

Abstract
Introduction
To understand the role of two interacting proteins LIMD1 and pRB in development of head and neck squamous cell carcinoma (HNSCC), alterations of these genes were analyzed in 25 dysplastic head and neck lesions, 58 primary HNSCC samples and two HNSCC cell lines.
Methods
Deletions of LIMD1 and RB1 were analyzed along with mutation and promoter methylation analysis of LIMD1. The genotyping of LIMD1 linked microsatellite marker, hmlimD1, was done to find out any risk allele. The mRNA expression of LIMD1 and RB1 were analyzed by Q-PCR. Immunohistochemical analysis of RB1 was performed. Alterations of these genes were correlated with different clinicopathological parameters.
Results
High frequency [94% (78/83)] of LIMD1 alterations was observed in the samples studied. Compare to frequent deletion and methylation, mutation of LIMD1 was increased during tumor progression (P = 0.007). Six novel mutations in exon1 and one novel intron4/exon5 splice-junction mutation were detected in LIMD1 along with a susceptible hmlimD1 (CA)20 allele. Some of these mutations [42% (14/33)] produced non-functional proteins. RB1 deletion was infrequent (27%). Highly reduced mRNA expression of LIMD1 (25.1 ± 19.04) was seen than RB1 (3.8 ± 8.09), concordant to their molecular alterations. The pRB expression supported this data. Tumors with LIMD1 alterations in tobacco addicted patients without HPV infection showed poor prognosis. Co-alterations of these genes led the worse patients' outcome.
Conclusions
Our study suggests LIMD1 inactivation as primary event than inactivation of RB1 in HNSCC development.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is an aggressive malignancy, accounts for 30-40% of all cancer types in Indian subcontinent [1]. Tobacco, betel nut leaf quid, alcohol, HPV-16/18 infection are well recognized carcinogenic risk factors for development of this cancer [2]. Despite significant progress in understanding molecular genetic events underlying the development of HNSCC, details mechanisms still remain unknown [3,4]. Suppression of tumorigenicity of oral cancer cell lines following introduction of chromosome 3p in microcell hybrid system, suggested the presence of at least one tumor suppressor gene (TSG) in this chromosome associated with HNSCC development [5]. Our previous study in HNSCC of Indian patients showed high frequency of loss of heterozygosity (LOH) in chromosomal (chr.) 3p21.31 region and its association with development of early dysplastic lesions [6]. Among the multiple TSGs localized in chr.3p21.31, our recent study demonstrated one of the candidate TSGs, LIMD1 alteration (deletion/methylation) was significantly associated with mild dysplastic lesions of head and neck [7]. Downregulation of this gene observed in HNSCC and lung cancer [7,8]. A recent study emphasized LIMD1 as a critical TSG showing frequent downregulation in expression due to genetic and epigenetic modification in human lung cancer [9]. But no coding region mutation of this gene was observed in lung cancer. Also a polymorphic dinucleotide cytosine-adenine [d(CA)] microsatellite repeat, hmlimD1 (Accession number EU125867) was located at 15 bp upstream of LIMD1 gene [7]. Susceptibility allele of this gene, if any, for HNSCC development was unknown. LIMD1 has 8 exons and encodes a 676 amino acid protein, with a leucine-rich nuclear export signal (NES) in its N-terminal Pre-LIM domain and in C-terminus harboring three LIM domains having nuclear localizing properties (NLS) [8-10]. It is a ZYXIN family protein, having tandem zinc fingers in its LIM domains facilitating protein-protein interactions [11]. LIMD1 was reported to inhibit cell growth and metastases, partly mediated through either an interaction of its N-terminal LEM domain (amino acid 18-68) with barrier-to-autointegration (BAF), a component of SWI/SNF chromatin-remodeling protein, or through interaction of its part of proline-serine rich domain (amino acid 326-608) with C-terminus of retinoblastoma protein, pRB (amino acid 763-928) followed by transcriptional repression of E2F target genes [8]. This might be due to the stabilization of pRB-E2F interaction. The retinoblastoma gene, RB1 was reported to be infrequently altered in HNSCC [12,13]. Our previous study showed RB1 gene deletions were mainly associated with later stages in HNSCC development [14,15]. However, alterations of LIMD1 and RB1 were not screened in same set of samples to understand their association together in development of the disease.
Thus in this study attempts have been made to analyze the alterations of LIMD1 and RB1 in 25 dysplastic head and neck lesions, 58 primary HNSCC samples and two HNSCC cell lines. We have screened LIMD1 mutation in the entire exon1 (1429 bp) and exon5 along with RB1 deletion and its protein expression (by immunohistochemistry, IHC) in the same set of samples. The frequency of LIMD1 mutation was then compiled with our previously reported [7] of its deletion, promoter methylation and mutation (in the SNP rs267236 site in exon1) frequencies in these samples for clinicopathological correlation. The alterations of LIMD1 were also correlated with RB1 alterations. A population-based case-control study was performed to find out any risk allele of hmlimD1. Our data demonstrated that inactivation of LIMD1 was primary event than RB1 in development of HNSCC.
Materials and methods
Patients, controls and cell lines
A total of 37 dysplastic lesions and 110 HNSCC tumors and their matched normal tissues were collected from 147 unrelated individuals after obtaining informed consent from patients according to hospital authorities of Chittaranjan National Cancer Institute and Cancer Center & Welfare Home, Kolkata, India. All tumors were graded and staged according to UICC TNM classification [16]. Freshly operated tissues were taken for isolation of DNA/RNA and immunohistochemical analysis. Among these samples deletion/methylation/mutation analysis of the genes was performed in 25 dysplastic lesions (mild, n = 3; moderate, n = 9; severe, n = 13), 58 HNSCC samples (stageI, n = 8; stageII, n = 11; stageIII, n = 21; stageIV, n = 18) and two HNSCC cell lines Hep2, UPCI: SCC084. However, the total samples pool was used for case-control study. Table 1 presented clinicopathological information of patient (n = 147) and unrelated controls (n = 187) having no previous and present history of HNSCC. Blood samples were collected from controls with informed consent, and Institutional Ethical Board approved the study. All patients and controls were an age and sex frequency-matched ethnically similar caste population from eastern India. Two HNSCC cell lines, Hep2 and UPCI: SCC084 were obtained from National Centre for Cell Sciences, Pune, India and from Prof. Susanne M. Gollin, University of Pittsburgh, USA, respectively.
Table 1.
Characteristics of patients and controls
| Controls(187) | Sex* | ||
|---|---|---|---|
| Male, N(%) | 157(83.9%) | ||
| Female,(N%) | 30(16.1%) | ||
| Mean Age# | 45.5 ± 5.2 | ||
| HNSCC & Dysplastic lesions(147) | Sex* | ||
| Male, N(%) | 110(74.8%) | ||
| Female,(N%) | 37(25.2%) | ||
| Mean Age# | 52 ± 6.4 | ||
| Lymp Node Positive | 26(17.7%) | ||
| Lymp Node Negative | 121(82.3%) | ||
| Tobacco User | 103(70%) | ||
| Tobacco free | 44(30%) | ||
| Alcohol User | 13(8.8%) | ||
| Alcohol free | 134(91.2%) | ||
| HNSCC (110) | Oral Cavity tumors(85) | Buccal Mucosa, N(%) | 32(29.1%) |
| Tongue, N(%) | 13(11.8%) | ||
| Cheek, N(%) | 19(17.3%) | ||
| Lip, N(%) | 3(2.7%) | ||
| Tonsil, N(%) | 4(3.6%) | ||
| Palate, N(%) | 2(1.8%) | ||
| Alveolus, N(%) | 11(10%) | ||
| Vocal cord, N(%) | 1(0.9%) | ||
| Laryngeal Tumors(12) & | Larynx, N(%) | 12(10.9%) | |
| Nasopharyngeal Tumor(3) | Nassopharynx, N(%) | 3(2.7%) | |
| Orofacial Tumors(10) | Maxilla, N(%) | 5(4.5%) | |
| Mandible, N(%) | 5(4.5%) | ||
| Histopathology(110) | StageI | 15(14%) | |
| StageII | 30(27%) | ||
| StageIII | 34(31%) | ||
| StageIV | 31(28%) | ||
| Dysplastic lesions (37) | Buccal Mucosa, N(%) | 37(100%) | |
| Histopathology(37) | Mild Dysplasia | 5(13.5%) | |
| Moderate Dysplasia | 19(51.4%) | ||
| Severe Dysplasia | 13(35.1%) | ||
* Difference in sex distribution: Control vs. HNSCC, P = 0.04,
# Mean age difference: Control vs. HNSCC, P = 0.01.
Microdissection and DNA Extraction
Cryosections (5 μm) were microdissected under dissecting microscope (Leica MZ16, Germany) using surgical knives to remove contaminant normal cells. Controls' blood and samples containing >60% tumor cells were taken for DNA isolation by phenol/chloroform extraction [17].
Mutation analysis
LIMD1 was screened for mutation in 25 dysplasias, 58 HNSCC samples and the two HNSCC cell lines by single strand conformation polymorphism (SSCP) analysis using [α-P32] dCTP [14]. For mutation analysis six sets of primers were designed to amplify whole exon1 and one set for the exon5 including their respective intron/exon borders (see Additional file 1: Table S1). In these samples deletion and promoter methylation of LIMD1 have already been reported [7], along with mutation status in exon1 at the SNP rs267236 site using LIMD1 Exon1.6 primer set (see Additional file 1: Table S1) in the 77/85 samples. Electrophoresis was done in 6% non-denaturing polyacrylamide gel with 10% glycerol at 2W for overnight and autoradiographed on X-ray film (Kodak, USA). Samples showing abnormal band shifts were sequenced using Genetic Analyzer (PE Applied Biosystems Inc, USA).
Deletion analysis
Deletion mapping of RB1 was done in the same set of 25 dysplasias, 58 HNSCC samples and two HNSCC cell lines (as mentioned in Mutation analysis) using one intragenic microsatellite marker, D13S153 (intron2 of RB1, 47.8 Mb from p-ter) [15].
Genotyping of hmlimD1 microsatellite marker
Genotyping of hmlimD1 was carried out by amplifying d(CA)n repeat in a standard polymerase chain reaction (PCR) using [γ-P32] ATP labeled forward primer. PCR products were electrophoresed on 7% denaturing polyacrylamide sequencing gel and autoradiographed [18]. Signal intensities of radio-labeled products were measured by densitometric scanner (Bio-Rad, USA). Amplified fragments ranged in size from 187-216 bp, depending on the number of CA repeats within amplified region. Representative homozygote for (CA)17, (CA)19, (CA)20, (CA)21, (CA)24 and (CA)32 genotypes were sequenced using Genetic Analyzer (PE Applied Biosystems Inc, USA) to validate CA repeats number.
mRNA expression analysis
The mRNA expression of LIMD1 and RB1 genes was analyzed by quantitative RT-PCR in paired primary HNSCC samples (n = 24) and two HNSCC cell lines using primers mentioned in Additional file 1: Table S1. The LIMD1 mRNA expression was already reported in 11 of the 24 HNSCC samples and the two cell lines [7]. Total RNA was isolated from the samples using TRIzol reagent (Invitrogen, USA) and complementary-DNA (cDNA) was synthesized using Random hexamer (Invitrogen, USA) and M-MuLV Reverse Transcriptase (Sibenzyme, Russia). Real-time quantification of LIMD1 and RB1 was performed in 40 cycles on an ABI Prism 7500 using Power SYBR Green PCR Master Mix (Applied Biosystems, USA) in a final volume of 25 μl human β2-microglobulin gene (B2M) was used as control. Each sample was loaded in triplicate. Relative level of gene expression was determined by comparative threshold cycle (ddCt) method [19] after normalization against B2M.
Immunohistochemical analysis
pRB expression was determined by immuno-staining in 5 dysplasias, 15 HNSCC samples and the two cell lines. Paraffin sections of primary tissues and cover slip cultures of the cell lines were reacted with 1:100 dilution of primary antibodies (rabbit polyclonal IgG, sc-7905, for pRB [M-153] Santa Cruz, USA, raised against amino acids 769-921 from C-terminus of pRB). HRP-conjugated (sc-2004 with 1:500 dilutions) and FITC-tagged (sc-2012 with 1:100 dilutions) goat anti-rabbit secondary antibodies (Santa Cruz, USA) were used for the primary tissues and cell lines respectively. For permanent staining of the primary tissues, the slides were developed using 3, 3'diaminobenzidine as the chromogen and counterstained with hematoxylin. The staining intensity (1 = weak, 2 = moderate, 3 = strong) and the percentage of positive cells (<1 = 0, 1-20 = 1, 20-50 = 2, 50-80 = 3 and >80 = 4) were detected by two observers independently and by combining the two scores, final evaluation of expression was done (= 2 = low, 3-4 = intermediate, 5-6 = normal, = 7 = high) [20]. The immunocytochemical stained slides of the cell lines was photographed using fluorescence microscope (Nikon Eclipse E600, Japan).
Detection of HPV-16 and HPV-18
Presence of HPV in HNSCC was detected by PCR using primers (MY09 and MY11) from consensus L1 region followed by typing of HPV 16/18 in L1 positive samples as described [6].
Statistical analysis
Fisher's exact test was used to determine different clinico-pathological association with tumors genetic profile, to evaluate case-control difference in distribution of genotypes and to measure the strength of association between LIMD1 d(CA)9-38 repeat polymorphisms and HNSCC risk. All statistical tests were 2-sided and considered significant at probability value, P < 0.05. Survival analysis was performed according to Kaplan-Meier method in 50 HNSCC samples (oral cavity n = 40, orofacial n = 4, larynx n = 4, nasopharynx n = 2). Post-operative overall survival was measured from date of surgery to date of last follow-up or death (upto 5 years). P-values were evaluated by log-rank test for censored survival data. Significance and relative risk of various potential prognostic factors was evaluated by Cox proportional hazards model and the hazard ratio (HR) for each parameter with 95% confidence interval (CI) in a multivariate fashion were estimated to predict joint impact of several prognostic factors on overall survival of patients with oral cavity cancer. Analysis of other sites in HNSCC was excluded due to small sample size. All statistical analysis was performed using EpiInfo 6.04b, SPSS 10.0 (SPSS Inc. Chicago, IL, USA).
Results
Alterations of LIMD1
To prove LIMD1 as a candidate TSG, mutation in exon1 and exon5 of this gene was screened in 83 of primary head and neck lesions (25 dysplasias and 58 HNSCC) and two HNSCC cell lines. The deletion and promoter methylation status of LIMD1 were screened in these sample previously [7]. Six novel mutations were observed in exon1 (Fig 1A-E) and one was in splice-junction of intron4/exon5 (C deletion 3nt upstream of exon5; Fig 1F, Table 2). Among the mutations in exon1, one transversion with aminoacid changes at 1003C→A (Pro335Thr; Fig 1E), three transition with aminoacid changes at 470C→T (Ser157Phe; Fig 1A) and without aminoacid changes at 882T→C (Thr294Thr; Fig 1C) and 1068T→C (Gly356Gly) [7] and two frameshift mutations due to single nucleotide deletions at 660A (Fig 1B) and 967T (Fig 1D) were observed. Two transition mutations were overlapped with the two SNPs (rs267237 and rs267236) (see additional file 2: Figure S1 and additional file 3: Figure legend for Figure S1.txt). Frameshift mutations led to premature terminations of proteins (Table 2, Fig 2). About 40% (33/83) of the primary head and neck lesions showed mutations in at least one site. Majority of mutations were clustered in proline and serine rich domain of LIMD1 (Fig 2). No mutation was observed in Hep2 and UPCI: SCC084 cells. The status and pattern of LIMD1 alterations (deletion/methylation/mutation) in the 83 of primary head and neck lesions and the two HNSCC cell lines were presented in the additional files (see Additional file 4: Table S2 and Additional file 5: Table S3). Mutation frequency of LIMD1 was low (8%, 2/25) in dysplastic lesions, but significantly (P = 0.007) increased in stage (I+II) tumors (42%, 8/19) (Fig 3A). Moreover, significant association has been seen between LIMD1 mutation and its methylation in HNSCC samples (Table 3).
Figure 1.

SSCP autoradiographs and respective chromatographs with resulting aminoacid sequences of representative samples showing mutations in LIMD1 exon 1 (A-E), and exon5 (F). Arrows indicated shifted bands and nucleotide changes. T, Tumor; N, corresponding normal.
Table 2.
Summary of LIMD1 mutations
| Mutations | ||||||||
|---|---|---|---|---|---|---|---|---|
| Exon1 | Exon5 | |||||||
| Nucleotide change | 470C → T | 660A del | 882C → T | 967T del | 1003C → A | 1068T → C | C del at intron4/exon5 | |
| TNM stage | Amino acid change | Ser157 Phe | Frame shift | Thr294Thr | Frame shift | Pro335Thr | Gly356Gly | Splice-juction |
| Samples showing mutations | ||||||||
| Moderate | L66 | - | - | - | - | - | - | - |
| Moderate | L139 | - | - | SNPa | - | - | + | - |
| Severe | L144 | - | - | SNPa | - | - | - | - |
| Severe | L126 | - | - | SNPa | - | - | + | - |
| Severe | L153 | - | - | SNPa | - | - | - | - |
| StageI | #292 | - | - | SNPa | - | - | - | - |
| StageI | #2642 | - | - | - | - | - | + | - |
| stageII | #2772 | - | - | + | - | - | - | - |
| stageII | #7216 | - | - | - | + | - | - | - |
| stageII | #2073 | + | - | SNPa | - | - | + | - |
| stageII | #5303 | - | + | - | - | - | - | - |
| stageII | #1234 | - | - | SNPa | - | - | SNPb | - |
| stageII | #2323 | - | - | + | - | - | - | - |
| stageII | #1108 | - | - | - | - | - | + | - |
| stageII | #7077 | + | - | - | - | - | - | - |
| StageIII | #821 | - | - | - | - | - | + | - |
| StageIII | #1367 | + | - | - | - | - | - | - |
| StageIII | #1552 | - | - | SNPa | - | + | - | - |
| StageIII | #615 | - | - | - | + | - | - | - |
| StageIII | #5090B | - | - | + | - | - | - | - |
| StageIII | #816 | - | - | + | - | - | - | - |
| StageIII | #326 | - | + | SNPa | - | - | - | - |
| StageIII | #1068 | - | - | SNPa | - | + | - | - |
| StageIII | #3893 | - | - | - | + | - | - | - |
| StageIII | #308 | - | + | SNPa | - | - | - | - |
| StageIII | #6433 | - | + | SNPa | - | - | - | - |
| StageIII | #1332 | + | - | SNPa | - | - | - | + |
| StageIII | #2398 | - | - | - | + | - | - | - |
| StageIII | #7059 | - | + | - | - | - | - | - |
| StageIV | #1087 | - | - | + | - | - | - | - |
| StageIV | #4075 | - | - | - | - | - | SNPb | - |
| StageIV | #2508 | - | + | - | - | - | + | - |
| StageIV | #5114 | - | - | + | - | - | - | - |
| StageIV | #5219 | - | + | - | - | - | - | - |
| StageIV | #1084 | + | - | - | - | - | SNPb | - |
| StageIV | #5184 | - | + | - | - | - | - | - |
| StageIV | #944 | - | - | - | - | - | - | + |
| StageIV | #2785 | - | - | SNPa | - | - | - | + |
| StageIV | #1774 | - | - | - | - | - | + | - |
| Cell lines | Hep2 | - | - | SNPa | - | - | SNPb | - |
| SCC084 | - | - | SNPa | - | - | SNPb | - | |
| Mutation frequency in HNSCC | 6% (5/85) | 9% (8/85) | 7% (6/85) | 4% (4/85) | 2% (2/85) | 9% (8/85) | 4% (3/85) | |
SNPa: rs267237; SNPb:rs267236; del: deletion; L: Dysplastic lesions; #: Invasive samples; '+': Presence; '-': Absence.
Figure 2.

Schematic diagram of LIMD1 gene and protein with mutational hotspot. Termination1,2represented premature termination of LIMD1 protein after 7aminoacids and 2 aminoacids respectively. Aminoacids 404-442: Reported pRB binding region [8].
Figure 3.
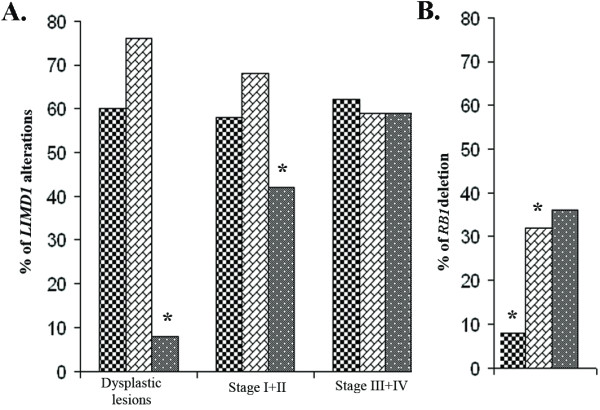
(A). Patterns of LIMD1 molecular alterations during HNSCC progression. Bars correspond as follows: *: indicated the level of significance. [Checkered line] Deletion; [brick patterned line] Methylation; [grey spotted line] Mutation. (B). RB1 deletion pattern in progression of HNSCC. Bars correspond as follows: [Checkered line] Dysplastic lesions; [brick patterned line] Stage I+II; [grey spotted line] StageIII+IV; *: indicated the level of significance.
Table 3.
Association study of LIMD1 different molecular alterations
| Dysplastic lesions | HNSCC | |||||
| Del+ | Del- | P | Del+ | Del- | P | |
| Meth+ | 11 | 8 | 0.7022 | 21 | 15 | 0.6887 |
| Meth- | 4 | 2 | 14 | 8 | ||
| Mut+ | 1 | 1 | 0.7634 | 18 | 13 | 0.7036 |
| Mut- | 14 | 9 | 17 | 10 | ||
| Mut+ | Mut- | Mut+ | Mut- | |||
| Meth+ | 1 | 18 | 0.3694 | 15 | 21 | 0.02139 |
| Meth- | 1 | 5 | 16 | 6 | ||
Del: Deletion; Meth: Methylation; Mut: Mutation; '+': Positive; '-': Negative.
Association of hmlimD1 polymorphism with risk of HNSCC
To find out LIMD1 susceptible allele, if any, associated with HNSCC risk, we analyzed allele polymorphism of hmlimd1 microsatellite marker in population based case-control study. Age and sex distribution between the cases and controls were found to be statistically significant (Table 1). A total of 12 CA repeat alleles were observed in our study population, ranging from 9 repeats [(CA)9] to 38 repeats [(CA)38] (Table 4). Among them, alleles (CA)17 and (CA)19 were relatively common (>10% in frequency) in control population. However, in cases >10% allele frequency was seen in (CA)19, (CA)20 and (CA)24 alleles. Overall, case-control difference in allele distribution was seen to be statistically significant (P = 0.0021). Comparing the frequency of each allele with that of all other alleles combined, case-control differences were significant for (CA)20 [P = 0.000005], (CA)32 [P = 0.0082] and borderline significance for (CA)24 allele [P = 0.05427]. The (CA)20 allele was over represented in cases, whereas (CA)17 and (CA)19 alleles were under represented in cases where >10% allele frequency of the alleles were seen. In comparison to homozygous alleles distribution among cases and controls, significant association of [(CA)20/(CA)20] allele (P = 0.00003) was seen with the cases. Alleles (CA)32 and (CA)38 were rare in this study population.
Table 4.
Allele frequency of (CA)n polymorphism in the LIMD1 gene in head and neck cancer cases and control
| Allele | No of (CA)n repeats | Case | Control | P# | ||
|---|---|---|---|---|---|---|
| No. of alleles | % | No. of alleles | % | |||
| (CA)9 | 9 | 7 | 2.38 | 11 | 2.94 | 0.6571 |
| (CA)13 | 13 | 4 | 1.36 | 35 | 9.36 | 0.00012 |
| (CA)17 | 17 | 23 | 7.82 | 46 | 12.29 | 0.0592 |
| (CA)19 | 19 | 97 | 32.99 | 169 | 45.19 | 0.00139 |
| (CA)20 | 20 | 66 | 22.45 | 36 | 9.63 | 0.000005 |
| (CA)21 | 21 | 21 | 7.14 | 15 | 4.01 | 0.075134 |
| (CA)24 | 24 | 30 | 10.2 | 23 | 6.15 | 0.05427 |
| (CA)26 | 26 | 2 | 0.68 | 15 | 4.01 | 0.0067 |
| (CA)27 | 27 | 19 | 6.46 | 13 | 3.48 | 0.0728 |
| (CA)30 | 30 | 10 | 3.4 | 6 | 1.6 | 0.1316 |
| (CA)32 | 32 | 14 | 4.76 | 5 | 1.34 | 0.0082 |
| (CA)38 | 38 | 1 | 0.34 | 0 | 0 | 0.259 |
| Total no. of alleles | 294 | 100 | 374 | 100 | ||
| χ2 = 29.175, df = 11, P = 0.0021 | ||||||
| Genotype | Case | % | Control | % | P# | |
| (CA)17/(CA)17 | 6 | 9.23 | 11 | 15.94 | 0.2434 | |
| (CA)19/(CA)19 | 33 | 50.76 | 56 | 81.16 | 0.0002 | |
| (CA)20/(CA)20 | 19 | 29.23 | 2 | 2.89 | 0.00003 | |
| (CA)21/(CA)21 | 5 | 7.69 | 0 | 0 | 0.0247 | |
| (CA)24/(CA)24 | 1 | 1.54 | 0 | 0 | 0.4851 | |
| (CA)32/(CA)32 | 1 | 1.54 | 0 | 0 | 0.4851 | |
# From Fisher's exact test.
OR, Odds ratio
CI, Confidence interval
Association of LIMD1 alterations with RB1 deletion in HNSCC
As LIMD1 was shown to interact with pRB [8], we studied the association of alterations of both genes. Compare to high LIMD1 alterations, RB1 deletion was very low (27%, 22/83) in the primary head and neck lesions (Fig 3B). Deletion frequency of RB1 was low in dysplastic lesions (8%, 2/25) and significantly (P = 0.045) increased to stage (I+II) tumors (32%, 6/19) consistence with our previous findings [13,14] (Fig 3B). No association between LIMD1 alterations and RB1 deletion was observed, however co-alteration of both genes was high (33%, 13/39) in the stage III+IV of head and neck lesions (Table 5).
Table 5.
Association study of LIMD1 alterations with RB1 deletion
| LIMD1 | |||||||
|---|---|---|---|---|---|---|---|
| Dysplasia | Stage I+II | Stage III+IV | |||||
| ALT+ | ALT- | ALT+ | ALT- | ALT+ | ALT- | ||
| RB1 | Del+ | 2 | 0 | 7 | 0 | 13 | 0 |
| Del- | 21 | 2 | 11 | 1 | 24 | 2 | |
| P | 0.6637 | 0.4326 | 0.3046 | ||||
Alt: Overall molecular alterations; Del: Deletion; '+': Positive; '-': Negative.
Comparison in the mRNA expression of LIMD1 and RB1
Quantitative RT-PCR analysis revealed high reduction of LIMD1 mRNA expression (25.1 ± 19.04) than RB1 (3.8 ± 8.09) (Fig 4). Reduced mRNA expression of LIMD1 showed significant correlation with the gene's methylation (see Additional file 6: Table S4). Half of these tumors (12/24) showed mean fold reduction in LIMD1 expression compare to 21% (5/24) in RB1. In Hep2 and UPCI: SCC084 cells RB1 expression was comparable to the normal tissues than that of reduced LIMD1 expression.
Figure 4.

(A). Quantitative RT-PCR analysis showing LIMD1 and RB1 expression in HNSCC samples (n = 24), Hep2 and UPCI: SCC084 cell lines. Bars represented the gene expression normalized to β2-microglobulin and relative to normal counterpart. Black bar represented RB expression and cross line filled bar represented LIMD1 expression.
Immuno-staining of pRB
In normal epithelium, immuno-staining revealed intense nuclear and cytoplasmic pRB expression at basal layer, followed by gradual low cytoplasmic expression in differentiated cells (Fig 5). In dysplastic lesions as well as in HNSCC, both nuclear and cytoplasmic expression of pRB was observed. Low/medium level of pRB expression was observed in 30% (6/20) of head and neck lesions contrary to frequent LIMD1 alterations (Table 6). In HPV18 positive Hep2 cells, pRB expression was mainly in cytosol, whereas in UPCI: SCC084 (HPV negative), it was expressed both in nucleus and cytoplasm. Concordance was seen between RB1 deletion with its RNA and protein expression (Fig 5).
Figure 5.

Immunohistochemical staining patterns of pRB in dysplastic lesions (L), HNSCC samples (T), normal tissues (Normal) and HNSCC cell lines (Hep2, UPCI: SCC084) samples showing high (a), low/negative (b) expression of pRB protein (indicated by arrows). In #797T, RB1 was not deleted whereas in #944T, this gene was deleted. The regions marked within the circle (20×) were magnified to 40× at inset.
Table 6.
Correlation between mRNA and protein expression of RB1 and their association with LIMD1 molecular alterations.
| RB1 | LIMD1 | ||||
|---|---|---|---|---|---|
| Sample No.; HPV status | Genetic alterations | mRNA status | Protein expression | Genetic/epigenetic alterations | mRNA status |
| Dysplastic lesions | |||||
| L127; HPV- | D- | ND | Normal | D+Me+Mu- | ND |
| L139; HPV- | D- | ND | Normal | D-Me+Mu- | ND |
| L154; HPV- | D- | ND | Normal | D+Me+Mu- | ND |
| L162; HPV- | D- | ND | Normal | D+Me+Mu- | ND |
| L158; HPV16 | D- | ND | Medium | D+Me+Mu- | ND |
| HNSCC samples | |||||
| #1004; HPV- | D- | Normal | Normal | D+Me+Mu- | ↓ |
| #5165; HPV- | D- | Normal | Normal | D+Me+Mu- | ↓ |
| #6817;HPV16 | D+ | ↓ | Medium | D+Me+Mu- | ↓ |
| #2884; HPV- | D- | Normal | Normal | D-Me-Mu- | Normal |
| #1108; HPV - | D+ | ↓ | Medium | D-Me+Mu+ | ↓ |
| #1774;HPV16 | D+ | ↓ | Low | D-Me+Mu+ | ↓ |
| #872; HPV- | D- | Normal | Normal | D-Me-Mu- | Normal |
| #5303; HPV- | D- | Normal | Normal | D-Me+Mu+ | ↓ |
| #6907;HPV16 | D- | ↓ | Low | D-Me+Mu- | ↓ |
| #1087; HPV- | D- | Normal | Normal | D-Me-Mu+ | Normal |
| #944; HPV- | D+ | ↓ | Low | D+Me+Mu+ | ↓ |
| #5733;HPV16 | D- | Normal | Normal | D+Me+Mu- | ↓ |
| #6392; HPV- | D- | Normal | Normal | D-Me+Mu- | ↓ |
| #7077; HPV- | D- | Normal | Normal | D-Me-Mu+ | ↓ |
| #797; HPV- | D- | Normal | Normal | D+Me+Mu- | ↓ |
| Hep2, HPV18 | D- | Normal | Normal | D-Me+Mu- | ↓ |
| UPCI:SCC084, HPV- | D- | Normal | Normal | D-Me+Mu- | ↓ |
L: Dysplastic lesions; # HNSCC samples; D: Deletion; Me: Methylation; Mu: Mutation; '+': Positive; '-': Negative; '↓': Down expression; ND: Not done.
Clinical and prognostic implications of LIMD1 and RB1 alterations
Using L1 primer, HPV DNA was detected in 56% (82/147) of head and neck lesions of which 84% (69/82) were HPV-16 positive, 11% (9/82) were HPV18 positive and the rest 5% (4/82) were positive for both HPV16/18. Univariate analysis showed significant association of RB1 deletion with HPV negativity (P = 0.02 for dysplastic lesions; P = 0.01 for HNSCC).
Log-rank test revealed significant poor survival in patients having alterations in at least one of these genes (Fig 6). Patients with both LIMD1 and RB1 alterations showed worse prognosis and absence of RB1 alterations did not change patients' survival considerably (Fig 6). Multivariate analysis showed that LIMD1 molecular alterations (P, 0.03; HR, 4.5; CI, 1.1-17.8) along with tobacco addiction (P, 0.05; HR, 4.5; CI, 0.96-20.8) in absence of HPV (P, 0.05; HR, 0.28; CI, 0.08-0.99) were significant predictor for poor survival of patients with oral cavity cancer (Table 7). Moreover, patients with advanced grade tumor (P, 0.50; HR, 1.49; CI, 0.46-4.89) having nodal metastasis (P, 0.42; HR, 1.63; CI, 0.49-5.34) along with RB1 deletion (P, 0.15; HR, 2.27; CI, 0.74-7.02) showed a trend to have poorer survival, albeit being statistically insignificant.
Figure 6.

Kaplan-Meier 5-year survival probability curves with cumulative survival of HNSCC patients by molecular alteration status in LIMD1 and RB1 loci. Solid line represented survival probability without molecular alterations of these two genes (wild type, denoted as '+') and dashed line represented the same probability with molecular alterations of either of these genes ('-' indicating altered state). n, total number of HNSCC samples studied in each case.
Table 7.
Multivariate analysis of overall survival of oral cavity cancer patients with different clinicopathological parameters.
| Variable | Overall survival | ||
|---|---|---|---|
| P-value | Hazard ratio(HR) | 95% CI for HR | |
| LIMD1 alteration | 0.0318 | 4.51 | 1.14-17.82 |
| RB deletion | 0.1535 | 2.27 | 0.74-7.02 |
| Stage | 0.5144 | 0.79 | 0.39-1.59 |
| Grade | 0.5044 | 1.49 | 0.46-4.89 |
| Age | 0.7786 | 0.84 | 0.25-2.85 |
| Node | 0.4228 | 1.63 | 0.49-5.34 |
| HPV | 0.0498 | 0.28 | 0.08-0.99 |
| Tobacco | 0.0559 | 4.49 | 0.96-20.89 |
| Alcohol | 0.2659 | 0.29 | 0.03-2.52 |
CI, Confidence interval.
Bold letters represent significance level.
Discussion
Differential alterations of LIMD1 and RB1 during HNSCC development
To understand the role of LIMD1 and RB1 in HNSCC development, alterations of these genes were analyzed in 25 dysplastic lesions of head and neck, 58 HNSCC samples and two HNSCC cell lines. In dysplastic lesions LIMD1 mutations was less frequent than deletion and methylation (see Additional file 4: Table S2 and Additional file 5: Table S3). However, during progression of the tumor comparable frequencies of these alterations were seen, suggesting the deletion and methylation of LIMD1 as early events in this tumorigenesis. About 80% (67/83) of the samples showed any two of the alterations in LIMD1 supporting the modified Knudson two hit hypothesis as a candidate TSG. High frequency (94%, 78/83) of LIMD1 alterations (deletion/methylation/mutation) than RB1 was seen in the samples. Majority of mutations in LIMD1 were present in and around the pRB binding domain of exon1. Unlike us, Huggins et al. detected very low frequency (4%, 6/165) of mutation in exon1 of LIMD1 at different positions in breast carcinoma [21]. In our samples, about 42% (14/33) of the mutations produced non-functional truncated LIMD1 proteins due to premature termination or splice-junction mutations (Table 2). The significance of synonymous mutations at two SNPs was not clear.
RB1 deletion was infrequent with much lower frequency in dysplastic lesions than invasive samples, suggests it less likely to be a candidate TSG in this tumor. Similar to other reports [22], there was a strong association between RB1 deletion and reduced pRB expression in our samples (P = 0.0006) (Table 6), indicating deletion as the main cause of RB1 inactivation. The differential downregulation in mRNA expression of LIMD1 and RB1 was concordant with their molecular alterations, reflecting genetic imbalances transmitted in transcript level. Intense pRB staining in basal layer of normal epithelial of oral cavity compare to the differentiated cells indicates differential regulation of pRB during differentiation. Similar reports were also found in normal epithelium of esophagus and cervix [23-25]. LIMD1 staining has not been done due to unavailability of commercial antibody. In dysplastic lesions and in HNSCC, pRB expression was seen both in nucleus and cytoplasm where RB1 deletion was absent, similar to basal layer of normal epithelium. However the functional status of pRB in tumor cells is not clear. The reduced level of LIMD1 in tumor cells might destabilize the pRB-E2F interaction and chromatin remodeling complex, resulting deregulation of cell cycle.
Identification of susceptible allele of LIMD1
As LIMD1 has been suggested to be a candidate TSG in HNSCC, our next attempt was to search for a susceptible allele of this gene, if any, associated with HNSCC risk. The case-control study identified (CA)20 as the risk allele both in its homozygous and heterozygous state for HNSCC development. Similar repeat length polymorphism in upstream of several genes has been reported [26-28]. The significance of the (CA)9-38 repeat variations at upstream of LIMD1 was not clear. Regarding the conformation, the (CA)19 repeat length might be a critical point. Though the significance of critical (CA)20 repeat as risk for this tumor development is not fully understood, it seems that this repeat length might destabilize Z-DNA conformation, causing transcriptional repression of LIMD1. Similar effect might also be imposed by (CA)21-(CA)38 alleles, but due to their low prevalence in our study population, statistical significance has not been observed. In light of the strength of associations and the increased risk for HNSCC, further research is warranted to explore the potential underlying mechanism(s) involved.
Association of LIMD1 and RB1 alterations with HNSCC progression and prognosis
Unlike methylation and deletion, significant association of LIMD1 mutation with tumor progression indicates that mutation might have some additive effect in inactivation of this gene. In multivariate analysis, association of LIMD1 alterations along with tobacco addictions HPV negativity and poor patients' outcome suggests LIMD1 as predictive clinical marker in progression of HNSCC. Agreed with similar findings [29-31], significant association was seen in RB1 deletion with HPV negative samples and with HNSCC progression. Likewise, a trend towards significance of RB1 deletion in advance grade tumors, with nodes of pathology has also been observed [31]. However, worse prognosis of the patients having both LIMD1 and RB1 alterations suggests RB1 inactivation might have some synergistic impact in HNSCC development. Comparatively better survival of patients with LIMD1+RB1- (LIMD1 unaltered &RB1 deleted) than LIMD1-RB1+ (LIMD1 altered &RB1 unaltered), indicates LIMD1 as a key regulator of the disease.
Conclusion
Thus, it can be concluded that LIMD1 is a susceptible gene for HNSCC development and its alterations, alone or with RB1 alterations acts as an important prognostic marker in this cancer.
Abbreviations
HNSCC: Head and neck squamous cell carcinoma.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SG carried out the molecular genetic studies, participated in the sequence alignment and drafted the manuscript. AG participated in the design of the study and performed the statistical analysis. GM, NM and SD provided the samples needed for this study. AR carried the histological studies and pathological screening. SR participated in its design and coordination and also helped to draft the manuscript. CKP conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors have read and approved the final manuscript.
Supplementary Material
Oligonucleotide primers used in the study. The data report the primers used in the study.
A representative chromatograph. The chromatograph represents C/T heterozygous in PBL at SNP rs267237 and C → T mutation in Tumor sample.
Legend for the Figure S1. Short explanation for the Figure S1
LIMD1 overall molecular alterations including deletion, methylation, mutation and mRNA expression. The data provided represent the overall molecular alterations ie deletion, promoter methylation, mutation of LIMD1 along with the mRNA expression in each of individual sample studied.
Pattern of LIMD1 alterations in HNSCC during progression of the disease. The data provided represent the frequencies of deletion, methylation and mutation during the progression of HNSCC.
Correlation between LIMD1 methylation and mRNA expression. The data provided represent the correlation between LMD1 promoter methylation and its mRNA expression in each individual sample.
Contributor Information
Susmita Ghosh, Email: psusmita01@gmail.com.
Amlan Ghosh, Email: amlan2006@gmail.com.
Guru P Maiti, Email: grprdm@gmail.com.
Nupur Mukherjee, Email: nupurmukherjee10@gmail.com.
Sankhadeep Dutta, Email: deepdshanko@yahoo.co.in.
Anup Roy, Email: anuproy@vsnl.net.
Susanta Roychoudhury, Email: susanta@iicb.res.in.
Chinmay K Panda, Email: ckpanda.cnci@gmail.com.
Acknowledgements
We are thankful to the Directors, Chittaranjan National Cancer Institute and Cancer Center & Welfare Home, Kolkata, India. We are also grateful to Prof. H. zur Hausen and Prof. E M. de Villiers for their generous gift of HPV-16/18 plasmids. We also thank Prof. Susanne M. Gollin for UPCI: SCC084 cell line. Financial support for this work was provided by grants from DST [SR/SO/BB-22/2003 dt. 02.11.04] and DBT [BT/PR/5524/Med/14/649/2004 of dt. 29.11.2005], Govt. of India to CKP and SR; UGC-NET Fellowship grant [F.2-3/2000 (SA-I)] to SG and CSIR-NET Fellowship grant [F.2-3/2000 (SA-I)] to AG.
References
- Tripathi A, Dasgupta S, Roy A. Sequential deletions in both Arms of Chromosome 9 are Associated with the Development of Head and Neck Squamous Cell Carcinoma in Indian Patients. J Exp Clin Cancer Res. 2003;22(3):289–297. [PubMed] [Google Scholar]
- Koch WM, Lango M, Sewell D, Zahurak M, Sidransky D. Head and neck cancer in nonsmokers: a distinct clinical and molecular entity. Laryngoscope. 1999;109:1544–51. doi: 10.1097/00005537-199910000-00002. [DOI] [PubMed] [Google Scholar]
- Forastiere A, Koch W, Trotti A. Head and neck cancer. N Engl J Med. 2001;345:1890–900. doi: 10.1056/NEJMra001375. [DOI] [PubMed] [Google Scholar]
- Williams HK. Molecular pathogenesis of oral squamous carcinoma. Mol Pathol. 2000;53:165–72. doi: 10.1136/mp.53.4.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzawa N, Yoshida MA, Oshimaura M, Ikeuchi T. Suppression of tumorigenicity in three different cell lines of human oral squamous cell carcinoma by introduction of chromosome 3p via microcell mediated chromosome transfer. Oncogene. 1995;11:1997–2004. [PubMed] [Google Scholar]
- Chakraborty SB, Dasgupta S, Roy A. Differential deletions in 3p are associated with the development of head and neck squamous cell carcinoma from Indian patients. Cancer Genetics and Cytogenetics. 2003;145:1–9. doi: 10.1016/S0165-4608(03)00105-5. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Ghosh A, Maiti GP. Alterations of 3p21.31 tumor suppressor genes in head and neck squamous cell carcinoma: Correlation with progression and prognosis. Int J Cancer. 2008;123(11):2594–2604. doi: 10.1002/ijc.23834. [DOI] [PubMed] [Google Scholar]
- Sharp TV, Munoz F, Bourboulia D. LIM domains-containing protein 1 (LIMD1), a tumor suppressor encoded at chromosome 3p21.3, binds pRB and represses E2F-driven transcription. Proc Natl Acad Sci. 2004;101:16531–16536. doi: 10.1073/pnas.0407123101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp TV, Al-Attar A, Foxler DE. The chromosome 3p21.3-encoded gene, LIMD1, is a critical tumor suppressor involved in human lung cancer development. Proc Natl Acad Sci. 2008;105:19932–19937. doi: 10.1073/pnas.0805003105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss H, Kedra D, Yang Y. A novel gene containing LIM domains (LIMD1) is located within the common eliminated region 1 (C3CER1) in 3p21.3. Hum Genet. 1999;105:552–559. doi: 10.1007/s004390051144. [DOI] [PubMed] [Google Scholar]
- Kadrmas JL, Beckerle MC. The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol. 2004;5:920–931. doi: 10.1038/nrm1499. [DOI] [PubMed] [Google Scholar]
- Blons H. Head and neck squamous cell carcinoma. Atlas Genet Cytogenet Oncol Haematol. 2002.
- Yoo GH, Xu HJ, Brennan JA. Infrequent Inactivation of the Retinoblastoma Gene Despite Frequent Loss of Chromosome 13q in Head and Neck Squamous Cell Carcinoma. Cancer Res. 1994;54:4603–4606. [PubMed] [Google Scholar]
- Tripathi A, Banerjee S, Chunder N. Differential alterations of the genes in the CDKN2A-CCND1-CDK4-RB1 pathway are associated with the development of head and neck squamous cell carcinoma in Indian patients. J Cancer Res Clin Oncol. 2003;129:642–650. doi: 10.1007/s00432-003-0485-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbir G, Roy A, Mandol S, Dam A, Roychoudhury S, Panda CK. Deletion mapping of chromosome 13q in head and neck squamous cell carcinoma in Indian patients: correlation with prognosis of the tumor. J Exp Path. 2006;87:151–161. doi: 10.1111/j.0959-9673.2006.00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer MH ed UICC TNM Classification of Malignant Tumors 19783Geneva: Union Internationale Contre le Cancer (UICC)76520 [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor, NY; Cold Spring Harbor, Laboratory; 1989. [Google Scholar]
- Dasgupta S, Mukherjee N, Roy S, Roy A, Sengupta A, Roychowdhury S, Panda CK. Mapping of candidate tumor suppressor genes' loci on human chromosome 3 in head and neck squamous cell carcinoma of Indian patient population. Oral Oncol. 2002;38:6–15. doi: 10.1016/S1368-8375(00)00131-7. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Perrone F, Suardi S, Dagrada G, Bossi P, Locati L, Pilotti S. Molecular and Cytogenetic Subgroups of Oropharyngeal Squamous Cell Carcinoma. Clin Cancer Res. 2006;12(22):6643–6651. doi: 10.1158/1078-0432.CCR-06-1759. [DOI] [PubMed] [Google Scholar]
- Huggins CJ, Gillb M, Andrulis IL. Identification of rare variants in the hLIMD1 gene in breast cancer. Cancer Genetics and Cytogenetics. 2007;178:36–41. doi: 10.1016/j.cancergencyto.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Xing EP, Yang GY, Wang LD, Shi ST, Yang CS. Loss of heterozygosity of the Rb gene correlates with pRb protein expression and associates with p53 alteration in human esophageal cancer. Clin Can Res. 1999;5:1231–1240. [PubMed] [Google Scholar]
- Hausen AZ, Sarbia M, Heep H, Willers R, Gabbert HE. Retinoblastoma-protein (prb) expression and prognosis in squamous-cell carcinomas of the esophagus. Int J Cancer. 1999;84(6):618–622. doi: 10.1002/(SICI)1097-0215(19991222)84:6<618::AID-IJC14>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Noraini MD, Siti-Aishah MA, Kwan SW. An immunohistochemical study of retinoblastoma gene product in normal, premalignant and malignant tissues of the uterine cervix. Malaysian Journal of Medical Sciences. 2003;10(1):52–59. [PMC free article] [PubMed] [Google Scholar]
- Edwards MJ, Thomas RC, Wong YL. Retinoblastoma gene expression in human non-melanoma skin cancer. J Cutan Pathol. 2003;30:479–485. doi: 10.1034/j.1600-0560.2003.00004.x. [DOI] [PubMed] [Google Scholar]
- Cai Q, Gao YT, Wen W. Association of breast cancer risk with a GT dinucleotide repeat polymorphism upstream of the estrogen recepter-A gene. Cancer Res. 2003;63:5727–5730. [PubMed] [Google Scholar]
- Wang B, Ren J, Ooi LLPJ, Chong SS, Lee CGL. Dinucleotide repeats negatively modulate the promoter activity of Cyr61 and is unstable in hepatocellular carcinoma patients. Oncogene. 2005;24:3999–4008. doi: 10.1038/sj.onc.1208550. [DOI] [PubMed] [Google Scholar]
- Cleveland RJ, Gammon MD, Edmiston SN. IGF1 CA repeat polymorphisms, lifestyle factors and breast cancer risk in the long island breast cancer study project. Carcinogenesis. 2006;27(4):758–765. doi: 10.1093/carcin/bgi294. [DOI] [PubMed] [Google Scholar]
- Wiest T, Schwarz E, Enders C, Flechtenmacher C, Bosch FX. Involvement of intact HPV16 E6/E7 gene expression in head and neck cancers with unaltered p53 status and perturbed pRb cell cycle control. Oncogene. 2002;21:1510–1517. doi: 10.1038/sj.onc.1205214. [DOI] [PubMed] [Google Scholar]
- Andi T, Kahn T, Pfuhl A. Etiological Involvement of Oncogenic Human Papillomavirus in Tonsillar Squamous Cell Carcinomas Lacking Retinoblastoma Cell Cycle Control. Cancer Res. 1998;58:5–13. [PubMed] [Google Scholar]
- Saranath D. In: Contemporary issues in oral cancer. Saranath D, editor. New Delhi: Oxford University Press; Integrated biology and molecular pathology of oral cancer; pp. 50–51. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oligonucleotide primers used in the study. The data report the primers used in the study.
A representative chromatograph. The chromatograph represents C/T heterozygous in PBL at SNP rs267237 and C → T mutation in Tumor sample.
Legend for the Figure S1. Short explanation for the Figure S1
LIMD1 overall molecular alterations including deletion, methylation, mutation and mRNA expression. The data provided represent the overall molecular alterations ie deletion, promoter methylation, mutation of LIMD1 along with the mRNA expression in each of individual sample studied.
Pattern of LIMD1 alterations in HNSCC during progression of the disease. The data provided represent the frequencies of deletion, methylation and mutation during the progression of HNSCC.
Correlation between LIMD1 methylation and mRNA expression. The data provided represent the correlation between LMD1 promoter methylation and its mRNA expression in each individual sample.