

Abstract
A subset of the mucosotropic human papillomaviruses (HPVs) including HPV16 are etiological agents for the vast majority of cervical cancer, other anogenital cancers, and a subset of head and neck squamous cell carcinomas. HPV16 encodes three oncogenes; E5, E6, and E7. While E6 and E7 have been well studied and clearly shown to be important contributors to these cancers, less is known about E5. In this study, we utilized E5 transgenic mice to investigate the role of E5 in cervical cancer. When treated for six months with estrogen, a cofactor for cervical carcinogenesis, E5 transgenic mice developed more severe neoplastic cervical disease than like-treated nontransgenic mice, though no frank cancers were detected. In addition, E5 when combined with either E6 or E7 induced more severe neoplastic disease than seen in mice expressing only one viral oncogene. Prolonged treatment of E5 transgenic mice with exogenous estrogen uncovered an ability of E5 to cause frank cancer. These data indicate that E5 acts as an oncogene in the reproductive tracts of female mice.
Keywords: E5, HPV, Cervical Cancer, E6, E7
INTRODUCTION
HPV16 E5 is an 83 amino acid hydrophobic membrane associated protein that localizes to the endoplasmic reticulum (1, 2). Studies with bovine papillomavirus initially identified E5 as a potent oncogene (3). HPV16 E5 was subsequently shown to transform murine fibroblasts and keratinocytes in tissue culture (4-6), enhance the immortalization potential of HPV16 E6 and E7 (7), and cooperate with HPV16 E7 to stimulate proliferation of human and mouse primary cells (8, 9). HPV16 E5 can activate the EGFR in a ligand-dependant manner (6, 10, 11) and associates with the 16 KDa subunit of the vacuolar ATPase. This latter interaction can block the acidification of endosomes that is necessary for the degradation of cell surface receptors including EGFR leading to increased recycling of these receptors back to the cell surface. This in turn could explain E5’s ability to increase EGFR signaling (6, 12). Other activities that could contribute to E5’s oncogenic abilities include the inhibition of apoptosis (13-15), inhibition of gap junction-mediated cell-cell communication (16), down-regulation of surface expression of MHC class I and II molecules (17, 18), interaction with EVER1/2 (19), and reduced transcription of p21 (20).
To examine the oncogenic properties of E5 in vivo, K14E5 transgenic mice were created in which expression of E5 was directed to the basal layer of the stratified squamous epithelia. These mice display epidermal hyperplasia, aberrant differentiation of the epithelium, and are susceptible to spontaneous skin tumors (21). E5 was shown to contribute to the promotion and progression stages in skin carcinogenesis similar to what was seen previously in E6 transgenic mice (22, 23). In contrast, E7 was found to only contribute to the promotion stage of skin carcinogenesis (23). Thus, in skin carcinogenesis, E5 and E6 are the more potent oncogenes. In contrast to the skin, we found that E7 is the more potent oncogene in cervical cancer compared to E6, using transgenic mice (24). Specifically, after 6 months treatment with exogenous estrogen, a cofactor in cervical carcinogenesis, E7 transgenic mice developed cancers throughout the reproductive tract while E6 transgenic mice did not. E6/E7 double transgenic mice developed larger cancers demonstrating a subtle role for E6 in cervical carcinogenesis (24). E6 mice do develop cancers when treated with 9 months of exogenous estrogen (25).
In this current study, we investigated whether E5 alone or in combination with the other HPV16 oncogenes, E6 and E7, contributes to the development of cervical cancer. Groups of HPV16 transgenic mice expressing one or more of the three viral oncoproteins in stratified epithelia, or control, nontransgenic mice, were treated for 6 or 9 months with exogenous estrogen and the reproductive tracts were analyzed for the presence of disease and cancer formation. Expression of E5 led to more severe neoplastic disease of the cervix compared to that observed in nontransgenic mice, though frank cancers only arose after the longer treatment with estrogen. This finding is similar to what was observed with mice expressing E6. E5 when expressed together with E6 or E7 led to greater tumor burden than seen with any oncogene alone; however E5’s contribution was lost when all three oncogenes were expressed. Together these findings indicate that E5 can contribute to cervical carcinogenesis alone or cooperatively with one other viral oncogene, and that its potency is similar to that of E6. However it remains unclear whether its oncogenic potency is manifest in the presence of all three oncogenes.
MATERIALS AND METHODS
Mouse lines and estrogen treatment
The K14E5 (line 32,(21)), K14E6 (line 5737,(26)), and K14E7 (line 2304,(27)) transgenic mouse strains carrying the HPV16 E5, E6 or E7 oncogenes, respectively, under the control of the human keratin 14 (K14) promoter were maintained on the inbred FVB/N genetic background. These lines were crossed to each other or to nontransgenic FVB/N mice to generate mice hemizygous for none, one, two or all three HPV16 transgenes. Mice were genotyped by PCR as previously described (21, 26, 27). To monitor for cervical carcinogenesis, five-week old virgin female transgenic, or nontransgenic FVB/N mice were either treated or not treated with 17-β estradiol (0.05mg, 60-day release pellets) for a period of six or nine months. All mice were bred and maintained in the American Association for Accreditation of Laboratory Animal Care-approved McArdle Laboratory Animal Care Facility in accordance with an institutionally approved animal protocol.
Analysis of reproductive tracts
Reproductive tracts of estrogen treated or nontreated female mice were harvested after 6 or 9 months of estrogen treatment and analyzed as described previously (24). Briefly, tissues were fixed in 4% paraformaldehyde, paraffin embedded, sectioned, and every tenth 5-μm section stained with hematoxylin and eosin (H&E) and histologically examined for tumors and/or dysplastic disease with the worst grade of lesion scored as the final diagnosis. Frank cancers were measured at the largest cross-sectional area using the Zeiss Axiovision (version 3.1) program (Zeiss, Thorwood, NY). Any cancer with a cross sectional area >0.5 mm2 was classified as a large invasive cancer (LIC). All other cancers were classified as micro-invasive cancers (MIC).
Statistical analysis
Fisher’s exact test was used to determine the significance in tumor incidence. Two-sided Wilcoxon rank-sum test was used to determine the significance of all other data. Statistical analysis was carried out using the Mstat program (www.mcardle.wisc.edu/mstat/).
Immunohistochemistry
Histological sections were deparaffinized in Xylenes, rehydrated in a series of alcohols, boiled in 10mM citrate buffer for 17 mins to unmask antigens, blocked in 10% horse serum in PBS for 1 hour, then incubated overnight at 4°C with primary antibody specific for either BrdU (Ab-2; Calbiochem, San Diego, CA) or p-ERK1/2 (Cell Signaling, Danvers, MA), each diluted1:100 in block. A universally biotinylated secondary antibody was applied for 30 minutes (Vectastain universal secondary), washed in PBS, and incubated in ABC (Vectastain, Vector labs, Burlingame, CA) reagent for 30 minutes. Sections were developed with DAB reagent for appropriate time, counterstained with hemotoxylin, dehydrated in a series of alcohols and cover slipped.
RESULTS
HPV16 E5 acts as an oncogene in the cervix
HPV16 E7 alone has been shown to induce cervical cancer in mice treated for 6 months with physiological levels of exogenous estrogen (17-β estradiol) that is sufficient to induce continuous estrus (24). In contrast, in mice treated for 6 months, HPV16 E6 was unable to induce frank cancer, but was able to contribute to the severity of disease by increasing the incidence of large invasive cancers (LIC) in E6E7 double transgenic mice compared to E7 mice alone (24). To determine what role HPV16 E5 may have in cervical carcinogenesis, female K14E5 transgenic mice were treated for 6 months with the same dose of 17-β estradiol used in prior studies of K14E6 and K14E7 mice, and reproductive tracts were harvested, embedded and sectioned throughout. Every tenth section was stained with Hemotoxylin and Eosin (H&E) and scored for the worst stage of neoplastic disease, ranging from hyperplasia, to dysplasia, to frank cancer (either microinvasive [MIC], or large invasive [LIC] cancers, the latter defined as having a cross sectional area greater than >0.5 mm2) present within the lower reproductive tract (Table 1, Table S1). As was the case for nontransgenic as well as E6 transgenic mice, none of the E5 transgenic mice (n=15) developed frank cancer of the cervix, cervico-vaginal junction or the vagina (Table 1). Interestingly, 3 of 19 E5E6 double transgenic mice did develop cancers (Table 1) and those arose within the cervix or cervico-vaginal junction, but this low incidence was not statistically significant when compared to nontransgenic mice (p=0.24).
Table 1.
Cervical histopathology in mice treated with exogenous estrogen for 6 months
| Genotype (total # mice) | Grade of Disease* |
% LRT** Cancer | Average #LRT Cancers/Mouse | Average LRT Cancer Size (mm2) | |||||
|---|---|---|---|---|---|---|---|---|---|
| H | CIN1 | CIN2 | CIN3 | MIC | LIC | ||||
| NTG (n=14)b | 11 | 2 | 1 | 0a | 0 | 0 | |||
| E5 (n=15)b,c,d | 3 | 6 | 6 | 0 | 0 | 0 | |||
| E6 (n=22)c | 10 | 9 | 3 | 0 | 0 | 0 | |||
| E5E6 (n=19)d | 2 | 5 | 8 | 1 | 3 | 16a | 0.26 | 0.12 | |
| E7 (n=16) | 1 | 1 | 1 | 4 | 7 | 2 | 56e | 2.0f | 0.12g |
| E5E7 (n=13) | 2 | 9 | 2 | 85e | 4.4f | 0.17g | |||
| E6E7 (n=9) | 2 | 4 | 3 | 78 | 4.2 | 0.41 | |||
| E5E6E7 (n=10) | 1 | 7 | 2 | 90 | 3.2 | 0.12 | |||
Footnotes:
indicated in each column labeled ‘H’ to ‘LIC’ is the number of mice for which the indicated stage of disease was the worst disease state found throughout the cervix.
LRT (lower reproductive tract: includes cancers found within the cervix, cervico-vaginal junction and vagina
% mice with cancers: E5E6 vs. NTG p=0.24, two-sided Fisher’s Exact test
severity of disease: E5 vs NTG p=0.002, two-sided Wilcoxon rank sum test; also p=0.002 by two-sided Fisher’s Exact test comparing incidence of hyperplasia to all CIN
severity of disease: E5 vs E6 p=0.05, two-sided Wilcoxon rank sum test
severity of disease: E5 vs E5E6 p=0.0006, two-sided Wilcoxon rank sum test
% mice with cancers: E5E7 vs E7 p=0.13, two-sided Fisher’s Exact test
average # cancers: E5E7 vs E7 p=0.04, two-sided Wilcoxon rank sum test
average cancer size: E5E7 vs E7 p=0.01, two-sided Wilcoxon rank sum test
As seen in women, cervical cancers arise in mice as a result of a progressive neoplastic disease characterized by the onset of benign lesions (CIN1-3) within the cervical epithelia that become progressively less differentiated and more dysplastic, leading ultimately to the development of microinvasive, then large invasive carcinomas. To assess more thoroughly the influence of viral oncogenes on the complete range of the progressive disease that arises within the HPV16 transgenic mice, we performed a two-sided Wilcoxon rank sum test in which each mouse was ranked by the worst stage of disease present in their cervix (i.e. hyperplasia was given a score of 1, CIN1 = 2, CIN2 = 3, CIN3 = 4, MIC = 5, and LIC = 6) as reported in Table 1. Using this test, we found that E5 significantly increased the severity of cervical disease over that observed in the nontransgenic mice (p = 0.002). The progressive disease was more severe in E5 mice than E6 mice (p = 0.05), and E5 contributed strongly to the increased severity of disease in E5E6 double transgenic mice (E6 vs. E5E6 p = 0.0006). A similar pattern of increased dysplastic disease in E5 transgenic mice was also observed in the vaginal epithelium (Table S1). Based upon these observations we conclude that HPV16 E5 contributes to the development of neoplasia in the lower reproductive tract to a degree that is similar if not greater than that of HPV16 E6.
E5 cooperates with E7 to cause cervical cancer
E7 has been previously shown to be a very potent oncogene in the reproductive tracts of female mice (24). Consistent with prior observations, cervical cancer incidence in 6-month estrogen-treated E7 transgenic mice in the present study was 56% (Table 1). E5E7 double transgenic mice were generated to determine if E5 could cooperate with E7 in inducing cervical cancers. We found a marginal (p=0.13) increase in the incidence of cervical cancer in E5E7 mice (85%) compared to E7 mice (56%). Tumor multiplicities and tumor sizes were also compared between groups of transgenic mice. The mean number of tumors in E5E7 (4.4) double transgenic mice was significantly higher that E7 (2.0) mice alone (p=0.04) (Figure 1, Table 1) and similar to that of E6E7 double transgenic mice (4.2). There also was a significant increase in the size of tumors between E5E7 (0.17 mm2) double transgenic mice and E7 (0.12 mm2) transgenic mice (p=0.01) (Figure 1, Table 1). To analyze whether E5 is somehow altering the levels of E7 protein thereby indirectly contributing to increased oncogenesis we monitored levels of expression of MCM7, a gene induced in its expression via E7’s inactivation of pRB and consequent activation of E2F transcription factors. E7 alone led to the potent induction of MCM7 throughout the cervical epithelium (supplemental Fig. 1), consistent with our prior studies (28). The levels of induction of MCM7 in the cervical epithelium of E5/E7 and E6/E7 double transgenic mice and E5/E6/E7 triple transgenic mice was indistinguishable from that observed in the E7 singly transgenic mice (supplemental Fig. 1). Thus E5 is not altering the activity levels of E7 protein in the cervix. In sum, these data show that E5 and E7, when expressed together, lead to increased tumor multiplicity and tumor size. This is similar to what is seen between E6 and E7 ((25); Table 1).
Figure 1.

Comparison of the tumor multiplicity (top) and tumor size (bottom) of reproductive tumors in mice treated with 6 months of exogenous estrogen.
We were also interested in learning if there was a further augmentation of carcinogenesis in E5/E6/E7 triply transgenic mice compared to double transgenic mice. We saw no difference in the cancer incidence between E6E7 mice and E5E6E7 mice. Interestingly, in the context of analyses of the E5E6E7 triple transgenic mice, it appeared that E5 was inhibitory to cancer growth because E5E6E7 tumors were on average smaller than E6E7 tumors (Table 1). However this difference in average tumor size reflected the fact that there were several very large outlier tumors amongst those arising in the E6/E7 mice (Fig. 1). Indeed the tumor size between the two groups (E6/E7 versus E5/E6/E7) of mice was not statistically significant (p=0.64). The fact that there was little effect of E5 on the frequency or growth rate of cancers in the presence of both E6 and E7 might explain why, in spite of its capacity in mice to cause cervical cancer when expressed alone or in combination with E6 or E7 (above data), it appears to be dispensable in a significant fraction human cervical cancers wherein both E6 and E7 are always found to be co-expressed.
E5 alone is sufficient to induce cervical cancer with longer estrogen treatment
As indicated in Table 1, neither E5 nor E6 alone was sufficient to induce frank cancer when these mice were treated for six months with exogenous estrogen, although both contributed to more severe overall disease, and both synergized with E7 to cause cancers in this time period. With longer estrogen treatment (9 months), E6 alone has been shown to be sufficient to induce frank cancer (25). We therefore investigated whether E5 transgenic mice when treated for 9 months with exogenous estrogen developed frank cancer. There were statistically significant increases in cervical cancer incidence (p=0.04) and tumor multiplicities (p=0.03) in E5 compared to nontransgenic mice treated nine months with 17β-estradiol (Table 2). Indeed, the incidence of cancer and cancer multiplicities were similar for the E5 and E6 mice (Table 2, Figure 2). There was no difference in tumor size between E5 and nontransgenic mice whereas E6 transgenic mice developed larger cancers than either E5 or nontransgenic mice (Table 2, Figure 2). Prior analyses indicated that the ability for E6 alone to induce large cancers largely correlates with its capacity to bind to subset of cellular targets with leucine rich motifs that includes the ubiquitin ligase E6AP, required for E6’s destabilization of p53 (25). Not surprisingly both the E5 and E6 mice treated for nine months showed significant increases (p <10-6 and p<10-6, respectively) in the overall severity of cervical disease compared to like-treated nontransgenic mice. These results in the nine month treated mice again demonstrate a similarity between E5 and E6 in terms of their oncogenic potency in the cervix.
Table 2.
Cervical histopathology of mice treated with exogenous estrogen for 9 months
| Genotype (total # mice) | Grade of Disease* |
% LRT Cancer | Average # LRT Cancers/Mouse | Average LRT Cancer Size (mm2) | |||||
|---|---|---|---|---|---|---|---|---|---|
| H | CIN1 | CIN2 | CIN3 | MIC | LIC | ||||
| NTG (n=23)c,d | 17 | 3 | 1 | 2 | 8.7a | 0.09b | 0.038 | ||
| E6 (n=39)d | 3 | 9 | 7 | 8 | 5 | 7 | 31 | 0.50 | 0.76 |
| E5 (n=20)c | 1 | 1 | 6 | 5 | 7 | 35a | 0.51b | 0.041 | |
Footnotes:
indicated in each column labeled ‘H’ to ‘LIC’ is the number of mice for which the indicated stage of disease was the worst disease state found throughout the cervix.
LRT (lower reproductive tract: includes cancers found within the cervix, cervico-vaginal junction and vagina
% cancers: E5 vs. NTG p=0.04, one-sided Fisher’s Exact Test
average # cancers: E5 vs NTG p=0.03, two-sided Wilcoxon rank sum test
severity of disease: E5 vs NTG p<10-6, two-sided Wilcoxon rank sum test
severity of disease: E6 vs NTG p<10-6, two-sided Wilcoxon rank sum test
Figure 2.

Comparison of the tumor multiplicity (top) and tumor size (bottom) of reproductive tumors in mice treated with 9 months of exogenous estrogen.
Effects of E5 on cell cycle progression in the cervical epithelium
The papillomavirus life cycle is intricately tied to the differentiation program of the host’s stratified epithelium. Progeny virus is produced in the suprabasal compartment wherein the virus reprograms terminally differentiated cells to support DNA synthesis (29). E5 and E7 have been shown previously to synergize in driving unscheduled DNA synthesis in the suprabasal compartment of human keratinocytes in organotypic tissue cultures (30, 31). To determine whether E5 and E7 act together to induce suprabasal DNA synthesis in vivo, we monitored the presence of cells supporting DNA synthesis within the cervical epithelium of the different groups of mice used in this study. Mice were injected with BrdU 1 hour prior to sacrifice and histological sections subjected to BrdU-specific immunohistochemistry. In the basal compartment E7 suppresses DNA synthesis in the cervical epithelium (32). This suppression was observed again in our hands (Fig 3). E5 also had a similar effect (Fig. 3). In the suprabasal compartment, however, E5 in combination with E7 increased the percentage of suprabasal cells supporting DNA synthesis over that seen in E7 alone mice and this increase was statistically significant (p=0.02, E7 versus E5E7) (Figure 3). The rather subtle, yet statistically significant effect of E5 in augmenting the level of suprabasal DNA synthesis in the mouse cervical epithelium is similar to what we observed in our prior studies on the role of E5 in the viral life cycle (30). These data corroborate the prior studies in tissue culture demonstrating a synergy between E5 and E7 in driving unscheduled DNA synthesis.
Figure 3.
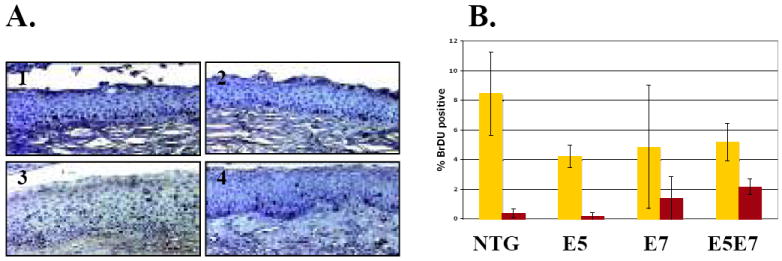
Characterization of the proliferative index in cervical epithelia. A. Representative pictures of BrdU-specific immunohistochemical staining of cervical epithelium from nontransgenic (1), E5 (2), E7 (3), and E5E7 (4) mice. B. Quantification of BrdU-labeling index of cells within distinct layers of cervical epithelia. The average percentage of basal (yellow) and suprabasal (red) BrdU-positive cells was obtained from ten (× 40) microscope fields per mouse. An average of at least three mice per genotype were used to calculate the percentage. The difference in % suprabasal DNA synthesis between E5E7 and E7 cervical epithelium was statistically significant (p=0.02, two-sided Wilcoxon rank sum test).
E5 activates the MAPK pathway in mice treated for 9 months with estrogen
HPV16 E5 has been shown to enhance ligand-dependant phosphorylation and activation of the EGFR suggesting that E5 stimulates mitogenic stimulation through the EGFR. To determine if mitogenic signaling is activated in the cervical epithelium of mice, reproductive tracts of transgenic mice treated with estrogen for 9 months were stained for the presence of phosphorylated ERK1/2. Phosphorylated ERK1/2 was not detected in cervical epithelium from nontransgenic and E6 transgenic mice (Figure 4). In contrast, nuclear phospho-ERK1/2 staining was easily detected in cervical epithelium of the E5 transgenic mice treated for 9 months (Figure 4). This same pattern of positive staining was observed in tumors from nine-month E5 transgenic mice but not in tumors from the nontransgenic or E6 mice (Figure 4). These data indicate that E5 causes an increased steady state level of activated ERK1/2 and is consistent with E5 being able to activate EGFR-signaling. However, this observation was limited to the nine-month treated mice; E5 did not cause a detectable increase in phospho-ERK1/2 in mice treated only for 6 months (data not shown). This finding is consistent with our prior studies in the skin of young E5 transgenic mice wherein we could not detect increased EGFR activity (21). The later age at which phosph-ERK1/2 could be selectively detected in the E5 transgenic tissue could point either to increased levels/activity of E5 in these older mice, or the loss of some negative regulator of MAPK activity in the cervix of older female mice.
Figure 4.
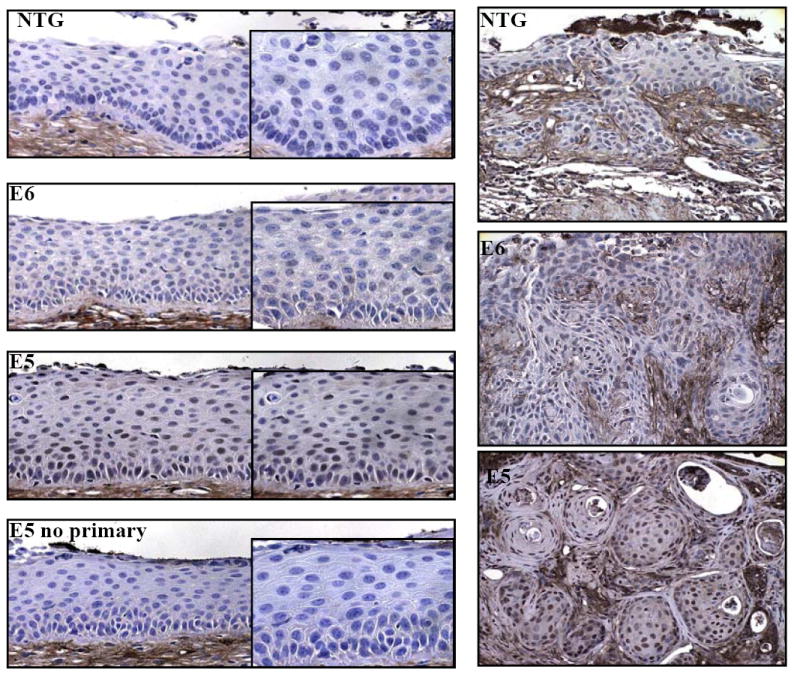
ERK1/2 activation in transgenic mice treated 9 months with exogenous estrogen. Sections from nontransgenic, E6, and E5 transgenic mice were immunohistochemically stained for the presence of phosphorylated ERK1/2. Shown are representative pictures of cervical epithelium (left panels) and tumors (right panels) from nontransgenic, E6, and E5 transgenic mice treated with exogenous estrogen for 9 months. Right panels insets: higher magnifications of epithelium. Bottom right panel: no primary antibody control.
DISCUSSION
In this study we provide evidence that HPV16 E5 can contribute to cervical carcinogenesis. In estrogen treated mice, E5 was able to induce cervical cancers on its own, and synergize with E6 or E7 to induce more severe cervical disease. The capacity to cooperate with E6 or E7 in cervical carcinogenesis correlates with its previously demonstrated cooperation with E6 or E7 in the immortalization of human keratinocytes (7), and is consistent with each of these HPV oncogenes contributing to cervical carcinogenesis.
Like E6, E5 was able to induce cancer in mouse reproductive tracts but only after nine months of treatment with estrogen; whereas, E7 could induce tumor formation at a high frequency with just six months of treatment. E5 transgenic mice did develop more severe disease compared to E6 transgenic mice. Nevertheless, E7 remains the most potent HPV oncogene in the context of cervical carcinogenesis. This potency contrasts to what was previously observed in the context of skin carcinogenesis, in which E5 and E6 are the more potent oncogenes (22, 23). In the skin, E5 and E6 both contribute to the promotion and malignant progression stages of carcinogenesis, whereas E7 only contributes to promotion. The underlying reason for the differences in potency of these oncogenes in these two stratified epithelial tissues remains elusive.
HPV16 E5 has previously been implicated in the productive stage of the viral life cycle, wherein it contributes to inducing unscheduled DNA synthesis in the normally quiescent suprabasal compartment of stratified epithelia (30). This induction of unscheduled DNA synthesis is thought to allow the vegetative amplification of the viral genome, synthesis of which relies upon the cellular DNA replication machinery. In organotypic cultures of human keratinocytes harboring the HPV16 genome, E5 and E7, when expressed together act synergistically to induce unscheduled DNA synthesis in the suprabasal compartment. It was therefore not surprising to find the same to be true in the context of the cervical epithelia of E5E7 double transgenic mice. It remains unclear, however, whether this capacity of E5 to alter suprabasal cells in combination with E7 relates to its carcinogenic properties.
Our studies also provide new insights into the possible mechanism(s) of action by which E5 contributes to cervical carcinogenesis. That we could see increased levels of phopho-ERK in the epithelia and cancers arising in the 9-month estrogen treated E5 transgenic mice is consistent with E5 being able to increase EGFR’s activity. However, the absence of such an effect at earlier time points raises the question whether EGFR activation is sufficient or necessary for E5-mediated oncogenesis at least at earlier time points. One study suggests that E5 is able to activate the ERK1/2 in an EGFR-independent manner leaving open the possibility that E5 may be able to activate ERK1/2 independent of EGFR activation (33). The ability of E5 to activate ERK1/2 at 9 months but not at 6 months may also be due to physiological differences reflective of the age of the mice. We currently are pursuing efforts to investigate whether EGFR is required for E5 induced phenotypes in our mice and if it is required for E5 induced carcinogenesis.
Mice transgenic for the HPV16 oncogene E5 have increased dysplastic disease in the cervical epithelium and when treated with estrogen for 9 months develop frank cancer. In addition E5 cooperated with E7 to increase tumor multiplicity and size. This data shows that E5 may be important in human cancers and should be looked at more closely. Previous work showing E5 is involved in the productive stage of the viral life cycle (30), increased suprabasal DNA synthesis in E5 transgenic mice (21), and ability to stimulate EGF dependant proliferation in human keratinocytes (6, 8, 9) suggests that E5 may play a role in expanding infected keratinocytes. Although we are not able to show whether the EGFR is required for these activities we did show that E5 activates the MAPK pathway consistent with E5 enhancing ligand dependant EGFR activation. Forty percent of human cancers do not express E5 protein, which correlates with the integration of the HPV genome into the host genome (34). In our studies, there was no appreciable effect of E5 on overall disease severity in mice that express both E6 and E7. This raises the possibility that E5 plays a minimal role in cervical carcinogenesis in the context of a natural infection, wherein both E6 and E7 are expressed. Alternatively, it could indicate that E5’s role is limited to a subset of human cervical cancers, perhaps a reflection of genetic differences amongst the human population. Regardless, the data presented in this paper demonstrates that E5 can increase the dysplastic environment and aid in inducing DNA synthesis, both of which could expand the population of infected cells within a patient.
Supplementary Material
Acknowledgments
We thank Bill Sugden and Norman Drinkwater for providing comments on this manuscript. This study was supported by grants from the NIH (CA022443 and T32 CA009135).
References
- 1.Conrad M, Bubb VJ, Schlegel R. The human papillomavirus type 6 and 16 E5 proteins are membrane-associated proteins which associate with the 16-kilodalton pore-forming protein. J Virol. 1993;67:6170–8. doi: 10.1128/jvi.67.10.6170-6178.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Disbrow GL, Sunitha I, Baker CC, Hanover J, Schlegel R. Codon optimization of the HPV-16 E5 gene enhances protein expression. Virology. 2003;311:105–14. doi: 10.1016/s0042-6822(03)00129-6. [DOI] [PubMed] [Google Scholar]
- 3.Talbert-Slagle K, DiMaio D. The bovine papillomavirus E5 protein and the PDGF beta receptor: it takes two to tango. Virology. 2009;384:345–51. doi: 10.1016/j.virol.2008.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leechanachai P, Banks L, Moreau F, Matlashewski G. The E5 gene from human papillomavirus type 16 is an oncogene which enhances growth factor-mediated signal transduction to the nucleus. Oncogene. 1992;7:19–25. [PubMed] [Google Scholar]
- 5.Pim D, Collins M, Banks L. Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene. 1992;7:27–32. [PubMed] [Google Scholar]
- 6.Straight SW, Hinkle PM, Jewers RJ, McCance DJ. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J Virol. 1993;67:4521–32. doi: 10.1128/jvi.67.8.4521-4532.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stoppler MC, Straight SW, Tsao G, Schlegel R, McCance DJ. The E5 gene of HPV-16 enhances keratinocyte immortalization by full-length DNA. Virology. 1996;223:251–4. doi: 10.1006/viro.1996.0475. [DOI] [PubMed] [Google Scholar]
- 8.Valle GF, Banks L. The human papillomavirus (HPV)-6 and HPV-16 E5 proteins co-operate with HPV-16 E7 in the transformation of primary rodent cells. J Gen Virol. 1995;76:1239–45. doi: 10.1099/0022-1317-76-5-1239. [DOI] [PubMed] [Google Scholar]
- 9.Bouvard V, Matlashewski G, Gu ZM, Storey A, Banks L. The human papillomavirus type 16 E5 gene cooperates with the E7 gene to stimulate proliferation of primary cells and increases viral gene expression. Virology. 1994;203:73–80. doi: 10.1006/viro.1994.1456. [DOI] [PubMed] [Google Scholar]
- 10.Crusius K, Auvinen E, Steuer B, Gaissert H, Alonso A. The human papillomavirus type 16 E5-protein modulates ligand-dependent activation of the EGF receptor family in the human epithelial cell line HaCaT. Exp Cell Res. 1998;241:76–83. doi: 10.1006/excr.1998.4024. [DOI] [PubMed] [Google Scholar]
- 11.Tomakidi P, Cheng H, Kohl A, Komposch G, Alonso A. Modulation of the epidermal growth factor receptor by the human papillomavirus type 16 E5 protein in raft cultures of human keratinocytes. Eur J Cell Biol. 2000;79:407–12. doi: 10.1078/0171-9335-00060. [DOI] [PubMed] [Google Scholar]
- 12.Straight SW, Herman B, McCance DJ. The E5 oncoprotein of human papillomavirus type 16 inhibits the acidification of endosomes in human keratinocytes. J Virol. 1995;69:3185–92. doi: 10.1128/jvi.69.5.3185-3192.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kabsch K, Mossadegh N, Kohl A, et al. The HPV-16 E5 protein inhibits TRAIL-and FasL-mediated apoptosis in human keratinocyte raft cultures. Intervirology. 2004;47:48–56. doi: 10.1159/000076642. [DOI] [PubMed] [Google Scholar]
- 14.Zhang B, Spandau DF, Roman A. E5 protein of human papillomavirus type 16 protects human foreskin keratinocytes from UV B-irradiation-induced apoptosis. J Virol. 2002;76:220–31. doi: 10.1128/JVI.76.1.220-231.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kabsch K, Alonso A. The human papillomavirus type 16 E5 protein impairs TRAIL-and FasL-mediated apoptosis in HaCaT cells by different mechanisms. J Virol. 2002;76:12162–72. doi: 10.1128/JVI.76.23.12162-12172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oelze I, Kartenbeck J, Crusius K, Alonso A. Human papillomavirus type 16 E5 protein affects cell-cell communication in an epithelial cell line. J Virol. 1995;69:4489–94. doi: 10.1128/jvi.69.7.4489-4494.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang B, Li P, Wang E, et al. The E5 protein of human papillomavirus type 16 perturbs MHC class II antigen maturation in human foreskin keratinocytes treated with interferon-gamma. Virology. 2003;310:100–8. doi: 10.1016/s0042-6822(03)00103-x. [DOI] [PubMed] [Google Scholar]
- 18.Ashrafi GH, Tsirimonaki E, Marchetti B, et al. Down-regulation of MHC class I by bovine papillomavirus E5 oncoproteins. Oncogene. 2002;21:248–59. doi: 10.1038/sj.onc.1205008. [DOI] [PubMed] [Google Scholar]
- 19.Lazarczyk M, Pons C, Mendoza JA, Cassonnet P, Jacob Y, Favre M. Regulation of cellular zinc balance as a potential mechanism of EVER-mediated protection against pathogenesis by cutaneous oncogenic human papillomaviruses. J Exp Med. 2008;205:35–42. doi: 10.1084/jem.20071311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsao YP, Li LY, Tsai TC, Chen SL. Human papillomavirus type 11 and 16 E5 represses p21(WafI/SdiI/CipI) gene expression in fibroblasts and keratinocytes. J Virol. 1996;70:7535–9. doi: 10.1128/jvi.70.11.7535-7539.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Genther Williams SM, Disbrow GL, Schlegel R, Lee D, Threadgill DW, Lambert PF. Requirement of epidermal growth factor receptor for hyperplasia induced by E5, a high-risk human papillomavirus oncogene. Cancer Res. 2005;65:6534–42. doi: 10.1158/0008-5472.CAN-05-0083. [DOI] [PubMed] [Google Scholar]
- 22.Maufort JP, Williams SM, Pitot HC, Lambert PF. Human papillomavirus 16 E5 oncogene contributes to two stages of skin carcinogenesis. Cancer Res. 2007;67:6106–12. doi: 10.1158/0008-5472.CAN-07-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song S, Liem A, Miller JA, Lambert PF. Human papillomavirus types 16 E6 and E7 contribute differently to carcinogenesis. Virology. 2000;267:141–50. doi: 10.1006/viro.1999.0106. [DOI] [PubMed] [Google Scholar]
- 24.Riley RR, Duensing S, Brake T, Munger K, Lambert PF, Arbeit JM. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63:4862–71. [PubMed] [Google Scholar]
- 25.Shai A, Brake T, Somoza C, Lambert PF. The human papillomavirus E6 oncogene dysregulates the cell cycle and contributes to cervical carcinogenesis through two independent activities. Cancer Res. 2007;67:1626–35. doi: 10.1158/0008-5472.CAN-06-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song S, Pitot HC, Lambert PF. The human papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals. J Virol. 1999;73:5887–93. doi: 10.1128/jvi.73.7.5887-5893.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herber R, Liem A, Pitot H, Lambert PF. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. J Virol. 1996;70:1873–81. doi: 10.1128/jvi.70.3.1873-1881.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brake T, Connor JP, Petereit DG, Lambert PF. Comparative analysis of cervical cancer in women and in a human papillomavirus-transgenic mouse model: identification of minichromosome maintenance protein 7 as an informative biomarker for human cervical cancer. Cancer Res. 2003;63:8173–80. [PubMed] [Google Scholar]
- 29.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nature Rev Cancer. 2002;2:342–50. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 30.Genther SM, Sterling S, Duensing S, Munger K, Sattler C, Lambert PF. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J Virol. 2003;77:2832–42. doi: 10.1128/JVI.77.5.2832-2842.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flores ER, Allen-Hoffmann BL, Lee D, Lambert PF. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J Virol. 2000;74:6622–31. doi: 10.1128/jvi.74.14.6622-6631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balsitis S, Dick F, Dyson N, Lambert PF. Critical roles for non-pRb targets of human papillomavirus type 16 E7 in cervical carcinogenesis. Cancer Res. 2006;66:9393–400. doi: 10.1158/0008-5472.CAN-06-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crusius K, Rodriguez I, Alonso A. The human papillomavirus type 16 E5 protein modulates ERK1/2 and p38 MAP kinase activation by an EGFR-independent process in stressed human keratinocytes. Virus Genes. 2000;20:65–9. doi: 10.1023/a:1008112207824. [DOI] [PubMed] [Google Scholar]
- 34.Chang JL, Tsao YP, Liu DW, Huang SJ, Lee WH, Chen SL. The expression of HPV-16 E5 protein in squamous neoplastic changes in the uterine cervix. J Biomed Sci. 2001;8:206–13. doi: 10.1007/BF02256414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.