also associated with the degree of baseline cognitive impairment that was independent of age, delirium, and concomitant cholinesterase use, suggesting a relationship between serum TNF-α levels and both rates of subsequent cognitive decline and long-term cognitive impairment. It is notable that a number of chronic low-grade inflammatory conditions, including atherosclerosis,20 periodontitis,21 diabetes,22 smoking,23 and obesity,24 increase chronic systemic levels of TNF-α25 and are also risk factors for AD. However, whether chronically increased serum TNF-α is a risk factor or simply a surrogate marker for disease progression in AD remains unclear.26,27
Acute systemic inflammation, in the form of a variety of systemic infections or tissue injury, occurred in around half of these elderly subjects with AD over a 6-month follow-up period. SIEs were associated with a 2-fold increase in the rate of cognitive decline and an increase in serum TNF-α levels over the 6-month follow-up period, which seemed to largely explain the association with cognitive decline. Despite the suggestion of a “dose” effect, it is possible that the relationship between cognitive decline and SIEs is explained by the hypothesis that patients with a more rapid rate of cognitive decline are more susceptible to infections or trauma. However, it is notable that at baseline there was no evidence to suggest that subjects with more severe dementia had a greater frequency of SIEs at or preceding baseline. Furthermore, animal studies are strongly supportive of the concept that experimentally induced systemic inflammation that causes increases in systemic proinflammatory cytokines such as TNF-α can drive neurodegeneration when the central innate immune cells are already primed by the disease.11,12,28,29 Thus, a plausible explanation of our observations is that in AD, acute systemic inflammation associated with increased TNF-α leads to increased cognitive decline rather than SIEs simply being a surrogate marker of disease progression.
The effect of having both acute and chronic inflammation on cognitive decline was marked. Thus, subjects with high levels of TNF-α at baseline and the presence of SIEs over the following 6 months had a 10-fold increased rate of cognitive decline compared with subjects with low levels of TNF-α at baseline and no SIEs over the following 6 months. Indeed, subjects who maintained low levels of TNF-α showed no cognitive decline at 6 months.
Delirium is associated with both short- and long-term cognitive impairment30,31 and was more likely to occur in subjects with increased TNF-α at baseline. In this study, SIEs were associated with increased cognitive decline independently of the presence of delirium. While it is possible that some episodes of subsyndromal delirium may have been missed, the lack of association of SIEs with the presence of delirium most likely reflects the mild nature of the majority of the SIEs recorded here. Although examination of different SIEs shows some variability in the effect sizes on cognitive decline, small numbers preclude any meaningful comparisons.
In keeping with other studies,32 high TNF-α levels in this study were associated with increased mortality. This may have led to an underestimate of the cognitive effects of TNF-α on cognitive decline.
The role of TNF-α within the brain is controversial, with evidence supportive of both deleterious and protective effects.33–36 However, if systemic inflammation has different CNS consequences depending on the existing relative activation state of the central innate immune system, dampening down systemic TNF-α may prove to be beneficial in AD.
DISCLOSURE
Prof. Holmes has received research support from the Alzheimer’s Society, the UK Department of Health, and the Medical Research Council. Dr. Cunningham has received non–industry-sponsored funding for travel and receives research support from the Wellcome Trust; his sister is an employee of Wyeth Pharmaceuticals. Prof. Perry has received honoraria from the Danish Research Council and UCB; and receives research support from the European Union, the Medical Research Council, the Biotechnology and Biological Sciences Research Council, and Wellcome Trust. Ms. Zotova, Ms. Woolford, Ms. Dean, Ms. Kerr, and Mr. Culliford report no disclosures.
Address correspondence and reprint requests to Prof. Clive Holmes, University of Southampton, Memory Assessment and Research Centre, Botley Rd., Southampton, UK, SO30 3JB ch4@soton.ac.uk
Supported by the Alzheimer’s Society.
Disclosure: Author disclosures are provided at the end of the article.
Received January 27, 2009. Accepted in final form June 16, 2009.
REFERENCES
- 1.Yaffe K, Kanaya A, Lindquist K, et al. The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA 2004;292:2237–2242. [DOI] [PubMed] [Google Scholar]
- 2.Marsland AE, Gianaros PJ, Abramowitch SM, Manuck SB, Hariri AR. Interleukin-6 covaries inversely with hippocampal grey matter volume in middle-aged adults. Biol Psychiatry 2008;64:484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engelhart MJ, Geerlings MI, Meijer J, et al. Inflammatory proteins in plasma and the risk of dementia: the Rotterdam study. Arch Neurol 2004 May;61:668–672. [DOI] [PubMed]
- 4.Tan ZS, Beiser AS, Vasan RS, et al. Inflammatory markers and the risk of Alzheimer disease: the Framingham Study. Neurology 2008;70:1222–1223. [DOI] [PubMed] [Google Scholar]
- 5.Bermejo P, Martin-Aragon S, Benedi J, et al. Differences of peripheral inflammatory markers between mild cognitive impairment and Alzheimer’s disease. Immunol Lett 2008;117:198–202. [DOI] [PubMed] [Google Scholar]
- 6.Bonotis K, Krikki E, Holeva V, Aggouridaki C, Costa V, Baloyannis S. Systemic immune aberrations in Alzheimer’s disease patients. J Immunol 2008;193:183–187. [DOI] [PubMed] [Google Scholar]
- 7.Ray S, Britschgi M, Herbert C, et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signalling proteins. Nat Med 2007;13:1359–1362. [DOI] [PubMed] [Google Scholar]
- 8.Holmes C, El-Okl M, Williams AL, Cunningham C, Wilcockson D, Perry VH. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer’s disease. J Neurol Neurosurg Psychiatry 2003;74:788–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lanzrein AS, Johnston CM, Perry VH, Jobst KA, King EM, Smith AD. Longitudinal study of inflammatory factors in serum, cerebrospinal fluid, and brain tissue in Alzheimer disease: interleukin-1beta, interleukin-6, interleukin-1 receptor antagonist, tumor necrosis factor-alpha, the soluble tumor necrosis factor receptors I and II, and alpha1-antichymotrypsin. Alzheimer Dis Assoc Disord 1998;12:215–227. [DOI] [PubMed] [Google Scholar]
- 10.Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol 2007;7:161–167. [DOI] [PubMed] [Google Scholar]
- 11.Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 2005;25:9275–9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunningham C, Campion S, Lunnon K, et al. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry 2009;65:304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheng JG, Mrak RE, Griffin WS. Neuritic plaque evolution in Alzheimer’s disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathol (Berl) 1997;94:1–5. [DOI] [PubMed] [Google Scholar]
- 14.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 15.Rosen WG, Mohs RC, Davis K. A new rating scale for Alzheimer’s Disease. Am J Psychiatry 1984;141:1356–1364. [DOI] [PubMed] [Google Scholar]
- 16.Inouye SK, van Dyck CH, Alessi CA, Balkin S, Siegal AP, Horwitz RI. Clarifying confusion: the confusion assessment method: a new method for detection of delirium. Ann Intern Med 1990;113:941–948. [DOI] [PubMed] [Google Scholar]
- 17.Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice—a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003;108:499–511. [DOI] [PubMed] [Google Scholar]
- 18.Alvarez A, Cacabelos R, Sanpedro C, Garcia-Fantini M, Aleixandre M. Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol Aging 2007;28:533–536. [DOI] [PubMed] [Google Scholar]
- 19.Bruunsgaard H, Pedersen M, Pedersen BK. Aging and proinflammatory cytokines. Curr Opin Hematol 2001;8:131–136. [DOI] [PubMed] [Google Scholar]
- 20.Cechetto DF, Hachinski V, Whitehead SN. Vascular risk factors and Alzheimer’s disease. Expert Rev Neurother 2008;8:743–750. [DOI] [PubMed] [Google Scholar]
- 21.Kamer AR, Dasanayake AP, Craig RG, Glodzik-Sobanska L, Bry M, de Leon MJ. Alzheimer’s disease and peripheral infections: the possible contribution from periodontal infections, model and hypothesis. J Alzheimers Dis 2008;13:437–449. [DOI] [PubMed] [Google Scholar]
- 22.Kodl CT, Seaquist ER. Cognitive dysfunction and diabetes mellitus. Endocr Rev 2008;29:494–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiu C, De RD, Fratiglioni L. The epidemiology of the dementias: an update. Curr Opin Psychiatry 2007;20:380–385. [DOI] [PubMed] [Google Scholar]
- 24.Luchsinger JA. Adiposity, hyperinsulinemia, diabetes and Alzheimer’s disease: an epidemiological perspective. Eur J Pharmacol 2008;585:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welsh P, Woodward M, Rumley A, Lowe G. Associations of plasma pro-inflammatory cytokines, fibrinogen, viscosity and C-reactive protein with cardiovascular risk factors and social deprivation: the fourth Glasgow MONICA study. Br J Haematol 2008;141:852–861. [DOI] [PubMed] [Google Scholar]
- 26.Licastro F, Pedrini S, Caputo L, et al. Increased plasma levels of interleukin-1, interleukin-6 and alpha-1-antichymotrypsin in patients with Alzheimer’s disease: peripheral inflammation or signals from the brain? J Neuroimmunol 2000;103:97–102. [DOI] [PubMed] [Google Scholar]
- 27.Zuliani G, Ranzini M, Guerra G, et al. Plasma cytokines profile in older subjects with late onset Alzheimer’s disease or vascular dementia. J Psychiatr Res 2008;41:686–693. [DOI] [PubMed] [Google Scholar]
- 28.Godoy MC, Tarelli R, Ferrari CC, Sarchi MI, Pitossi FJ. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson’s disease. Brain 2008;131:1880–1894.18504291 [Google Scholar]
- 29.Nguyen MD, D’Aigle T, Gowing G, Julien JP, Rivest S. Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J Neurosci 2004;24:1340–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.WHO. The ICD-10 Classification of Mental and Behavioural Disorders. Geneva: World Health Organization; 1992. [Google Scholar]
- 31.McCusker J, Cole M, Dendukuri N, Belzile E, Primeau F. Delirium in older medical inpatients and subsequent cognitive and functional status: a prospective study. CMAJ 2001;165:575–583. [PMC free article] [PubMed] [Google Scholar]
- 32.Bruunsgaard H, Ladelund S, Pedersen AN, Schroll M, Jorgensen T, Pedersen BK. Predicting death from tumour necrosis factor-alpha and interleukin-6 in 80-year-old people. Clin Exp Immunol 2003;132:24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pickering M, Cumiskey D, O’Conner JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol 2008;90:663–670. [DOI] [PubMed] [Google Scholar]
- 34.Zhao X, Bausano B, Pike BR, et al. TNF-alpha stimulates caspase-3 activation and apoptotic cell death in primary septo-hippocampal cultures. J Neurosci Res 2001;64:121–131. [DOI] [PubMed] [Google Scholar]
- 35.Beattie EC, Stellwagen D, Morishita W, et al. Control of synaptic strength by glial TNFα. Science 2002;295:2285. [DOI] [PubMed] [Google Scholar]
- 36.Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci 2005;25:3219–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]