

Abstract
The zinc-ejecting aldehyde dehydrogenase (ALDH) inhibitory drug disulfiram (DSF) was found to be a breast cancer-associated protein 2 (BCA2) inhibitor with potent antitumor activity. We herein describe our work in the synthesis and evaluation of new series of zinc-affinic molecules to explore the structural requirements for selective BCA2-inhibitory antitumor activity. An N(C=S)S-S motif was found to be required, based on selective activity in BCA2-expressing breast cancer cell lines and against recombinant BCA2 protein. Notably, the DSF analogs (3a and 3c) and dithio(peroxo)thioate compounds (5d and 5f) were found to have potent activity (submicromolar IC50) in BCA2 positive MCF-7 and T47D cells but were inactive (IC50 >10 μM) in BCA2 negative MDA-MB-231 breast cancer cells and the normal breast epithelial cell line MCF10A. Testing in the isogenic BCA2 +ve MDA-MB-231/ER cell line restored antitumor activity for compounds that were inactive in the BCA2 negative MDA-MB-231 cell line. In contrast, structurally related dithiocarbamates and benzisothiazolones (lacking the disulfide bond) were all inactive. Compounds 5d and 5f were additionally found to lack ALDH-inhibitory activity, suggestive of selective E3 ligase-inhibitory activity and worthy of further development.
Introduction
The balance between the production of new cellular proteins and their targeted degradation is part of the normal choreographed life cycle of the cell, and numerous previous studies have elucidated the role of the ubiquitin-proteosome system in the highly regulated degradation of >80% of cellular proteins.1 Polyubiquitination (tagging by the small protein ubiquitin) of target protein substrates is carried out by three classes of enzymes, of which the diverse and abundant ubiquitin E3 ligase family catalyse the final mechanistic step of ubiquitin transfer to specific lysyl residues of target proteins prior to proteosomal degradation.2 Ubiquitin E3 ligase biology presents a number of therapeutic targets, since deregulated E3 ligase activity is known to be a feature of proliferative diseases such as cancer.3 An example of a well-studied E3 ligase whose deregulation has been exploited for potential therapeutic benefit is murine double minute 2 protein (Mdm2), which targets the tumor suppressor protein p53 for destruction.4 Inhibition of the protein-protein interaction between Mdm2 and p53 for therapeutic gain is illustrated by the development of the Nutlin class of antitumor agents currently being studied in clinical trials for cancer.5 Other components of the ubiquitin-proteosome system have provided additional targets for cancer therapy, leading to the development of the 26S proteosome inhibitor bortezomib as an approved agent for the treatment of multiple myeloma.6
BCA2 is an E3 ubiquitin ligase that was isolated from an invasive breast cancer cell line, and has been shown to be highly expressed in 56% (530/945) of invasive breast cancers, but not in most normal tissues examined.7 Down-regulation of BCA2 has been shown to inhibit breast cancer cell growth and invasiveness.7 BCA2 is expressed in the cytoplasm and nucleus, and nuclear BCA2 correlates with positive estrogen receptor status (p < 0.004). The classification of BCA2 as a Really Interesting New Gene (RING)-finger protein8 with ubiquitin E3 ligase activity implicates the presence of a double Zn2+-binding motif arranged in a cross-brace structure (Figure 1), termed RING-finger, as being essential for ubiquitin E3 ligase catalytic activity. The critical role of the Zn2+-containing RING domain of BCA2 has been demonstrated by point mutation of key zinc-binding cysteine residues leading to the complete loss of enzyme activity.7,9 BCA2 (also known as Rabring7) has been found to complex with a cytoplasmic binding partner Rab7, a small GTPase involved in cellular endocytosis and trafficking of oncogenic receptor tyrosine kinases (such as epidermal growth factor receptor (EGF-R)) for destruction in the lysosome.10,11 Hence targeting BCA2 within breast cancer cells may allow Rab7 to fulfill its function in tyrosine kinase receptor degradation, preventing the receptor recycling and sustained mitogenic signaling known to contribute to the development of resistance to tyrosine kinase-inhibitory therapeutics.10 Taken together, the data outlined above suggest that BCA2 could prove an important therapeutic target within the E3 ubiquitin ligase class for the future treatment of breast cancer.
Figure 1.
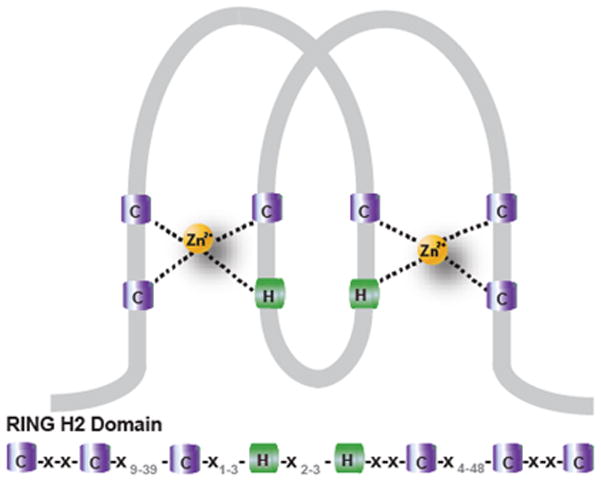
Cross brace structure of the RING-H2 domain as found in the ubiquitin E3 ligase BCA2. C, cysteine (purple); H, histidine (green); Zn2+, zinc ions (yellow); grey, number of other amino acids between the cysteine and histidine motifs.
Given the crucial role of Zn2+ ions in the catalytic RING domain of BCA2, a series of “zinc-ejecting” compounds from the National Cancer Institute database have been screened for their ability to inhibit BCA2 activity in BCA2-expressing breast cancer cell lines such as MCF-7. These studies have led to the identification of DSF (NSC25953),10,12,13 a registered drug for the treatment of alcoholism by virtue of additional ALDH1 activity, as a potent BCA2-inhibitory antitumor agent. In this paper we describe the synthesis and antitumor evaluation of four series of novel “zinc-affinic” agents, in order to optimize selective activity against recombinant BCA2 and BCA2-expressing breast cancer cell lines.14 From this antitumor data, we derived SAR insight into BCA2-inhibitory pharmacophore requirements, leading to the identification of potent and selective BCA2-inhibitory antitumor agents without accompanying ALDH-inhibitory activity.
Chemistry
Rationale for Choice of Candidate Compound Series
Four series of molecules having the potential ability to remove Zn2+ ions from the RING domain of BCA2 were synthesised, based on structures where the core group is known for zinc-binding activity (Figure 2). Initial studies focused on agents structurally related to the lead compound DSF (bis(diethylaminothiocarbonyl)-disulfide; tetraethylthiuram disulfide), and derived from different secondary amine starting materials, in order to explore the structure-activity relationships (SAR) around DSF with respect to BCA2-inhibitory antitumor activity.
Figure 2.
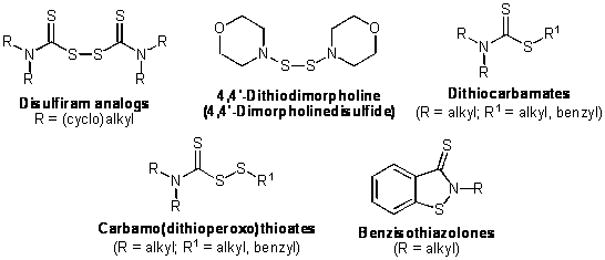
Potential zinc-binding BCA2 inhibitors.
The ability of compounds such as DSF (and its pyrrolidine and piperidine analogs) to form chelation complexes with Zn2+ has previously been reported.15,16 An alternative mechanism for removal of Zn2+ ions from the RING domain of BCA2 is the process of zinc-ejection, where modification of active-site cysteine residues causes ejection of Zn2+ from the active site. In the case of DSF and close analogs, the zinc-ejection process appears to be favoured for interactions with zinc-binding proteins. For example, the inhibition of the histone demethylase JMJD2A by zinc-ejection, following treatment with a range of potential Zn2+ ejectors including DSF, has recently been described.17
A further “symmetrical” disulfide structurally related to the DSF-like compounds – 4,4′-dithiodimorpholine (4,4′-dimorpholinedisulfide) – was synthesised in order to explore the structural requirement of a dithioester function for BCA2-inhibitory antitumor activity. 4,4′-Dithiodimorpholine has previously been studied as a potential lead compound for the treatment of neoplastic lesions of the cervix by targeting zinc-binding domains of the E6 oncoprotein of human papillomavirus.18
The carbamo (dithioperoxo) thioate series was designed as a series of “unsymmetrical” disulfides containing a cleavable S-S bond and the N(C=S)S-S group characteristic to DSF. This series presented the opportunity to vary the R and R1 groups (Fig. 2) to explore SAR requirements for BCA2-inhibitory antitumor activity. It is noteworthy that carbamo(dithioperoxo)thioates (mixed disulfides derived from DSF) have previously been reported as inhibitors of human mitochondrial19 and sheep liver20 ALDH, most likely via zinc-ejection. The registered drug status of DSF itself is based on its inhibitory activity against ALDH.
In order to further explore the structural requirements for BCA2-mediated antitumor activity, we synthesised a diverse series of dithiocarbamates. The dithiocarbamates bear close structural similarities with DSF analogs, but lack the S-S single bond, making this series an ideal candidate for exploring the importance of the disulfide bond in mediating antitumor activity.
Benzisothiazol-3-one derivatives have been reported to have a zinc-ejection effect, and have been tested for antiretroviral activity and their ability to eject Zn2+ from the HIV nucleocapsid zinc-finger protein NCP7.21 In addition we were interested in the benzisothiazolone structure as a known zinc-ejector lacking a labile S-S bond.
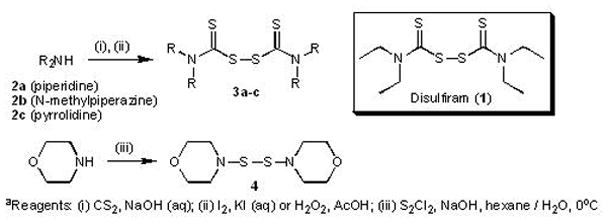
Synthesis of DSF Analogs (Bis(dialkylthiocarbamoyl)disulfides; 3a–c) and 4,4′-Dithiodimorpholine (4)
DSF (1) is commercially available (Sigma-Aldrich). Other DSF analog thiuram disulfides (3a–c) were synthesised in low to moderate yields via a one-pot reaction (General Method A, Experimental Section, Scheme 1), by mixing the appropriate secondary amine (2a–c) with carbon disulfide under basic conditions (aqueous sodium hydroxide). Without further purification, the (presumed) intermediate dithiocarbamic acid was oxidised to the required thiuram disulfide in low to moderate yields as previously described using either aqueous iodine/potassium iodide22 (compounds 3a and 3b) or hydrogen peroxide23 in acetic acid (compound 3c). More recently an efficient synthesis of thiuram disulfides has been reported using carbon tetrabromide to promote reaction of amines with carbon disulfide and subsequent disulfide bond formation.24 Alternative methods for the chemical oxidation of thiols to disulfides are also available, for example the use of bromine on hydrated silica.25 The structurally related dithiodimorpholine (4) was synthesised as previously described,18 via treatment of morpholine with sulfur monochloride under basic conditions (Scheme 1).
Scheme 1a.
Synthesis of Carbamo(dithioperoxo)thioates
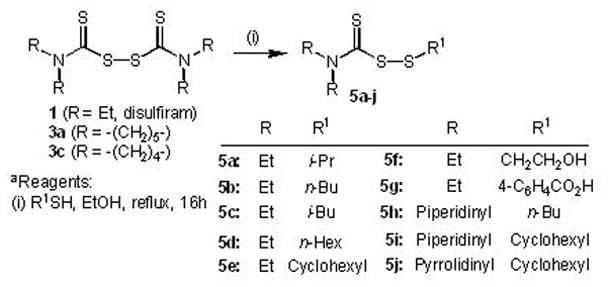
A series of carbamo(dithioperoxo)thioates (5a–j) that are structurally related to DSF analogs were synthesised in order to extend our understanding of SAR within the class of BCA2-inhibitory zinc-ejectors and to further explore whether an S-S single bond is necessary for BCA2-inhibitory antitumor activity. The one-pot synthesis of carbamo(dithioperoxo)thioates is essentially straightforward, and is based on previous described methods (General Method B, Experimental Section, Scheme 2).26,27 Briefly, stirring commercially available DSF (or synthetic analogs 3a or 3c) with the appropriate thiol in ethanol under refluxing conditions (16 h) yielded the required carbamo(dithioperoxo)thioate product (5a–j, Scheme 2) in low to moderate yield following purification by column chromatography using mixtures of ethyl acetate and hexane as eluent.
Scheme 2a.
Synthesis of Dithiocarbamates
Dithiocarbamates (9a-ac) were readily synthesised in a one-pot reaction according to the method of Azizi et al.28,29 (General Method C, Experimental Section; Scheme 3). The appropriate secondary amine (2a, 2c, 6a–c) was mixed with carbon disulfide and the required alkyl halide (7a–e) or Michael acceptor (8a–b) in equimolar ratios in water (5–18 h). In some instances, extraction of the aqueous phase using ethyl acetate gave rise to the pure product directly after solvent evaporation, whereas on other occasions it was necessary to purify products by column chromatography using mixtures of ethyl acetate and hexane as eluent. Spectroscopic and analytical data of dithiocarbamate products 9a–ac are recorded in Supporting Information.
Scheme 3a.
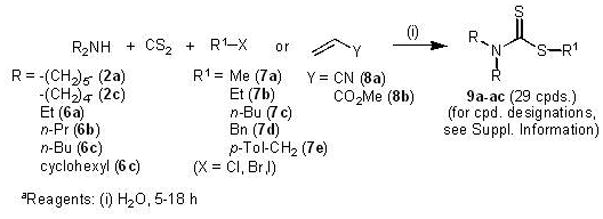
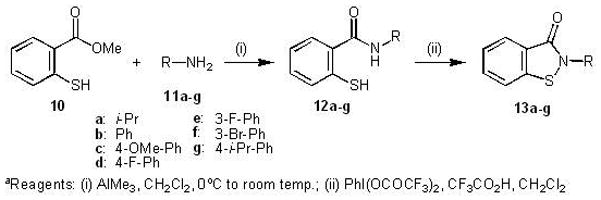
Synthesis of Benzisothiazolones
The general procedure for the synthesis of benzisothiazolones was adopted from the previously reported method of Correa et al. (General Method D, Experimental Section; Scheme 4).30 The first step of the synthesis involves formation of the amide (12a–g) by reacting methyl thiosalicylate (10) with different primary amines (11a–g), followed by oxidative ring closure using phenyliodonium di(trifluoroacetate) (PIFA) to give the required benzisothiazolones (13a–g) in low to moderate overall yield.
Scheme 4a.
Biological Results
Screening of Zinc Affinic Compounds in BCA2+ and BCA2low/− Cells
A total of 50 compounds representing the four different classes of sulfur-based structures described above were evaluated for antiproliferative activity by methyltetrazolium (MTT) assay in the BCA2-positive breast cancer cell lines MCF-7, T47D, MDA-MB-231/ER, the BCA2-low/negative (low/−) breast cancer cell line MDA-MB-231 and the normal breast epithelial cell line MCF10A (Fig. 3A–B). MDA-MB-231/ER is an isogenic subclone of MDA-MB-231 generated by transfection with ERα. ERα expression activates BCA2 expression via transcriptional regulatory mechanisms.31 MCF10A was used as a surrogate for assessing normal tissue toxicity of the zinc ejection agents. A compound was considered active if the IC50 was <10 μM. We selected 10 μM as a cut off point, because our lead compound DSF has an IC50 in BCA2 positive cells between 0.1–0.3 μM. Potential selectivity for BCA2 inhibition by a candidate compound was considered if growth inhibition was observed in BCA2 positive cells, but not MDA-MB-231 or MCF10A cells. None of the 29 tested dithiocarbamates (9a-9ac) or the seven benzisothiazolones (13a–g) showed activity in any of the breast cancer cell lines (data not shown). All of the ten dithioperoxothioates (5a–j) were active with IC50’s ranging from 0.15 to 2.75 μM in BCA2+ cells, and 0.9 to >10 μM in MDA-MB-231 or MCF10A cells (Table 1, Fig. 3B). However, only 5d and 5f were active in BCA2+, but inactive in BCA2low/− cells; 5d was more potent than 5f (Table 1). Similarly, DSF analogs (3a–c) were very active in BCA2+ cells and inactive in BCA2 low/− cells with the exception of 3b (Table 1). 4,4′-Dithiomorpholine (4) was inactive in both BCA2+ and BCA2low/− cells. The most potent DSF derivative was 3a with an IC50 in MCF-7 of 0.03 μM (Table 1, Figure 3A). Based on their activity profile compounds 3a, 3c, 5d and 5f were considered for further biological testing in comparison to DSF. Representative growth curves showing the antiproliferative activity of 3a and 5d in the BCA2-positive and BCA2-negative cell lines are depicted in Figures 3A and 3B.
Figure 3.
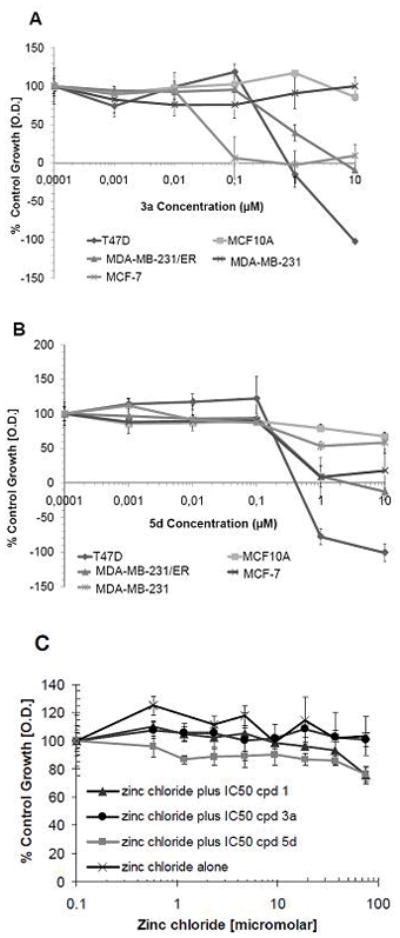
A. Representative growth curves for compound 3a in BCA2-positive and BCA2-negative cell lines from Table 1. B. Representative growth curves for compound 5d in BCA2-positive and BCA2-negative cell lines from Table 1 q. C. Effects of compounds 1, 3a and 5d in the presence of zinc chloride. To assess whether the addition of Zn2+ can rescue the growth inhibitory effects of zinc ejecting compounds in BCA2-positive MCF-7 cells, various concentrations of ZnCl2 were added together with IC50 concentrations of 1, 3a or 5d in MCF-7 cells. The value set as 100% is vehicle control treated MCF-7 cells without ZnCl2 and drug. ZnCl2 did prevent growth inhibition by compounds 1, 3a and 5d at all concentrations tested.
Table 1.
Activity of DSF analogs and carbamo(dithioperoxo)thioates against human breast cancer and normal cell linesa
| Mean IC50 ±SD (μM)b in cell linesc | |||||
|---|---|---|---|---|---|
| Compound | MCF-7 | MDA-MB-231 | MDA-MB-231/ER | T47D | MCF10A |
| 1 (DSF) | 0.1 ± 0.01 | >10 | 0.32 ± 0.14 | 0.17 ± 0.03 | 10 ± 0.2 |
| 3a | 0.03 ± 0.001 | >10 | 0.59 ± 0.12 | 0.27 ± 0.02 | >10 |
| 3b | 0.35 ± 0.05 | 3.0 ± 0.4 | 0.29 ± 0.1 | 0.33 ± 0.02 | >10 |
| 3c | 0.38 ± 0.03 | >10 | 0.25 ± 0.1 | 0.28 ± 0.02 | >10 |
| 4 | > 10 | >10 | >10 | >10 | >10 |
| 5a | 0.39 ± 0.28 | 0.96 ± 0.03 | 0.25± 0.1 | 0.23 ± 0.01 | >10 |
| 5b | 0.4 ± 0.3 | 7.4 ± 2.6 | 0.3 ±0.13 | 0.15 ± 0.02 | >10 |
| 5c | 0.45 ± 0.25 | 6.0 ± 2.8 | 2.1 ± 0 .43 | 0.20 ± 0.02 | >10 |
| 5d | 0.43 ± 0.1 | >10 | 0.35 ± 0.28 | 0.23 ± 0.02 | >10 |
| 5e | 0.3 ± 0.28 | 0.96 ± 0.01 | 0.28 ± 0.17 | 0.18 ± 0.02 | >10 |
| 5f | 0.5 ± 0.24 | >10 | 2.75± 0.17 | 0.30 ± 0.03 | >10 |
| 5g | 0.5 ± 0.2 | 6.3 ± 2.6 | 0.55± 0.12 | 0.15 ± 0.01 | >10 |
| 5h | 0.65 ± 1.2 | 8 ± 1.6 | 0.39 ± 0.17 | 0.35 ± 0.03 | >10 |
| 5i | 0.6 ± 0.1 | 6.7± 2.5 | 2.3 ± 0.22 | 0.25 ± 0.03 | >10 |
| 5j | 1.4 ± 1.8 | 4.7 ± 3.9 | 0.25±0.08 | 1.35 ±0.02 | >10 |
Determined by MTT assay (72 hour drug exposure), see ref 42 for details.
Compounds tested in triplicate, data expressed as mean values of three independent experiments.
Cancer cell line origin: MCF-7 (breast; BCA2 +ve; ER +ve), MDA-MB-231 (breast; BCA2 low/−ve; ER −ve), MDA-MB-231/ER (isogenic stable ER transfected clone of MDA-MB-231, BCA2+ve, ER +ve), T47D (breast, BCA2 +ve, ER +ve). MCF10A, normal breast epithelial cell line.
To test our hypothesis that the DSF analog sand carbamo (dithioperoxo) thioates act through a zinc ejection mechanism, MCF-7 cells were treated with compounds 1, 3a and 5d in the presence of zinc (II) chloride or with zinc chloride alone (Fig. 3C). When zinc chloride was added at a range of concentrations between 600 nM and 75 μM to MCF-7 cells exposed to the respective IC50s of 1, 3a and 5d (Table 1), their growth inhibitory activity was completely abolished (Fig. 3C). Zinc chloride alone had no effect on the growth of MCF-7 cells. These results are consistent with our working hypothesis that the active DSF and carbamo (dithioperoxo) thioate analogs are causing inhibition of BCA2-mediated cell growth via a reversible zinc ejection mechanism.
Evaluation of Lead Compounds for Inhibition of BCA2 E3 Ligase Activity/Autoubiquitination
Lead compounds from the cell-based screening procedure were tested for their potential to inhibit the E3 ligase activity of BCA2. A hallmark of RING-finger ubiquitin E3 ligases is the capability of autoubiquitination, hence the ubiquitination efficacy of such ligases can be studied in absence of a functional substrate.7,9 Recombinant wild type human BCA2 produced in E. coli was used together with ATP, a rabbit E1 and the recombinant human E2 UbcH5b. Compounds active towards breast cancer cell lines expressing BCA2 (MCF-7, T47D, MDA-MB-231/ER), but inactive in BCA2low/− MDA-MB-231 breast cancer and normal MCF10A cells (Fig. 4A), plus DSF were added to the ubiquitination reaction at concentrations of 5 and 50 μM (Fig. 4B). Because this assay employs highly purified, recombinant BCA2, E1 and E2 enzymes and drugs are only incubated with the enzyme mixture for 30 min., therefore drug concentrations higher than those inhibiting cell growth to 50% or 100% in a 5 day assay were used. Autoubiquitination of BCA2 was detected with anti-ubiquitin antibodies. In the event of the autoubiquitination, a high molecular weight polyubiquitin signal is seen (Fig. 4B, 5 μM reactions) and low molecular weight ubiquitin bands are weak or absent. In contrast, when autoubiquitination is inhibited, ubiquitin is not attached to the E3 ligase and shows strong bands, whereas polyubiquitin signals are absent (Fig. 4B, 50 μM reactions). All putative BCA2 inhibitory compounds were able to inhibit the autoubiquitination of recombinant BCA2 at a concentration of 50 μM, but not 5 μM (Fig. 4B). Identical experiments performed using the recombinant RING-E3 ligase Mdm2 did not show inhibition of Mdm2 autoubiquitination (data not shown). These data suggest that our cell-based screening system was predictive of direct BCA2 enzyme inhibition.
Figure 4.
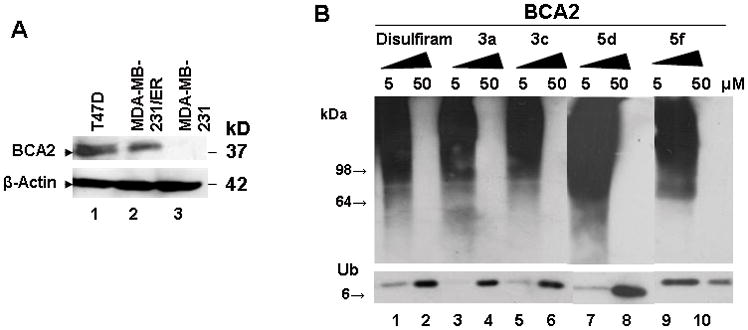
A. Western blot showing expression of endogenous BCA2 in T47D and the isogenic MDA-MB-231 parental and MDA-MB-231/ER breast cancer cell lines. Anti-BCA2 antibodies were developed by us and used as previously described.7 Beta-actin was used as equal loading control. B. Western blot analysis of BCA2 autoubiquitination in the presence of disulfide analogs at 5 and 50 μM of drug dissolved in dimethylsulfoxide. The membrane was probed with anti-ubiquitin antibodies. Shown are the ubiquitin signal (8kD) and high molecular weight polyubiquitinylated BCA2 (>98 kD). Polyubiquitinated proteins are typified by a high molecular weight smear.
Figure 5.
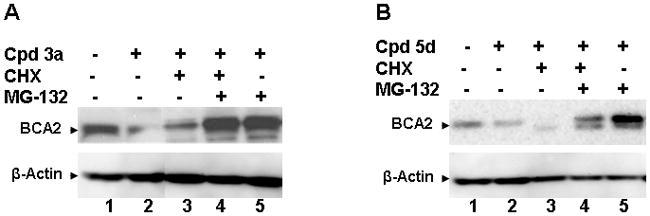
Western blots of endogenous BCA2 protein expression in MCF-7 breast cancer cells treated for 16 hrs with the BCA2 ligase inhibitors 3a (A) and 5d (B) in the presence and absence of the protein synthesis inhibitor CHX and the proteasome inhibitor 14. A. MCF-7 cells treated with vehicle only (DMSO, lane 1); compound (cpd) 3a at its 100% growth inhibitory concentration (0.25 μM, lane 2); 3a and CHX (lane 3); 3a, CHX and 14 (lane 4); and 3a plus 14 (lane 5). Beta-actin was used as equal loading control. B. MCF-7 cells treated with vehicle only (DMSO, lane 1); compound (cpd) 5d at its 100% growth inhibitory concentration (1 μM, lane 2); 5d and CHX (lane 3); 5d, CHX and 14 (lane 4); and 5d plus 14 (lane 5). Beta-actin was used as equal loading control.
Effects of Zinc Ejecting Compounds 3a and 5d on BCA2 Protein Stability in Cells
To confirm that inhibition of the BCA2 catalytic E3 ligase activity by zinc ejecting compounds seen in the autoubiquitination experiments is of relevance under physiological conditions, we performed cellular assays with the BCA2+ MCF-7 breast cancer cell line (Fig. 5). We selected the most potent compound from each of the DSF analog (3a) and dithioperoxothioate series (5d). MCF-7 cells, when exposed for 16 hrs (approximate half-life of BCA27) to both compounds at their IC100 concentrations, showed a marked reduction of BCA2 protein levels (Fig. 5A and 5B, lanes 2). The addition of the protein synthesis inhibitor cycloheximide (CHX) to 3a or 5d showed steady state levels (Fig. 5A) or further reduction of BCA2 (Fig. 5B). Importantly, when the proteasome inhibitor N-(benzyloxycarbonyl)-leucinylleucinylleucinal (MG-132)32 was added to the zinc ejector and CHX combination or the zinc ejector treatment alone (Fig. 5A–B, lanes 4–5), BCA2 was stabilized and accumulated even in absence of protein synthesis (Fig. 5A–B, lanes 4). These studies show that treatment with 3a or 5d leads to down-regulation of BCA2 protein, and that this process is dependent on the ubiquitin-proteasome pathway. If proteasomal degradation of ubiquitinated BCA2 protein is inhibited by 14 (MG-132), the E3 ligase accumulates (Fig. 5).
Testing for ALDH Inhibition
To study whether novel BCA2 inhibitory disulfide analogs 3a and 3c, or the dithioperoxothioates 5d and 5f, retained the ability to inhibit ALDH1 like DSF, we performed an ALDH1 inhibition test. To detect ALDH1 positive cancer cells, a flow cytometry based method was recently developed using the leukemia cell line K562, known to have high levels of ALDH1.33 Thus, we analyzed the ability of DSF, 3a, 3c, 5d and 5f to inhibit the ALDH1-positive cell fraction in K562 cells compared to a known inhibitor of ALDH activity, diethylaminobenzaldehyde (DEAB) (Table 2). While DSF and 3a inhibited ALDH1 to 57 and 79 % respectively, 3c, 5d and 5f showed little inhibition of this enzyme. DSF analog 3c inhibited only a negligible fraction of ALDH1 (3.4 %) and is therefore the most selective BCA2 E3 ligase inhibitor amongst the series of disulfides tested in this study. Dithio(peroxo)thioate compounds 5d and 5f also exhibited low levels of ALDH inhibition (9.8 % and 8.6 % relative to control, respectively).
Table 2.
ALDH inhibition in K562 cells for lead compounds.
| Compound (15 μM) | ALDH+ K562 cells (%) | ALDH Inhibition (%) |
|---|---|---|
| BAAA (Positive Control) | 40.39 ± 2.21 | 0 |
| DEAB (Negative Control) | 0.53 ± 0.11 | 98.68 |
| 1 (disulfiram) | 17.26 ± 2.21 | 57.26 |
| 3a | 8.3 ± 1.7 | 79.45 |
| 3c | 39.01 ± 0.8 | 3.4 |
| 5d | 36.47 ± 3.82 | 9.8 |
| 5f | 36.9 ± 3.15 | 8.6 |
Discussion
During this study we aimed to identify potent and selective BCA2-inhibitory antitumor agents devoid of the ALDH-inhibitory activity characteristic to DSF. Considerable progress has been made towards these goals. In terms of the identification of potent and selective activity in BCA2-expressing breast cancer cell lines, the thiuram disulfide compound 3a was found to be the most active (IC50 = 30 nM) in the MCF-7 cell line, with no activity (IC50 > 10 μM) in the BCA2 negative MDA-MB-231 breast cancer cell line and MCF10A normal breast epithelial cells. The activity of 3a in MCF-7 cells was found to exceed that of DSF itself (IC50 = 100 nM). The similar activity profile of the DSF analog 3a to DSF is perhaps not surprising given its structural similarity, however it is notable that the other closely related DSF analogs were substantially less potent and/or less selective (3b) compared to DSF and 3a. The lack of activity for dithiodimorpholine (4) is suggestive of the need for a dithioester function for BCA2 inhibitory activity.
Amongst the series of dithio(peroxo)thioates (5a–j) structurally related to DSF, two compounds (5d and 5f) stand out as having selective activity in the sub-micromolar IC50 range against the BCA2-expressing breast cancer cell lines compared to BCA2-negative/low MDA-MB-231 and normal MCF10A cells. Surprisingly, however, other dithioperoxothioate compounds more closely related in structure to DSF (such as 5a and 5e) did not display selective activity against BCA2-expressing cancer cell lines. The selectively active compounds 5d and 5f reinforce our hypothesis that an N(C=S)S-S group is required for selective activity in the BCA2-expressing breast cancer cell lines, and the cellular data was further validated by the ability of compounds 3a, 3c, 5d and 5f, plus DSF, to abolish the autoubiquitination activity of recombinant BCA2 at 50 μM (Figure 4). Moreover, for the most potent compounds of each class, 3a and 5d, we could show that inhibition of isolated enzyme activity translates into down-regulation of endogenous BCA2 at physiologically relevant drug concentrations (IC100) that is dependent on its degradation through the ubiquitin-proteasome system (Fig. 5). The importance of reversible zinc ejecting properties of 3a and 5d similar to that of DSF (compound 1) for their mode of action, was demonstrated by the zinc chloride rescue studies shown in Figure 3C. When Zn2+ ions were added to 1, 3a or 5d concentrations that inhibit the growth of the BCA2 positive cell line MCF-7 to 50% under physiological Zn2+ levels, their growth inhibitory activity was abolished even at the lowest concentration tested.
At the outset of our studies, we were interested in the discovery of selective BCA2-inhibitory antitumor agents that lacked the ALDH-inhibitory activity that is the basis for the registered drug status of DSF in the treatment of alcoholism. Although DSF is a safe drug, the ingestion of alcohol causes a “hangover” effect leading to symptoms such as flushing of the skin, accelerated heart rate, shortness of breath, nausea, vomiting, and circulatory collapse. Interestingly, studies on the role of ALDH1 as a marker of normal and malignant mammary stem cells associated with a poor clinical outcome34 suggest that retention of ALDH-inhibitory activity alongside BCA2 inhibition may not necessarily be an adverse property for the development of new anticancer agents.
By virtue of its zinc-12 and copper-35 binding properties, DSF has been shown to exhibit a number of interesting antitumor properties. The anti-proliferative and pro-apoptotic effects of DSF have been attributed (in various cancer model systems) to include inhibition of proteasome activity,35,36 nuclear factor kB,37 ATP Binding Cassette (ABC) drug transporter protein activity,38,39 and angiogenesis.40 Pharmacological profiling of DSF using human tumor cell lines and tumor cells from patients has provided further evidence of antitumor potential.41
The literature around the antitumor activity of DSF is strongly indicative of a diversity of molecular targets and drug properties, mediated largely through the binding of metal ions (zinc and copper) and the interaction of DSF with key cysteine residues in target proteins.42 Given that the literature on interactions of thiuram disulfides and dithioperoxothioates with zinc-dependant proteins is dominated by zinc-ejection effects,17,42 we hypothesize that inhibition of BCA2 is likely to be related to reversible zinc-ejection from the enzyme active site via modification of key cysteine residues. This hypothesis is supported by our zinc chloride rescue data shown in Figure 3C. Despite its rather indiscriminate biochemical and pharmacological profile, DSF is notable for being very well tolerated following long term treatment at relatively high doses (routinely 300–500 mg per day), and for its high oral bioavailability (>80% bioavailability, with approximately 20% of drug remaining in the body for 1–2 weeks post-ingestion).42 It is likely that the most active thiuram disulfides (3a and 3c) and dithio(peroxo)thioates (5d and 5f) synthesized in this study will also exhibit a diverse spectrum of antitumor activity, given their structural similarity to DSF. Further studies are ongoing to further define the antitumor potential of the lead compounds synthesized here, and their associated stability and toxicology.
Conclusions
The synthesis and evaluation of a variety of experimental agents based on the lead compound DSF has uncovered new antitumor agents with potent and selective antiproliferative activity against the E3 ubiquitin ligase BCA2. Potent and selective activity was observed in BCA2-expressing breast cancer cell lines (e.g. compound 3a, 5d), and biochemical and cellular studies with recombinant and endogenous BCA2 respectively confirmed the ability of active compounds to inhibit the RING E3 ligase. In two cases (5d and 5f) new BCA2-inhibitory molecules were found that lacked the ALDH-inhibitory activity characteristic to DSF and analogs.
Experimental
Chemistry
Melting points were measured on a Griffin apparatus and are uncorrected. Mass spectra were recorded on a Bruker MicroTOF LC instrument or at the EPSRC National Mass Spectrometry Centre (Swansea, U.K.). NMR spectra were recorded on a Bruker AVANCE 500 MHz instrument; coupling constants (J values) are in Hz. Merck silica gel 60 (40–60 μM) was used for column chromatography. All commercially available starting materials were used without further purification. Following purification, all new compounds were determined to possess ≥95% purity as determined by melting point/NMR/mass spectrometry and comparison with published data (for previously reported compounds), or a combination of spectroscopic analysis and combustion analysis (% CHN tested in duplicate) for new compounds. All compounds described were synthesised in the laboratory except the initial lead compound DSF, which is commercially available (Sigma-Aldrich, T1132, >97%).
General Method (A) for the Synthesis of Thiuram Disulfides (3a–b).22
A solution of sodium hydroxide (160 mg, 4.0 mmol) in water (4 mL) was added to a mixture of secondary amine (4.0 mmol) in water (5 mL). After stirring at room temperature for 20 min., carbon disulfide (0.24 mL, 4.0 mmol) was added, and the mixture was stirred for a further 90 min. A solution of iodine in potassium iodide was prepared by adding potassium iodide (3.19 g, 19.2 mmol) and water (8 mL) to iodine (1.02 g, 4.0 mmol). The I2/KI(aq) solution was then added dropwise to the reaction mixture, whereupon the solution initially turned yellow followed by formation of a precipitate. After stirring for a further 3 hr. the precipitate was collected by filtration in vacuo, and washed with excess water and 1M aqueous sodium thiosulfate. The crude product was purified by recrystallization from petroleum ether and toluene to give the required thiuram disulfide (3a–b) as a yellow solid in low to moderate yield.
Bis(piperidinylthiocarbonyl)disulfide (3a)
From piperidine. Yield = 28%. Mp 124 °C (lit.24 124–126 °C). 1H NMR (CDCl3) δ 4.25 (8H, m, NCH2), 1.78 (12H, m). 13C NMR (CDCl3) δ 192.88 (C=S), 55.92 (NCH2), 45.68, 24.41. m/z (EI+) 320 (M+), 147.
Synthesis of Bis(pyrrolidinylthiocarbonyl)disulfide (3c).23
A solution of pyrrolidine (0.49 mL, 6.0 mmol) in THF (1 mL) was added to a solution of sodium hydroxide (240 mg, 6.0 mmol) in water (2 mL) with stirring at 0 °C. Carbon disulfide (0.36 mL, 6.0 mmol) was then added dropwise and the mixture stirred at 0 °C for 30 min. Crushed ice (4.5 g) and acetic acid (0.9 mL) were then added, leading to the formation of a white precipitate. After a further 30 min., hydrogen peroxide (0.37 mL of 27.5% solution, 3.0 mmol) was added dropwise whilst maintaining the reaction temperature at 0 °C. n-Hexane (2.5 mL) was then added leading to the formation of precipitate after 30 min. that was collected by filtration and washed with hexane and 2% aqueous acetic acid. The crude product was recrystallized (toluene/hexane) to give the product as a white solid in 39% yield. Mp 135–137 °C (lit.43 137–139 °C). 1H NMR (CDCl3) δ 3.98 (8H, m, NCH2), 2.17 (4H, m), 2.01 (4H, m). 13C NMR (CDCl3) δ 189.31 (C=S), 57.12 (NCH2), 51.10 (NCH2), 26.75, 24.42. m/z (EI+) 292 (M+), 146.
Synthesis of Dithiodimorpholine (4).18
A solution of sulfur monochloride (3.20 mmol) in hexane (10 mL) was slowly added to a rapidly stirred two-phase mixture of morpholine (3.20 mmol), water (20 mL), hexane (10 mL) and sodium hydroxide (4.22 mmol) at 0 °C. The reaction mixture was then stirred at room temperature for a further 1 h, then the separated aqueous layer was washed with hexane (20 mL) and the combined organic layers washed with 1N HCl (aq) (10 mL) then water (10 mL). The organic layers were dried (Na2SO4), followed by concentration in vacuo (<40 °C) and recrystallization from n-hexane to give the product as a pale yellow solid in 65% yield. Mp 117–118 °C (lit.18 118–120 °C). 1H NMR (CDCl3) δ 3.74 (8H, m, OCH2), 2.81 (8H, m, NCH2). 13C NMR (CDCl3) δ 67.44 (OCH2), 55.92 (NCH2). m/z (CI+) 237 (M+ + 1).
General Method (B) for the Synthesis of Carbamo(dithioperoxo)thioates (5a–j)
An equimolar mixture of the appropriate thiol (9.0 mmol) and thiuram disulfide (9.0 mmol) in ethanol (20 mL) were heated under reflux for 16 h. Following concentration of the reaction mixture in vacuo, the pure product was obtained in low yield after purification by column chromatography using hexane/ethyl acetate as eluant.
Hexyl diethylcarbamo(dithioperoxo)thioate (5d).19
From DSF and n-hexanethiol. Yellow oil, 9% yield. 1H NMR (DMSO-d6) δ 4.02 (2H, q, J = 6.8 Hz, NCH2), 3.83 (2H, q, J = 6.8 Hz, NCH2), 2.78 (2H, t, J = 6.7 Hz, SCH2), 1.59 (2H, m), 1.40 (2H, m), 1.11–1.35 (10H, m), 0.92 (3H, t, J = 6.5 Hz, CH3CH2CH2). 13C NMR (DMSO-d6) δ 51.08 (NCH2), 46.78 (NCH2), 37.97 (SCH2), 30.82 (CH2), 27.83 (CH2), 27.47 (CH2), 21.97 (CH2), 13.80 (CH2), 12.93 (CH3), 11.15 (CH3). m/z (CI+) 266.1 (M+ + 1).
2-Hydroxyethyl diethylcarbamo(dithioperoxo)thioate (5f).19
From DSF and β-mercaptoethanol. Thick green oil, 12% yield. 1H NMR (DMSO-d6) δ 4.84 (1H, t, J = 6.0 Hz, OH), 3.99 (2H, q, J = 6.8 Hz, NCH2), 3.83 (2H, q, J = 6.8 Hz, NCH2), 3.60 (2H, q, J = 6.4 Hz, CH2OH), 2.89 (2H, t, J = 6.4 Hz, SCH2), 1.25 (6H, m, 2 × CH3). 13C NMR (DMSO-d6) δ 59.15 (CH2OH), 51.18 (NCH2), 46.82 (NCH2), 40.70 (SCH2), 12.95 (CH3), 11.17 (CH3). m/z (CI+) 226.0 (M+ + 1).
General Method (C) for the Synthesis of Dithiocarbamates (9a-ac).28,29
A mixture of secondary amine (10 mmol), carbon disulfide (10 mmol) and alkyl halide/Michael acceptor in water (50 mL) were stirred at room temperature for 16 h. The crude product was extracted using ethyl acetate (3 × 50 mL), dried (MgSO4) and concentrated in vacuo. Purification by column chromatography using ethyl acetate/hexane as eluant gave the crude product (9a-9ac) in moderate yield. See Supplementary Information for details of spectroscopic data.
General Method (D) for the Synthesis of Benzisothiazolones (13a–g).30
A solution of AlMe3 (11.52 mmol, 2.0 M in toluene) was added dropwise to a cooled (0 °C) suspension of the amine (11.52 mmol) in dichloromethane (30 mL). When the addition was complete, the reaction mixture was allowed to warm to room temperature and stirring was continued for 30 min. Methylthiosalicylate (0.7ml, 5.76 mmol) was added and the mixture was heated under reflux (16 h), then allowed to cool and slowly quenched with 5% aqueous HCl (20 mL). The resulting precipitate was removed by vacuum filtration, and the filtrate transferred to a separating funnel, where the organic phase was collected. The aqueous phase was extracted using dichloromethane (3 × 15 mL), and the organic extracts were combined and washed with a saturated solution of NaHCO3 (15 mL) and brine (15 mL), followed by drying (Na2SO4) and concentration in vacuo. Column chromatography (ethyl acetate/hexane) gave the intermediate amides (12a–g), which were used in the oxidative ring closure step without further purification. A solution of phenyliodonium di(trifluoroacetate) (PIFA) (249 mg, 0.58 mmol) in CH2Cl2 (9 mL) was added at 0 °C to a solution of the intermediate benzamide (0.39 mmol) and TFA (0.09 mL, 1.16 mmol) in CH2Cl2 (6 mL). The reaction mixture was stirred for 1 h, then the solvent was removed in vacuo to yield a residue that was purified using a column chromatography (ethyl acetate/hexane). See Supplementary Information for details of spectroscopic data.
Biology
Cell Culture
The human breast cancer cell lines T47D, MDA-MB-231, and the human chronic myelogenous leukemia cell line K562 were obtained from American Type Culture Collection (Manassas, VA). The breast cancer cell line MCF-7 and the normal mammary epithelial cell line MCF10A were obtained from the Karmanos Cancer Institute (formerly known as Michigan Cancer Foundation) Cell Repository. MDA-MB-231/ER was generated by transfecting an estrogen receptor alpha (ERα) pcDNA3 construct into parental MDA-MB-231 following the Lipofectamine™ method as described by the manufacturer (Invitrogen, Carlsbad). Stable clones were derived by G418 selection. ERα expression was confirmed by immunostaining.7 All cancer cell lines were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA) containing 10% heat-inactivated fetal bovine serum (FBS, Hyclone from Fisher Scientific, Pittsburgh, PA). MDA-MB-231/ER was cultured in the presence of 1mg/mL G418 (Invitrogen, Carlsbad, CA). MCF10A cells were grown in mammary epithelial growth medium (MEGM Bullet Kit, Cambrex Bio Science, East Rutherford, NJ). Cells were passaged routinely and kept at 37 °C and 5% CO2. Exponentially growing cells were used in all experiments.
MTT Assay
The MTT proliferation assay was performed to assess at which concentration these compounds would produce 50% growth inhibition (IC50) in tumor cells in order to identify leads that could be developed as anticancer agents. The doses employed ranged from 0.00001 to 10 μM. Compounds requiring IC50 concentrations > 10 μM were considered inactive. The MTT assay was essentially performed as previously described by us in detail.44 For compounds found to be active (IC50 < 10 μM), three independent experiments were performed and IC50 values determined as mean ± standard deviation (SD). Growth curves were generated and IC50 and IC100 concentrations (100% growth inhibition) were delineated from the growth curves as reported.44 The MTT assay was further used to assess whether the addition of ZnCl2 can rescue the growth inhibitory activity of compounds 1, 3a and 5d in BCA2-positive MCF-7 cells. In brief, BCA2-positive MCF-7 cells were treated with vehicle control (growth set as 100%) and the IC50 concentrations of 1, 3a and 5d in the presence of a range of zinc chloride concentrations between 600 nM and 75 μM for 5 days followed by MTT assay as described above.
Western blotting
Western blots were done for assaying the expression of endogenous BCA2 in the breast cancer cell lines MCF-7 and MDA-MB-231, MDA-MB-231/ER, and T47D. Exponentially growing cells were collected to generate cell lysates and Western blotting was performed as described by us previously.7
Effects of zinc ejecting compounds on recombinant BCA2 autoubiquitination
Drug stocks (10 mM) were prepared in DMSO and diluted to 100 μM in 1× ubiquitination assay reaction buffer. Ubiquitination assays were essentially performed as described previously by using recombinant wild-type His-BCA2 in the presence and absence of E2 conjugating enzyme UbcH5b extract, and with or without specific zinc ejecting compounds.7 In brief, 10 ng recombinant His-BCA2 was used with diluted drugs to obtain a final concentration of 5 or 50 μM zinc ejector in 30 μL reaction volume, and the mixtures were incubated at room temperature for 30 minutes before performing the ubiquitination assay. The protein-drug solutions were then mixed with 3 μL of 20mM ATP, 1 μL ubiquitin (1 μg, Sigma, St. Louis, MO), 1 μL E1 (20 ng, rabbit E1, Calbiochem, San Diego, CA), and 1 μL E2 (20 ng, recombinant human UbcH5b, Boston Biochem, Boston MA) in 50mM Tris HCl pH 8.0 plus 3 μL 10X reaction buffer (500 mM Tris HCl pH 8.0, 20mM DTT, 50mM MgCl2) and 21 μL H2O to obtain a final reaction volume of 30 μL. Each mixture was incubated at 30 °C for 1h, then 10 μL 4X SDS-gel loading buffer was added and the mixture was boiled for 3 min., followed by separation of reactions on a 4–20% gradient SDS-PAGE gel (Novex from Invitrogen) and immunoblotting.
Effects of zinc ejecting compounds on endogenous BCA2 protein stability
Experiments were essentially performed as in described by us previously.7 In brief, 1×106 MCF-7 cells were seeded into 6-well plates and grown overnight. Compounds 3a (IC100 = 0.25 μM) and 5d (IC100 = 1 μM) were then added at the 100% growth inhibitory concentrations for 16 h alone or in combination with the protein synthesis inhibitor CHX (Sigma, 100 μM) and/or the proteasome inhibitor 14 (Calbiochem, 10 μM). Control cells were treated with vehicle (DMSO) for 16 h. Whole cell lysates were analyzed for BCA2 protein expression by Western blotting.
Determination of ALDH1 Inhibition
K562 cells (human immortalized myelogenous leukaemia line) are known to express very high levels of ALDH1. Thus, these cells were used to analyze effects of our lead BCA2 inhibitory compounds and DSF for the inhibition of ALDH1. The ALDH assay kit ALDEFLUOR (StemCell Technologies, Vancouver, BC) was used according to the manufacturers instructions. Briefly, K562 cells were suspended in ALDEFLUOR assay buffer at concentration of 1×106 cells/mL. To prepare a substrate for ALDH1, bodipy-aminoacetaldehyde diethyl acetal (BAAA-DA) was activated to bodipy-aminoacetaldehyde (BAAA) in 1N HCl. Cells were incubated with BAAA in the presence or absence of DEAB, an inhibitor for ALDH1 for 45 min at 37 °C. DEAB was added at concentrations of 15 μM from a 1.5 mM stock prepared in ethanol. Thus, we compared the effects of DEAB on ALDH1 activity to those of our lead compounds at the same concentration and by preparing dilutions in ethanol. Cells were washed once with ALDEFLUOR assay buffer, and then cellular fluorescence based on bodipy-aminoacetate converted BAAA by ALDH1 was measured by BD LSR I through channel 1 (FL1, BD Biosciences, San Jose, CA). ALDH1+ cells were analyzed by a FL1 (horizontal) versus side scatter (SSC) dot plots by comparing fluorescence obtained from cells incubated with DEAB. The percentage of ALDH1+ cells in presence and absence of DEAB and compounds was determined and the percentage of ALDH1 inhibition calculated.
Supplementary Material
Acknowledgments
We acknowledge the support of the Algerian Consulate (PhD studentship to GB), and the EORTC Pharmacology and Molecular Mechanisms (PAMM) group for a mini-grant to initiate initial chemistry studies (to ADW and AMB). BCA2 biology and compound testing was supported by Award Number R01CA127258 from the National Cancer Institute, Developmental Therapeutics Program (AMB). We thank Mrs. Lin Xiong for excellent technical support. We also acknowledge the support of the EPSRC National Mass Spectrometry Centre, Swansea, U.K.
Footnotes
Abbreviations: ABC, ATP binding cassette; ALDH, aldehyde dehydrogenase; BAAA, bodipy-aminoacetaldehyde; BCA2, Breast Cancer Associated protein 2; CHX, cycloheximide; DEAB, diethylaminobenzaldehyde; DSF, disulfiram; EGF-R, epidermal growth factor receptor; Mdm2, murine double minute 2 protein; MTT, methyltetrazolium; PIFA, phenyliodonium di(trifluoroacetate); RING, Really Interesting New Gene.
Supporting Information Available: Spectroscopic data for thiuram disulfide (3b), carbamo(dithioperoxo)thioate (5a, 5b, 5c, 5e, 5g, 5h, 5i, 5j), dithiocarbamate (9a-9ac) and benzisothiazolone (13a–g) compounds.
References
- 1.Burger AM, Seth A. The ubiquitin-mediated protein degradation pathway in cancer: therapeutic implications. Eur J Cancer. 2005;40:2217–2229. doi: 10.1016/j.ejca.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 2.Hershko A, Ciechanover A. The ubiquitin system. Ann Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 3.Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov. 2006;5:596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- 4.Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao XL, Vu BT, Qing WG, Packman K, Myklebost O, Heimbrook DC, Vassilev LT. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci USA. 2006;103:1888–1893. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Secchiero P, di Lasio MG, Gonelli A, Zauli G. The MDM2 inhibitor Nutlins as an innovative therapeutic tool for the treatment of haematological malignancies. Curr Pharm Des. 2008;14:2100–2110. doi: 10.2174/138161208785294663. [DOI] [PubMed] [Google Scholar]
- 6.Adams J, Kauffman K. Development of the proteasome inhibitor Velcade™ (Bortezomib) Cancer Invest. 2004;22:304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- 7.Burger AM, Gao YG, Amemiya Y, Kahn HJ, Kitching R, Yang YL, Sun P, Narod SA, Hanna WM, Seth AK. A novel RING-type ubiquitin ligase breast cancer-associated gene 2 correlates with outcome in invasive breast cancer. Cancer Res. 2005;65:10401–10412. doi: 10.1158/0008-5472.CAN-05-2103. [DOI] [PubMed] [Google Scholar]
- 8.Joazeiro CA, Weissman AM. RING finger proteins: mediators of ubiquitin ligase activity. Cell. 2000;102:549–552. doi: 10.1016/s0092-8674(00)00077-5. [DOI] [PubMed] [Google Scholar]
- 9.Amemiya Y, Azmi P, Seth A. Autoubiquitination of BCA2 RING E3 ligase regulates its own stability and affects cell migration. Mol Cancer Res. 2008;6:1385–1396. doi: 10.1158/1541-7786.MCR-08-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burger AM, Amemiya Y, Kitching R, Seth AK. Novel RING E3 ubiquitin ligases in breast cancer. Neoplasia. 2006;8:689–695. doi: 10.1593/neo.06469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakane A, Hatakeyama S, Sasaki T. Involvement of Rabring7 in EGF receptor degradation as an E3 ligase. Biochem Biophys Res Comm. 2007;357:1058–1064. doi: 10.1016/j.bbrc.2007.04.052. [DOI] [PubMed] [Google Scholar]
- 12.Burger AM, Phatak P, Wilson M, Seth AK. Disulfiram inhibits the E3 ligase activity of breast cancer associated gene 2 (BCA2) and the growth of BCA2-expressing breast cancers in vitro and in vivo. EJC Suppl. 2006;4:118. [Google Scholar]
- 13.Burger AM, Amemiya Y, Seth AK. Disulfiram inhibits the ubiquitin E3 ligase activity of the novel breast cancer associated gene 2 BCA2 and the growth of BCA2 expressing breast cancer cell lines. Proc Amer Assoc Cancer Res. 2006;47:5513. [Google Scholar]
- 14.Brahemi G, Fiasella A, Brancale A, Westwell A, Burger A. Design and synthesis of BCA2 inhibitors. EJC Suppl. 2008;6:44. [Google Scholar]
- 15.Bond AM, Hollenkamp AF. Exchange and other reactions associated with zinc(II) dithiocarbamate oxidation and reduction processes observed at mercury and platinum electrodes in dichloromethane. Inorg Chem. 1990;29:284–289. [Google Scholar]
- 16.Saravanan M, Prakasam BA, Ramalingam K, Bocelli G, Cantoni A. M(S)2(I)2 (M=Zn, Cd) and Hg(S)3I coordination environment of transition metal complexes – synthesis, spectral, and single crystal X-ray structural investigations. Z Anorg Allg Chem. 2005;631:1688–1692. [Google Scholar]
- 17.Sekirnik R, Rose NR, Thalhammer A, Seden PT, Mecinovic J, Schofield CJ. Inhibition of the histone demethylase JMJD2A by ejection of structural Zn(II) Chem Comm. 2009:6376–6378. doi: 10.1039/b916357c. [DOI] [PubMed] [Google Scholar]
- 18.Beerheide W, Sim MM, Tan YJ, Bernard HU, Ting AE. Inactivation of the human papillomavirus-16 E6 oncoprotein by organic disulfides. Bioorg Med Chem. 2000;8:2549–2560. doi: 10.1016/s0968-0896(00)00193-0. [DOI] [PubMed] [Google Scholar]
- 19.Mackerell AD, Jr, Vallari RC, Pietruszko R. Human mitochondrial aldehyde dehydrogenase inhibition by diethyldithiocarbamic acid and methanthiol mixed disulfide: a derivative of disulfiram. FEBS Lett. 1985;179:77–81. doi: 10.1016/0014-5793(85)80195-2. [DOI] [PubMed] [Google Scholar]
- 20.Kitson TM. Effect of disulfiram on aldehyde dehydrogenases of sheep liver. Biochem J. 1975;151:407–412. doi: 10.1042/bj1510407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loo JA, Holler TP, Sanchez J, Gogliotti R, Maloney L, Reilly MD. Biophysical characterization of zinc ejection from HIV nucleocapsid protein by anti-HIV 2,2-dithiobis[benzamides] and benzisothiazolones. J Med Chem. 1996;39:4313–4320. doi: 10.1021/jm960253w. [DOI] [PubMed] [Google Scholar]
- 22.Kitson TM. Effect of some analogs of disulfiram on aldehyde dehydrogenases of sheep liver. Biochem J. 1976;155:445–448. doi: 10.1042/bj1550445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song QL, Wang ZW, Sanghvi YS. A short, novel, and cheaper procedure for oligonucleotide synthesis using automated solid phase synthesizer. Nucleosides, Nucleotides & Nucleic Acids. 2003;22:629–633. doi: 10.1081/NCN-120021968. [DOI] [PubMed] [Google Scholar]
- 24.Liang F, Tan J, Piao C, Liu Q. Carbon tetrabromide promoted reaction of amines with carbon disulfide: facile and efficient synthesis of thioureas and thiuram disulfides. Synthesis. 2008;(22):3579–3584. [Google Scholar]
- 25.Ali MH, McDermott M. Oxidation of thiols to disulfides with molecular bromine on hydrated silica gel support. Tetrahedron Lett. 2002;43:6271–6273. [Google Scholar]
- 26.Gilmore WF, Clark RN. New alkylsulfenyl N, N-dialkyldithiocarbamates. J Chem Eng Data. 1969;14:119–120. [Google Scholar]
- 27.Field L, Buckman JD. Organic disulfides and related substances. 25. Thiocarbamoyl and imidocarbamoyl disulfides. J Org Chem. 1968;33:3865–3871. [Google Scholar]
- 28.Azizi N, Aryanasab F, Torkiyan L, Ziyaei A, Saidi MR. One-pot synthesis of dithiocarbamates accelerated in water. J Org Chem. 2006;71:3634–3635. doi: 10.1021/jo060048g. [DOI] [PubMed] [Google Scholar]
- 29.Azizi N, Aryanasab F, Saidi MR. A straightforward and highly efficient catalyst-free one-pot synthesis of dithiocarbamates under solvent-free conditions. Org Lett. 2006;8:5275–5277. doi: 10.1021/ol0620141. [DOI] [PubMed] [Google Scholar]
- 30.Correa A, Tellitu I, Dominguez E, San Martin R. Novel alternative for the N-S bond formation and its application to the synthesis of benzisothiazol-3-ones. Org Lett. 2006;8:4811–4813. doi: 10.1021/ol061867q. [DOI] [PubMed] [Google Scholar]
- 31.Burger AM, Kona FR, Amemiya Y, Gao Y, Bacopulos S, Seth AK. Role of the BCA2 ubiquitin E3 ligase in hormone responsive breast cancer. The Open Cancer Journal. (Special Issue on “Transcriptional and posttranscriptional regulation in hormonedependent cancer”) doi: 10.2174/1874079001003010116. [in press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Domingo-Domenech J, Pippa R, Tapia M, Gascon P, Bachs O, Bosch M. Inactivation of NF-kappaB by proteasome inhibition contributes to increased apoptosis induced by histone deacetylase inhibitors in human breast cancer cells. Breast Cancer Res Treat. 2008;112:53–62. doi: 10.1007/s10549-007-9837-8. [DOI] [PubMed] [Google Scholar]
- 33.Burger AM. Targeting Leukemic Stem Cells. In: Bagley RG, Teicher BA, editors. Stem Cells and Cancer. Humana Press; New York, NY: 2009. pp. 263–273. [DOI] [Google Scholar]
- 34.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006;66:10425–10433. doi: 10.1158/0008-5472.CAN-06-2126. [DOI] [PubMed] [Google Scholar]
- 36.Lövborg H, Oberg F, Rickardson L, Gullbo J, Nygren P, Larsson R. Inhibition of proteasome activity, nuclear factor-KappaB translocation and cell survival by the antialcoholism drug disulfiram. Int, J Cancer. 2006;118:1577–1580. doi: 10.1002/ijc.21534. [DOI] [PubMed] [Google Scholar]
- 37.Liu GY, Frank N, Bartsch H, Lin JK. Induction of apoptosis by thiuramdisulfides, the reactive metabolites of dithiocarbamates, through coordinative modulation of NFkappaB, c-Fos/c-jun, and p53 proteins. Mol Carcinogenesis. 1998;22:235–246. doi: 10.1002/(sici)1098-2744(199808)22:4<235::aid-mc5>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 38.Loo TW, Clarke DM. Blockage of drug resistance in vitro by disulfiram, a drug used to treat alcoholism. J Natl Cancer Inst. 2000;92:898–902. doi: 10.1093/jnci/92.11.898. [DOI] [PubMed] [Google Scholar]
- 39.Sauna ZE, Peng XH, Nandigama K, Tekle S, Ambudkar SV. The molecular basis of the action of disulfiram as a modulator of the multidrug resistance-linked ATP binding cassette transporters MDR1 (ABCB1) and MRP (ABCC1) Mol Pharmaol. 2004;65:675–684. doi: 10.1124/mol.65.3.675. [DOI] [PubMed] [Google Scholar]
- 40.Shian SG, Kao YR, Wu FY, Wu CW. Inhibition of invasion and angiogenesis by zinc-chelating agent disulfiram. Mol Pharmacol. 2003;64:1076–1084. doi: 10.1124/mol.64.5.1076. [DOI] [PubMed] [Google Scholar]
- 41.Wickström M, Danielsson K, Rickardson L, Gullbo J, Nygren P, Isaksson A, Larsson R, Lövborg H. Pharmacological profiling of disulfiram using human tumor cell lines and human tumor cells from patients. Biochem Pharmacol. 2007;73:25–33. doi: 10.1016/j.bcp.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 42.Sauna ZE, Shukla S, Ambudkar SV. Disulfiram, an old drug with new potential therapeutic uses for human cancers and fungal infections. Mol Biosyst. 2005;1:127–134. doi: 10.1039/b504392a. [DOI] [PubMed] [Google Scholar]
- 43.Kotali E, Varvoglis A. (Dialkyldithiocarbamoyl)diaryliodanes. J Chem Soc, Perkin Trans. 1987;1(12):2759–2763. [Google Scholar]
- 44.Phatak P, Cookson JC, Dai F, Smith V, Gartenhaus RB, Stevens MFG, Burger AM. Telomere uncapping by the G-quadruplex ligand RHPS4 inhibits clonogenic tumour cell growth in vitro and in vivo consistent with a cancer stem cell targeting mechanism. Brit J Cancer. 2007;96:1223–1233. doi: 10.1038/sj.bjc.6603691. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.