

Abstract
Our recent studies have shown that the FoxM1B transcription factor is overexpressed in human glioma tissues and that the level of its expression correlates directly with glioma grade. However, whether FoxM1B plays a role in the early development of glioma, i.e., in transformation, is unknown. In this study, we found that the FoxM1B molecule causes cellular transformation and tumor formation in normal human astrocytes (NHAs) immortalized by p53 and pRB inhibition. Moreover, brain tumors that arose from intracranial injection of FoxM1B-expressing immortalized NHAs displayed glioblastoma multiforme (GBM) phenotypes, suggesting that FoxM1B overexpression in immortalized NHAs not only transforms the cells but also leads to GBM formation. Mechanistically, our results showed that overexpression of FoxM1B up-regulated NEDD4-1, an E3 ligase that mediated the degradation and down-regulation of PTEN in multiple cell lines. Decreased PTEN in turn resulted in the hyperactivation of Akt, which led to phosphorylation and cytoplasmic retention of FoxO3a. Blocking Akt activation with PI3K/Akt inhibitors inhibited FoxM1B-induced transformation of immortalized NHAs. Furthermore, overexpression of FoxM1B in immortalized NHAs increased the expression of survivin, cyclin D1, and cyclin E, which are important molecules for tumor growth. Collectively, these results indicated that overexpression of FoxM1B, in cooperation with p53 and pRB inhibition in NHA cells, promoted astrocyte transformation and GBM formation through multiple mechanisms.
Keywords: FoxM1B, NEDD4-1, PTEN, Akt, transformation, tumorigenicity, glioblastoma multiforme
Introduction
Malignant glioma, including the most common subtype, astrocytoma, is aggressive, highly invasive, and neurologically destructive. The prognosis for patients with malignant glioma is poor (1, 2). A number of pathways have been correlated with human gliomas, such as pRb, p53, EGFR, PDGF/PDGFR, p16INK4a, p19ARF, telomerase, and Akt (3–6). Activation of the Akt pathway is strongly implicated in glioma development and progression. In human studies, Akt is activated in approximately 80% of GBMs (3). Activation of the Akt pathway in glioma is often achieved by underexpression of its negative regulator phosphatase and tensin homolog (PTEN). Studies have shown that PTEN is underexpressed in almost all GBM tumors, compared with levels in normal brain tissue (7). Several mechanisms may be associated with the underexpression of PTEN in gliomas, such as genetic alterations or promoter methylation (8–11). However, gene deletion, mutation, LOH, or promoter methylation in the PTEN gene is present in only about 50% of GBMs (7–11). Also, although PTEN genetic alterations are rare in low-grade gliomas, these tumors exhibit lower PTEN protein levels than does normal brain (7). Forthermore, underexpression of PTEN in the absence of genetic alterations or promoter methylation is seen frequently in gliomas as well (8). Thus, mechanisms for PTEN control at the transcriptional and posttranslational levels may be altered in glioma.
Ubiquitin-mediated degradation of PTEN has recently been reported, and NEDD4-1 (neural precursor cell–expressed, developmentally down-regulated 4-1) has been identified as the E3 ligase for PTEN (12). NEDD4-1 negatively regulates PTEN stability by catalyzing PTEN polyubiquitination. In fact, overexpression of NEDD4-1 has been shown to increase PTEN polyubiquitination in a dose-dependent manner (12). Consistent with the tumor-suppressive role of PTEN, NEDD4-1 was reported to be an oncogenic protein that regulates tumorigenesis (12). Overexpression of NEDD4-1 increased cellular transformation by K-Ras. Elimination of NEDD4-1 expression inhibited xenotransplanted tumor growth. Furthermore, NEDD4-1 regulates tumorigenesis by regulating Akt signaling via PTEN, and the oncogenic activity of NEDD4-1 is PTEN-dependent (12). However, little is known about the regulation of NEDD4-1.
FoxM1 is a member of the Forkhead box (Fox) transcription factor family known for its function as a regulator of the cell cycle (13–17). Results from recent studies have revealed that FoxM1 is substantially elevated in several aggressive human carcinomas and can contribute to oncogenesis in many tissue types (18–25). For example, using transgenic and knockout mouse models, studies have shown that FoxM1B plays essential roles in the developments of hepatocellular carcinoma (20), prostate carcinomas (21), lung cancer (22, 23) and colorectal cancer in mice (24). Moreover, FoxM1 is an essential molecule in the regulation of oxidative stress, which contributes to malignant transformation and tumor cell survival (16).
Increasing evidence indicates that FoxM1 may also contribute to oncogenesis in glioma. For example, several studies found that aberrant expression of FoxM1 was a common molecular alteration in malignant glioma (25–27). Another study that analyzed expression data from 201 GBM clinical specimens derived from TCGA showed the overexpression of FoxM1 in GBM specimens compared with nontumor controls (28). In our own studies of the functional importance of this factor, we found that FoxM1B was the predominant FoxM1 isoform in human gliomas (25). FoxM1 levels in human glioma tissue directly correlated with glioma grade and inversely correlate with the survival of GBM patients. In animal models, increased levels of FoxM1B expression in glioma cells enhanced the tumorigenicity, invasiveness, and angiogenesis of these cells (25, 29, 30).
In this study, we sought to determine whether overexpression of FoxM1B induces the transformation of human astrocytes into glioma cells, since FoxM1 expression is detected in low-grade glioma. We used immortalized normal human astrocyte (NHA)-E6/E7/hTERT cells because human astrocytes appear to be more genetically stable than do rodent cells (31), and individual events believed to be important in gliomagenesis might be more readily studied in the human cells (31–33). Moreover, the fact that NHA cells were not transformed by E6/E7/hTERT suggests that inactivation of the p53 and pRb pathways was not sufficient to create malignant astrocytes and that additional molecular events are needed (31). We found, to our knowledge for the first time, that FoxM1B expression in NHA-E6/E7/hTERT cells resulted in cellular transformation and GBM formation in nude mice, at least in part due to its role in the regulation of NEDD4-1, the activation of Akt, and the induced expression of survivin, cyclin D1, and cyclin E.
Materials and Methods
Cell lines and culture conditions
The NHA-E6/E7/hTERT cell line and Hs683 and HFU-251 glioma cell lines were described previously (25, 31, 33). COS-1 cells were obtained from the American Type Culture Collection. All of the cell lines were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum.
Retrovirus production and transduction
Retrovirus was produced with pLXSN-FoxM1B or pLXSN plasmids using the Phoenix A retrovirus production system. NHA-E6/E7/hTERT cells were infected with the virus (10 MOI) with 4 μg/mL Polybrene for 24 hours. To generate stable transduced cell lines, some infected cells were cultured with neomycin for 2 weeks.
Northern blot analysis
Northern hybridization was performed by using a [32P]dCTP-radiolabeled FoxM1 cDNA probe (25). Equal mRNA loading was monitored by hybridizing the same membrane with a β-actin cDNA probe.
Western blot analysis
Standard Western blotting was performed with antibody against FoxM1, cyclin E, cyclin D1, PTEN (Santa Cruz Biotechnology) or NEDD4-1, Akt, phospho-Akt, FoxO3a, phospho-FoxO3a (Cell Signaling Technology), and survivin (Novus Biologicals).
Animals
Female athymic BALB/c nude mice were purchased from the National Cancer Institute (Frederick, MD) and were maintained in institutional facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care.
Colony formation in soft agar assay
Cells (5 × 103) were mixed with 0.3% agar solution in DMEM containing 10% fetal bovine serum and 200 μg/ml of neomycin and poured this solution on top of a 0.60% agar layer in six-well tissue culture plates. Plates were incubated at 37°C in a humidified atmosphere containing 5% CO2 for 21 days. Colonies were then stained with p-iodonitrotetrazolium violet (1 mg/ml) and examined microscopically.
Subcutaneous tumor growth
Cells (1 × 107) were injected subcutaneously into nude mice (25). The length (L), width (W), and height (H) of the resulting tumors were measured once every 5 days. The volume of each tumor was calculated by using the formula 1/2 × L × W × H at every time point.
Intracranial human glioma xenograft model
Cells (1 × 106) were injected intracranially into nude mice as described previously (25). Animals showing general or local symptoms were killed; the remaining animals were killed 90 days after glioma-cell injection.
Histologic and immunohistochemical analysis of tumors
Tissue sections of brain tissue specimens were stained with H&E according to standard protocols. Tissue sections were also used for immunostaining with antibodies against CD31 (BD Biosciences), glial fibrillary acidic protein (GFAP, Dako), phospho-Akt (Cell Signaling Technology), PTEN, cyclin E, cyclin D1 (Santa Cruz Biotechnology), and survivin (Novus Biologicals).
Cycloheximide chase experiments
5 × 105 cells were plated into 6-well plates. Twenty-four hours later, 50 mg/ml of cycloheximide (CHX) was added to the cells, and the CHX treatment was terminated at the indicated time points. Cell lysates were then prepared, and the expression of PTEN was analyzed by Western blotting.
Chromatin immunoprecipitation assay
5 × 106 cells were prepared for the ChIP assay as according to the manufacturor’s instructions with the ChIP assay kit (Cell signaling). Resulting DNA complexes were first immunoprecipitated using indicated antibodies and then DNA was purified from the complexes. The DNA was then subjected to PCR to amplify the putative FoxM1 binding region 1 (−642 to −828 bp) or region 2 (−841 to −993 bp) of the NEDD4-1 promoter using the primers 5′-TCTTCGTCACTGCCTTCTG-3′ and 5′-TTGGACTCGGGATCTGAAA -3′ or the primers 5′-TATGGATGCCTTATTTGGTG-3′ and 5′-TGAACTAGGACCTCCTGTAGAAT-3′, respectively.
Statistical analyses
The significance of the in vitro results was determined by using Student’s t test (two-tailed), whereas the significance of the in vivo data was determined by using the Mann-Whitney U test.
Results
FoxM1 induces malignant transformation of immortalized NHAs
We infected NHA-E6/E7/hTERT cells with pLXSN-FoxM1B or control pLXSN retrovirus. The infected cells were plated for growth in soft agar. Both NHA-E6/E7/hTERT cells and NHA-E6/E7/hTERT cells infected with pLXSN were unable to grow in soft agar (Fig. 1A, Table 1). However, colonies were obtained with NHA-E6/E7/hTERT cells infected with the virus containing FoxM1B (Fig. 1A, Table 1). These results suggest that although inactivation of the p53 and pRb pathways is not sufficient to convert normal astrocytes into malignant glioma cells, such inactivation acts cooperatively with FoxM1B to achieve cellular transformation.
Figure 1. Effects of FoxM1B expression on transformation and glioma growth in vivo.
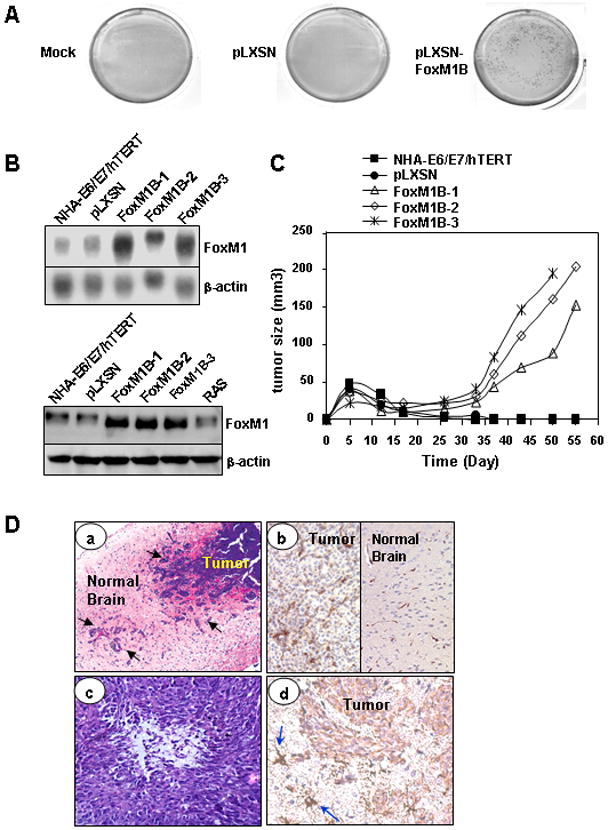
A, Colony-formation assays were performed with the NHA-E6/E7/hTERT cells infected with pLXSN-FoxM1B or pLXSN retrovirus, and without retroviruses (Mock), as indicated. Figure shows the typical colony-formation assay result. B, Determination of overexpression of FoxM1B in three independent pLXSN-FoxM1 retrovirus–transduced E6/E7/hTERT cell lines by Northern (upper panel) and Western (lower panel) blot analyses. C, Cells (1 × 107 cells/mouse) from NHA-E6/E7/hTERT, pLXSN, and three independent pLXSN-FoxM1B cell lines were injected subcutaneously into nude mice (n = 5), and tumor formation was determined. The results shown are for one representative experiment of two. D, Histological features of NHA-E6/E7/hTERT-FoxM1B brain tumors. a, A highly invasive phenotype of the tumors (arrows indicate invasive cells; H&E stains, magnification ×100). b, Increased microvessel proliferation in the tumors (CD34 stains, magnification ×100). c, Microscopic necrosis with pseudopalisading (H&E staining, magnification ×100). d, Brain tumor cells are GFAP positive (arrows indicate astrocytes in the surrounding tissue; GFAP stains, magnification ×200).
Table 1.
Effect of FoxM1B on transformation of immortalized normal human astrocytes (NHAs) and tumor growth in the brain of nude mice
|
Transformation | |
|---|---|
| Cell types | Colony numbersa |
| NHA-E6/E7/hTERT | 0 |
| NHA-E6/E7/hTERT/pLXSN | 0 |
| NHA-E6/E7/hTERT/FoxM1B | 1512 ± 91 |
|
Tumor growth in the brains of nude mice | ||
|---|---|---|
| Cell line | Incidence of tumor formation b | Median days of survival (range) |
| NHA-E6/E7/hTERT | 0/5 | 90 (All >90) |
| NHA- E6/E7/hTERT/pLXSN | 0/5 | 90 (All >90) |
| NHA- E6/E7/hTERT/FoxM1B-1 | 5/5 | 51 (47–58) * |
| NHA- E6/E7/hTERT/FoxM1B-2 | 5/5 | 54 (52–64) * |
| NHA- E6/E7/hTERT/FoxM1B-3 | 4/5 | 63 (52–>90) * |
NHA-E6/E7/hTERT cells were infected with retroviral constructs containing the FoxM1 expression unit (pLXSN-FoxM1B) or control pLXSN. Parental and infected NHA-E6/E7/hTERT cells (1 × 104) were plated in soft agar, and colonies were scored after 3 weeks. Results represent mean ± SD from three separate experiments.
Cells (1 × 106 cells) were implanted intracranially into nude mice. Mice were killed when they were moribund or on day 90. Brains were harvested, and tumor formation was histologically analyzed.
P < 0.001.
Results were shown for one representative experiment of two.
Immortalized NHAs that express FoxM1 are tumorigenic
Next, we created a series of cell lines immortalized by the expression of E6/E7/hTERT and expressing FoxM1B. To avoid clonal selection and variation, we carried out three independent infections of pLXSN-FoxM1B in NHA-E6/E7/hTERT cells and pooled G418-resistant colonies to establish stable cell lines, designated as FoxM1B-transduced cell lines (NHA-E6/E7/hTERT/FoxM1B-1, -2, and -3). FoxM1 protein expression was increased in FoxM1B-transduced NHA-E6/E7/hTERT cell lines (Fig. 1B).
We then evaluated FoxM1-infected NHA-E6/E7/hTERT cells for their ability to form subcutaneous tumors in nude mice; tumors formed within 3 weeks (Fig. 1C). No tumors developed by 8 weeks in mice injected with parental NHA-E6/E7/hTERT or pLXSN–infected cells. Thus, the results show that NHA-E6/E7/hTERT/FoxM1 cells are tumorigenic.
FoxM1 induces GBM formation of immortalized NHAs
We used an orthotopic xenograft model of human glioma to study the growth of these cells in the brains of nude mice. Intracranially implanted NHA-E6/E7/hTERT or pLXSN–infected cells did not form tumors by 90 days after injection (Table 1). In contrast, intracranially injected FoxM1B-infected NHA-E6/E7/hTERT cells did form brain tumors in nude mice and significantly shortened their survival duration (Table 1). We also examined the histologic features of brain tumors that arose from NHA-E6/E7/hTERT/FoxM1 cells in the brains of nude mice. The lesions were characterized by high cellularity and invasiveness, were pleomorphic, and exhibited microvessel proliferation and necrosis (Fig. 1D), all distinct histologic features of human GBM. In addition, the brain tumor cells were positive for GFAP, but the level of expression was less than in the astrocytes in the surrounding tissue, indicating a human astrocytic origin of the tumors (Fig. 1D). These findings suggest that the overexpression of FoxM1B in immortalized NHAs cells lead to the formation of GBM in nude mice.
Increased FoxM1 expression leads to Akt activation
In animal studies, activation of Akt appears to be essential for the formation of GBMs from genetically modified neural progenitors and normal human astrocytes (33, 34). Thus, we examined whether overexpression of FoxM1 affects the activation status of Akt in NHA-E6/E7/hTERT cells. We found that pLXSN-FoxM1B infections induced more phosphorylation and activation of Akt than did infection with retroviral pLXSN or Ras (Fig. 2A). In contrast, overexpression of FoxM1 did not affect ERK-1/2 activation, a different pathway from Akt (Fig. 2A). NHA-E6/E7/hTERT/FoxM1B brain tumors also expressed phosphorylated Akt (p-Akt) (Fig. 2B). Furthermore, overexpression of FoxM1 in NHA-E6/E7/hTERT by transient pLXSN-FoxM1B infection induced the nuclear translocation of Akt and the cytoplasmic retention of FoxO3a protein (Fig. 2C), which have been reported as significant predictors of p-Akt status in gliomas (35). These results indicate that increased FoxM1 expression leads to Akt activation and subsequently inhibits FoxO3a function.
Figure 2. Overexpression of FoxM1 increased p-Akt but not p-ERK-1/2.

A, The expression of FoxM1, p-Akt, total Akt, FoxO3a, p-FoxO3a, ERK-1/2, and p-ERK-1/2 on NHA-E6/E7/hTERT/FoxM1B cells or parental and NHA-E6/E7/hTERT/pLXSN cells was analyzed by using Western blotting. B, Paraffin sections from brain injected with pLXSN- and pLXSN-FoxM1B-transduced NHA-E6/E7/hTERT cells (tumors) were stained with antibody against p-Akt. C, Immunofluorescent microscopic analyses of Akt activation and FOXO3a localization. NHA-E6/E7/hTERT cells were transiently infected with pLXSN-FoxM1B or pLXSN. Then the cells were immunostained for Akt (green), and their nuclei were stained with DAPI (blue). The cells were also immunostained for FOXO3a (green), and their nuclei were stained with DAPI (blue). This is one representative experiment of three. D, Blocking Akt activation decreased the colony formation induced by FoxM1. NHA-E6/E7/hTERT cells were infected with retroviral-pLXSN or pLXSN-FoxM1B for 24 hours and then treated with LY294002 (25 μM) or wortmannin (0.1 μM) for 2 hours. Left, The expression of FoxM1, p-Akt, total Akt, FoxO3a, and p-FoxO3a was analyzed by using Western blotting. Right, The colony-formation ability of the cells was determined by a colony-formation assay. Each bar represents the mean ± standard deviation of the colony numbers from a representative experiment in triplicate. *P < 0.01 compared with the no-treatment group.
Blocking Akt activation inhibits FoxM1-induced transformation of immortalized NHAs
To determine whether FoxM1 induces transformation of immortalized NHAs through the Akt pathway, we treated NHA-E6/E7/hTERT/FoxM1 cells with PI3K inhibitors LY294002 and wortmannin. Both treatments inhibited the expression of p-Akt in the cells (Fig. 2D, left). Furthermore, blockade of Akt activation by LY294002 and wortmannin suppressed the transformation of immortalized NHAs, as determined by a colony-formation assay (Fig. 2D,right). Therefore, these data suggest that Akt activation is an important step in the FoxM1-mediated transformation of immortalized NHAs into glioma cells.
Increased FoxM1 expression leads to PTEN reduction and Akt activation
It is known that PTEN is an important molecule that regulates Akt activity by inhibiting PI3K (36, 37). To further define how FoxM1 activates the Akt pathway in NHA cells, we examined PTEN expression in these cells. Levels of PTEN expression were substantially lower in NHA-E6/E7/hTERT/FoxM1B cells than in NHA-E6/E7/hTERT or NHA-E6/E7/hTERT/pLXSN control cells (Fig. 3A,left). The observed decrease in PTEN expression in these cells was accompanied by enhanced Akt pathway signaling, as was evident by concomitant increases in the level of p-Akt, but not in the level of total Akt (Fig. 3A,left). NHA-E6/E7/hTERT/FoxM1B brain tumors also had lower levels of PTEN expression than did brain tissues from the mice injected with NHA-E6/E7/hTERT/pLXSN cells (Fig. 3A, right). Moreover, knockdown of FoxM1 in NHA-E6/E7/hTERT/FoxM1B cells restored PTEN expression and reversed the up-regulation of p-Akt in the cells (Fig. 3B, left). Furthermore, the overexpression of FoxM1 in Hs683 glioma cells down-regulated PTEN and up-regulated p-Akt expression (Fig. 3B, right).
Figure 3. FoxM1 down-regulated PTEN expression in NHA cells.
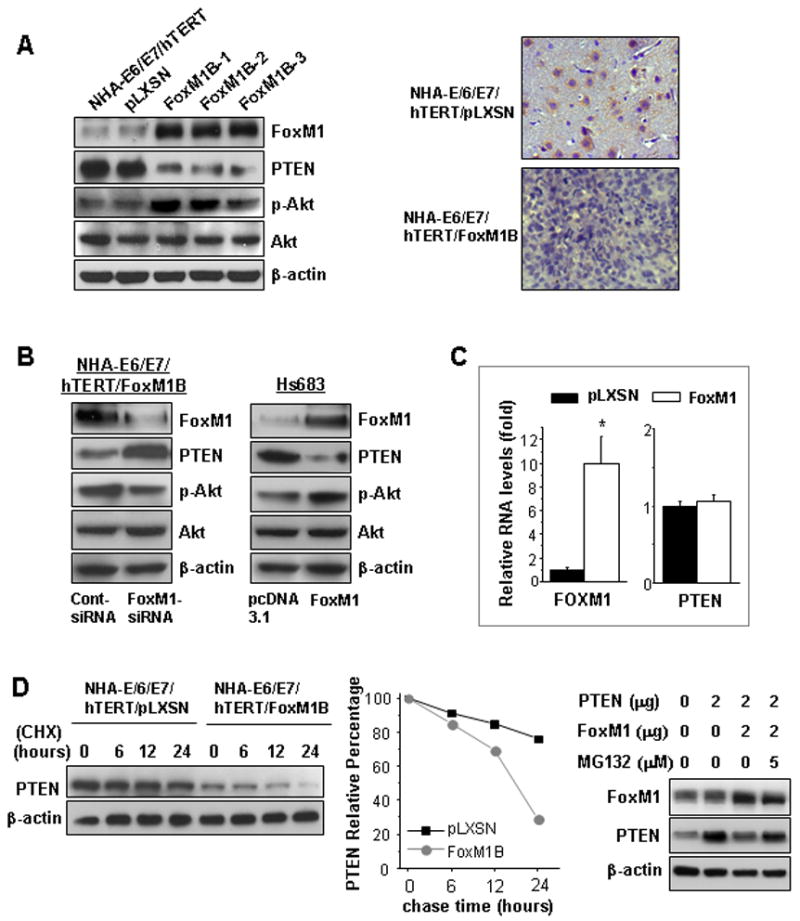
A, The expression of FoxM1, PTEN, p-Akt, and Akt in NHA-E6/E7/hTERT/FoxM1B or parental and NHA-E6/E7/hTERT/pLXSN cells was analyzed by using Western blotting (left). Paraffin sections from brain injected with pLXSN- and pLXSN-FoxM1B–transduced NHA-E6/E7/hTERT cells were stained with antibodies against PTEN (right). B, NHA-E6/E7/hTERT-FoxM1B cells were transfected with FoxM1 siRNA or control siRNA (50 nM) for 48 hours; the expression of FoxM1, PTEN, p-Akt and Akt was analyzed by using Western blotting (left). Hs683 cells were transfected with pcDNA3.1-FoxM1 and pcDNA3.1 control vector, and the expression of FoxM1, PTEN, p-Akt, and Akt was analyzed by using Western blotting (right). C, Real-time PCR analysis of relative mRNA level of FoxM1 and PTEN in pLXSN- and FoxM1B-transduced NHA-E6/E7/hTERT cells. Each bar represents the mean ± standard deviation in triplicate. *P < 0.01. D. Cycloheximide chase experiment (left). FoxM1B-transduced NHA-E6/E7/hTERT cells and pLXSN-transduced NHA-E6/E7/hTERT cells were treated with CHX (50 mg/ml), and cells were collected at the indicated time for western blotting analysis of PTEN expression. Relative PTEN protein stability (middle). Intensities of the bands in the Western blot were analyzed by using NIH Image software, and the integrated optical densities of the bands at time zero were set as 100%. Blockade of PTEN decreasing by MG132 (right). pLNCX-PTEN plasmid was co-transfeced with pcDNA3.1-FoxM1 or pcDNA3.1 plasmids into COS-1 cells for 24 hours. MG132 (0–5 μM) was added to the medium as indicated. The cells were cultured for another 24 hours, and Western blotting was performed.
FoxM1 regulates PTEN expression on the posttranscriptional level
To study the mechanism that may be responsible for the down-regulation of PTEN by FoxM1, we first examined the RNA levels of PTEN and found that the transcriptional levels of PTEN were similar in both the NHA-E6/E7/hTERT/FoxM1 and NHA-E6/E7/hTERT/pLXSN control cells (Fig. 3C). Next, we inhibited protein translation in cells with use of CHX treatment and analyzed PTEN protein levels at the indicated times. As shown in Figure 3D (left and middle), overexpression of FoxM1 in NHA-E6/E7/hTERT/FoxM1 cells resulted in an increase in the endogenous PTEN degradation rate, compared with NHA-E6/E7/hTERT/pLXSN control cells. Overexpression of FoxM1 in COS-1 cells also resulted in a decreased level of exogenous PTEN protein (Fig. 3D, right). Such a decrease in PTEN protein level can be blocked by proteasomal inhibitor MG132 (Fig. 3D, right). These results indicated that the decreased PTEN expression in FoxM1 overexpression cells was due, at least in part, to the increased stability of PTEN protein.
FoxM1 down-regulates PTEN expression by regulating NEDD4-1
To study how FoxM1 regulates PTEN degradation, we first performed protein-protein binding experiments on FoxM1 and PTEN. We did not find direct binding of FoxM1 with PTEN (data not shown), indicating that the regulation of PTEN by FoxM1 is probably an indirect mechanism. On the other hand, NEDD4-1 has been identified as an E3 ligase that can promote PTEN ubiquitination and degradation. We determined that endogenous NEDD4-1 can indeed regulate PTEN in NHA-E6/E7/hTERT/FoxM1 by showing that the knockdown of NEDD4-1 by siRNA in the cells caused an increase in endogenous PTEN (Fig. 4A). Therefore, this result confirmed the role of NEDD4-1 as PTEN E3 in ligase in the cells.
Figure 4. FoxM1 down-regulated PTEN expression by regulating NEDD4-1.
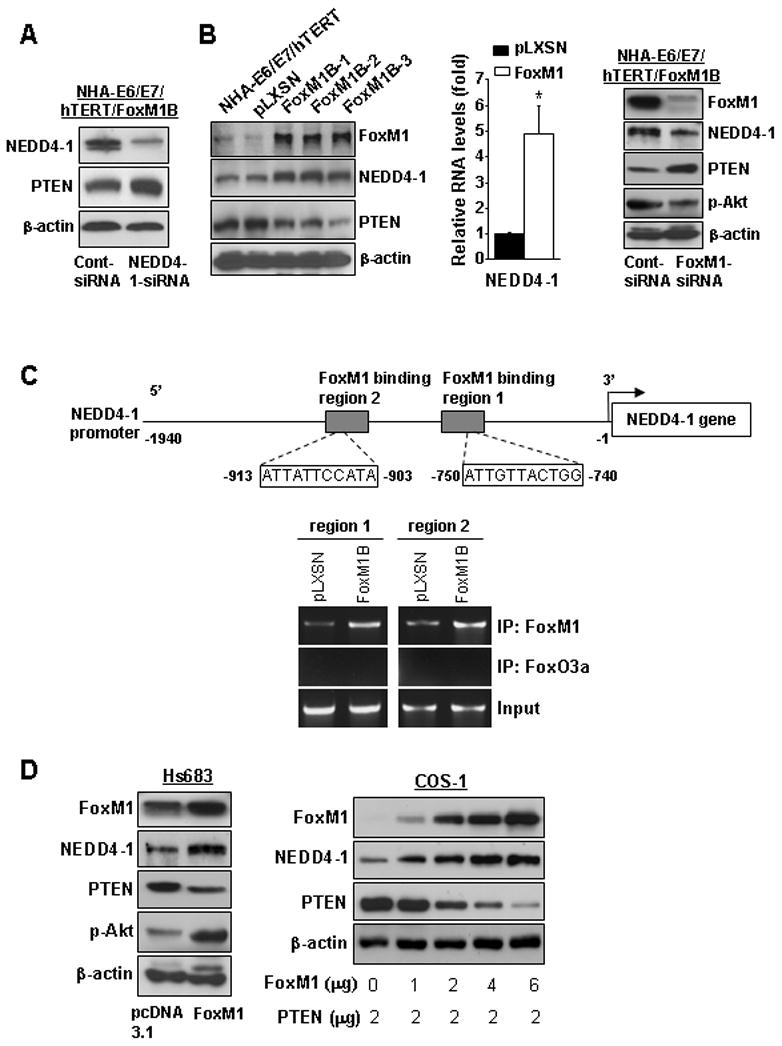
A, NHA-E6/E7/hTERT-FoxM1B cells were transfected with NEDD4-1-siRNA (50 nM) for 24 hours and then treated with CHX (50 mg/ml) for 6 hours; cells were collected for Western blotting of NEDD4-1 and PTEN expressions. B. The expression of FoxM1, NEDD4-1, and PTEN in parental and pLXSN- or FoxM1B-transduced NHA-E6/E7/hTERT cells was analyzed by using Western blotting (left). Real-time PCR analysis of relative mRNA level of NEDD4-1 in NHA-E6/E7/hTERT/pLXSN cells and FoxM1B cells (middle). NHA-E6/E7/hTERT-FoxM1B cells were transfected with FoxM1-siRNA or control siRNA (50 nM) for 48 hours and Western blottings were performed using the antibodies as indicated (right). C, Upper panel, the schematic structure of NEDD4-1 promoter shows the sequences and positions of putative FoxM1-binding sits on the promoter. The grey boxes indicate the putative FoxM1 binding region 1 and 2 in Chip assay. Lower panel, ChIP assays were performed with NHA-E6/E7/hTERT/pLXSN and FoxM1B cells using an anti-FoxM1 antibody or anti-FoxO3a antibody (as a control). We subjected 1% of the total cell lysates to PCR before immunoprecipitation as inputs. D. Hs683 cells were transfected with pcDNA3.1-FoxM1 and control vector (left). COS1 cells were co-transfected with 2 μg pLNCX-PTEN plasmid with different dosages of pcDNA3.1-FoxM1 plasmids (right). Cell lysates were collected 48 hours after the transfections for Western blotting using the antibodies as indicated.
We next examined the level of NEDD4-1 in the NHA cells. As shown in Figure 4B, the protein level of NEDD4-1 was up-regulated in NHA-E6/E7/hTERT/FoxM1B cells compared with levels in NHA-E6/E7/hTERT and NHA-E6/E7/hTERT/pLXSN cells, whereas the protein level of PTEN was down-regulated in the NHA-E6/E7/hTERT/FoxM1B cells. Moreover, the mRNA level of NEDD4-1 was up-regulated in NHA-E6/E7/hTERT/FoxM1B cells (Fig. 4B, middle). In contrast, knocking down FoxM1 from NHA-E6/E7/hTERT/FoxM1B cells inhibited the expression of NEDD4-1, restored PTEN expression, and inhibited p-Akt in the cells (Fig. 4B, right).
To determine whether NEDD4-1 could be a direct transcriptional target of FoxM1, we scanned ~2-kb promoter regions of NEDD4-1 gene with the FoxM1 DNA binding consensus sequence (13), and two FoxM1 putative binding sites (−740 to −750 bp and −903 to −913bp) were found in the promoter (Fig. 4C). To determine whether FoxM1 binds directly to the human NEDD4-1 promoter, we performed ChIP assays using two pairs of PCR primers to DNA sequences (−642/−828 bp or −841/−993 bp) in the vicinity of these potential FoxM1 binding sites. Indeed, ChIP assays in NHA-E6/E7/hTERT/pLXSN or FoxM1B cells with antibodies specific to either FoxM1 or FoxO3a (control) showed that FoxM1 protein is directly bound to the endogenous NEDD4-1 promoter compared to the FoxO3a control (Fig. 4C). Moreover, in the NHA-E6/E7/hTERT/FoxM1B cells that expressed high FoxM1 levels, there is an increased binding of FoxM1 protein to the promoter as compared to that in NHA-E6/E7/hTERT/pLXSN (Fig. 4C). These findings demonstrate that FoxM1 bound specifically to FoxM1-binding sites in the NEDD4-1 promoter in vivo, suggesting NEDD4-1 could be a direct transcriptional target of FoxM1.
To examine whether FoxM1 regulated NEDD4-1 in other cells, Hs683 glioma cells were transfected with FoxM1. Overexpression of FoxM1 in Hs683 glioma cells up-regulated NEDD-4, decreased PTEN, and increased p-Akt expressions (Fig. 4D, left). Furthermore, COS-1 cells were cotransfected with PTEN in the presence or absence of FoxM1 overexpression. Overexpression of FoxM1 in COS-1 also caused an increase in NEDD4-1 and a decrease in exogenous PTEN protein (Fig. 4D, right). Taken together, the above data indicated that FoxM1 regulated NEDD4-1 expression, hence PTEN expression, in the cells.
FoxM1 induces multiple occurrences of cancer-related gene expression
To further explore the molecular mechanisms involved in FoxM1-mediated GBM formation, we measured the expression of cyclin D1, cyclin E, and survivin in FoxM1B–overexpressed cell lines and in the brain tumor tissues of mice. Some of these molecules have been previously reported to be the downstream targets of Akt and/or FoxM1 and to be important factors for glioma proliferation (20, 24, 25, 38, 39). Our results showed that cyclin D1, cyclin E, and survivin are also highly expressed in the FoxM1B–overexpressed cells and in the brain tumors that arose from injection of NHA-E6/E7/hTERT/FoxM1B cells (Fig. 5A and 5B). In addition, knockdown of FoxM1 expression down-regulated these genes significantly in HFU-251 GBM cells (Fig. 5C). These results indicated that the overexpression of FoxM1 in NHA cells not only inhibited PTEN and activated the Akt pathway, but also induced the expression of multiple cell cycle–related genes, which are important molecules for tumor growth.
Figure 5. FoxM1 induced multiple occurrences of cancer-related gene expression.

A, The expression of FoxM1, survivin, cyclin D1, and cyclin E in parental, NHA-E6/E7/hTERT/pLXSN or FoxM1B cells was analyzed by using Western blotting. B, Paraffin sections from brain injected with pLXSN- or pLXSN-FoxM1B–transduced NHA-E6/E7/hTERT cells were stained with antibodies against survivin, cyclin D1, and cyclin E (magnification ×200). C, HFU-251 cells were transfected with FoxM1-siRNA (50 μM), and the expression of FoxM1, survivin, cyclin D1, and cyclin E was analyzed by using Western blotting.
Discussion
Although our previous studies had indicated that aberrant FoxM1B expression is critical to the regulation of tumorigenicity, invasion, and angiogenesis of human glioma cells, it was not known whether FoxM1B plays an important role in the transformation of human astrocytes into glioma cells. In this study, we found that overexpression of FoxM1 in immortalized human astrocytes not only transforms the cells but also causes them to progress into GBM cells in the brain. We also provided evidence that FoxM1 up-regulated the expression of NEDD-4, which resulted in increased PTEN protein degradation. Furthermore, the increased PTEN protein degradation in turn led to the up-regulation of activation of Akt pathway, a potential mechanism for the development of GBM.
Glioma formation and progression are widely regarded as multistep processes resulting from complex interplay between multiple genetic and epigenetic events (3–6, 31–33, 40, 41). Studies have shown that the loss of p53 and pRb pathways is not sufficient to induce glioma and that additional molecular events are needed such as Ras activation and PTEN loss (40, 41). Our results support this finding; specifically, we found that a combinationof FoxM1 overexpression, loss of p53 and pRb pathways, and human telomerase reverse transcriptase overexpression is sufficient to induce NHA cells to transform into glioma.
Moreover, our results support the involvement of PTEN underexpression in transformation and in GBM formation. Brain-specific inactivation of PTEN in a mouse model caused increased astrocyte proliferation that may render these cells susceptible to neoplastic transformation (42, 43). PTEN heterozygous knockout accelerated astrocytoma development in mice in which the Rb family proteins were inactivated (44). Moreover, it has been shown that successive loss of each PTEN allele may contribute to de novo formation of high-grade astrocytoma and progression into glioblastoma, respectively (45).
Numerous studies have suggested that PTEN can be inactivated not only by gene deletion and mutation but also by defects in posttranslational regulation during tumorigenesis (12, 46–47). Recent studies postulated that PTEN is subject to ubiquitination mediated by NEDD4-1, an E3 ligase, leading to PTEN degradation (12, 47). Moreover, the overexpression of NEDD4-1 was shown to induce PTEN degradation and promote K-Ras–mediated transformation (12). In bladder cancers, the PTEN level was inversely correlated with the level of NEDD4-1 (12). In breast cancer, NEDD4-1 was shown to be critical for targeting PTEN for degradation (47). Therefore, these studies suggest that NEDD4-1 shows oncogenic activity that is PTEN-dependent.
In the current study, we found that knockdown of NEDD4-1 in NHA-E6/E7/hTERT/FoxM1B cells caused an increase in endogenous PTEN, suggesting the role of NEDD4-1 in PTEN regulation in glioma cells. Furthermore, we demonstrated that the overexpression of FoxM1 down-regulated PTEN expression in NHA-E6/E7/hTERT, Hs683, and COS-1 cells (all with wild-type PTEN). The mechanism for the overexpression of FoxM1 that accelerates PTEN degradation is as follows: FoxM1 regulates the expression of NEDD4-1, which promotes PTEN ubiquitination and degradation in these cells.
PTEN inactivation could result in uncontrolled PI3K/Akt signaling activation, a mechanism of tumor formation. We have observed that increased expression of FoxM1 in NHA-E6/E7/hTERT caused a significant increase in the level of p-Akt and induced tumor colony formation in a soft agar assay and tumor formation in a mouse brain tumor model. Moreover, using the PI3K-specific inhibitors LY294002 and wortmannin can inhibit FoxM1-induced colony formation in vitro. Therefore, the results indicate that PI3K/Akt activation plays an important role in the glioma formation.
Several studies have reported that cyclin D1 and cyclin E expression correlate significantly with the degree of malignancy in astrocytomas (49). Survivin expression is also increased in GBMs and correlates with patient survival (50). In this study, we found that FoxM1B overexpression in glioma cells significantly increased survivin, cyclin D1, and cyclin E expression, consistent with findings in other types of cells (5, 11). Thus, the molecular mechanisms by which FoxM1 regulates the growth of glioma cells are associated with increasing the expression of these molecules that function as cell-cycle regulators and/or inhibitors of tumor cell apoptosis and are critical for tumor cell proliferation.
In summary, our findings demonstrated that FoxM1 plays an important role in promoting astrocyte transformation and GBM formation by the regulation of multiple factors. Of more importance, we have provided a new mechanism of FoxM1-induced transformation and GBM formation involving the overexpression of FoxM1 that up-regulates the expression of NEDD4-1 and hence destabilizes PTEN and activates the Akt pathway. Our findings strongly suggest that FoxM1 plays an important role in glioma transformation and may be a therapeutic target for glioma.
Acknowledgments
Grant support: This work was supported in part by National Cancer Institute grants R01-CA-116528 and P50-CA-127001, a research grant from the Brain Tumor Society, and a grant from the Center for Targeted Therapy at The University of Texas M. D. Anderson Cancer Center.
We thank Tamara Locke for editorial comments.
References
- 1.American Cancer Society. Cancer Facts and Figures. 2009 www.cancer.org.
- 2.Surawicz TS, Davis F, Freels S, Laws ER, Jr, Menck HR. Brain tumor survival: results from the National Cancer Data Base. J Neurooncol. 1998;40:151–60. doi: 10.1023/a:1006091608586. [DOI] [PubMed] [Google Scholar]
- 3.Maher EA, Furnari FB, Bachoo RM, et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–33. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 4.Uhrbom L, Kastemar M, Johansson FK, Westermark B, Holland EC. Cell type-specific tumor suppression by Ink4a and Arf in Kras-induced mouse gliomagenesis. Cancer Res. 2005;65:2065–9. doi: 10.1158/0008-5472.CAN-04-3588. [DOI] [PubMed] [Google Scholar]
- 5.Bachoo RM, Maher EA, Ligon KL, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–77. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 6.Westermark B, Heldin CH, Nistér M. Platelet-derived growth factor in human glioma. Glia. 1995;15:257–63. doi: 10.1002/glia.440150307. [DOI] [PubMed] [Google Scholar]
- 7.Rasheed BK, Stenzel TT, McLendon RE, et al. PTEN gene mutations are seen in high-grade but not in low-grade gliomas. Cancer Res. 1997;57:4187–90. [PubMed] [Google Scholar]
- 8.Ermoian RP, Furniss CS, Lamborn KR, et al. Dysregulation of PTEN and protein kinase B is associated with glioma histology and patient survival. Clin Cancer Res. 2002;8:1100–6. [PubMed] [Google Scholar]
- 9.Maier D, Zhang Z, Taylor E, et al. Somatic deletion mapping on chromosome 10 and sequence analysis of PTEN/MMAC1 point to the 10q25–26 region as the primary target in low-grade and high-grade gliomas. Oncogene. 1998;16:3331–5. doi: 10.1038/sj.onc.1201832. [DOI] [PubMed] [Google Scholar]
- 10.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiencke JK, Zheng S, Jelluma N, et al. Methylation of the PTEN promoter defines low-grade gliomas and secondary glioblastoma. Neuro Oncol. 2007;9:271–9. doi: 10.1215/15228517-2007-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Trotman LC, Koppie T, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129–39. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye H, Kelly TF, Samadani U, et al. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol Cell Biol. 1997;17:1626–41. doi: 10.1128/mcb.17.3.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laoukili J, Kooistra MR, Brás A, et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7:126–36. doi: 10.1038/ncb1217. [DOI] [PubMed] [Google Scholar]
- 15.Wierstra I, Alves J. FOXM1, a typical proliferation-associated transcription factor. Biol Chem. 2007;388:1257–74. doi: 10.1515/BC.2007.159. [DOI] [PubMed] [Google Scholar]
- 16.Park HJ, Carr JR, Wang Z, et al. FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO J. 2009;28:2908–18. doi: 10.1038/emboj.2009.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petrovic V, Costa RH, Lau LF, Raychaudhuri P, Tyner AL. FoxM1 regulates growth factor-induced expression of kinase-interacting stathmin (KIS) to promote cell cycle progression. J Biol Chem. 2008;283:453–60. doi: 10.1074/jbc.M705792200. [DOI] [PubMed] [Google Scholar]
- 18.Teh MT, Wong ST, Neill GW, Ghali LR, Philpott MP, Quinn AG. FOXM1 is a downstream target of Gli1 in basal cell carcinomas. Cancer Res. 2002;62:4773–80. [PubMed] [Google Scholar]
- 19.Madureira PA, Varshochi R, Constantinidou D, et al. The Forkhead box M1 protein regulates the transcription of the estrogen receptor alpha in breast cancer cells. J Biol Chem. 2006;281:25167–76. doi: 10.1074/jbc.M603906200. [DOI] [PubMed] [Google Scholar]
- 20.Kalinichenko VV, Major ML, Wang X, et al. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004;18:830–50. doi: 10.1101/gad.1200704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalin TV, Wang IC, Ackerson TJ, et al. Increased levels of the FoxM1 transcription factor accelerate development and progression of prostate carcinomas in both TRAMP and LADY transgenic mice. Cancer Res. 2006;66:1712–20. doi: 10.1158/0008-5472.CAN-05-3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim IM, Ackerson T, Ramakrishna S, et al. The Forkhead Box m1 transcription factor stimulates the proliferation of tumor cells during development of lung cancer. Cancer Res. 2006;66:2153–61. doi: 10.1158/0008-5472.CAN-05-3003. [DOI] [PubMed] [Google Scholar]
- 23.Wang IC, Meliton L, Tretiakova M, Costa RH, Kalinichenko VV, Kalin TV. Transgenic expression of the forkhead box M1 transcription factor induces formation of lung tumors. Oncogene. 2008;27:4137–49. doi: 10.1038/onc.2008.60. [DOI] [PubMed] [Google Scholar]
- 24.Yoshida Y, Wang IC, Yoder HM, Davidson NO, Costa RH. The forkhead box M1 transcription factor contributes to the development and growth of mouse colorectal cancer. Gastroenterology. 2007;132:1420–31. doi: 10.1053/j.gastro.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 25.Liu M, Dai B, Kang SH, et al. FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res. 2006;66:3593–602. doi: 10.1158/0008-5472.CAN-05-2912. [DOI] [PubMed] [Google Scholar]
- 26.van den Boom J, Wolter M, Kuick R, et al. Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. Am J Pathol. 2003;163:1033–43. doi: 10.1016/S0002-9440(10)63463-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rickman DS, Bobek MP, Misek DE, et al. Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer Res. 2001;61:6885–91. [PubMed] [Google Scholar]
- 28.Hodgson JG, Yeh RF, Ray A, et al. Comparative analyses of gene copy number and mRNA expression in glioblastoma tumors and xenografts. Neuro Oncol. 2009;11:477–87. doi: 10.1215/15228517-2008-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai B, Kang S-H, Gong W, et al. Aberrant FoxM1B expression increases matrix metalloproteinase-2 transcription and enhances the invasion of glioma cells. Oncogene. 2007;26:6212–9. doi: 10.1038/sj.onc.1210443. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Zhang N, Dai B, et al. FoxM1B transcriptionally regulates vascular endothelial growth factor expression and promotes the angiogenesis and growth of glioma cells. Cancer Res. 2008;68:8733–42. doi: 10.1158/0008-5472.CAN-08-1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sonoda Y, Ozawa T, Hirose Y, et al. Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res. 2001;61:4956–60. [PubMed] [Google Scholar]
- 32.Rich JN, Guo C, McLendon RE, Bigner DD, Wang XF, Counter CM. A genetically tractable model of human glioma formation. Cancer Res. 2001;61:3556–60. [PubMed] [Google Scholar]
- 33.Sonoda Y, Ozawa T, Aldape KD, Deen DF, Berger MS, Pieper RO. Akt pathway activation converts anaplastic astrocytoma to glioblastoma multiforme in a human astrocyte model of glioma. Cancer Res. 2001;61:6674–8. [PubMed] [Google Scholar]
- 34.Holland EC, Celestino J, Dai C, et al. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55–7. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- 35.Choe G, Horvath S, Cloughesy TF, et al. Analysis of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003;63:2742–6. [PubMed] [Google Scholar]
- 36.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3, 4, 5-trisphosphate. J Biol Chem. 1998;273:13375–8. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 37.Stambolic V, Suzuki A, de la Pompa JL, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 38.Alvarez B, Garrido E, Garcia-Sanz JA, Carrera AC. Phosphoinositide 3-kinase activation regulates cell division time by coordinated control of cell mass and cell cycle progression rate. J Biol Chem. 2003;278:26466–73. doi: 10.1074/jbc.M300663200. [DOI] [PubMed] [Google Scholar]
- 39.Asanuma H, Torigoe T, Kamiguchi K, et al. Survivin expression is regulated by coexpression of human epidermal growth factor receptor 2 and epidermal growth factor receptor via phosphatidylinositol 3-kinase/AKT signaling pathway in breast cancer cells. Cancer Res. 2005;65:11018–25. doi: 10.1158/0008-5472.CAN-05-0491. [DOI] [PubMed] [Google Scholar]
- 40.Zheng H, Ying H, Yan H, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129–33. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Read RD, Cavenee WK, Furnari FB, Thomas JB. A drosophila model for EGFR-Ras and PI3K-dependent human glioma. PLoS Genet. 2009;5:e1000374. doi: 10.1371/journal.pgen.1000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Furnari FB, Lin H, Huang HS, Cavenee WK. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci U S A. 1997;94:12479–84. doi: 10.1073/pnas.94.23.12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fraser MM, Zhu X, Kwon CH, Uhlmann EJ, Gutmann DH, Baker SJ. Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res. 2004;64:7773–9. doi: 10.1158/0008-5472.CAN-04-2487. [DOI] [PubMed] [Google Scholar]
- 44.Xiao A, Wu H, Pandolfi PP, Louis DN, Van Dyke T. Astrocyte inactivation of the pRb pathway predisposes mice to malignant astrocytoma development that is accelerated by PTEN mutation. Cancer Cell. 2002;1:157–68. doi: 10.1016/s1535-6108(02)00029-6. [DOI] [PubMed] [Google Scholar]
- 45.Kwon CH, Zhao D, Chen J, et al. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. 2008;68:3286–94. doi: 10.1158/0008-5472.CAN-07-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276:993–8. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- 47.Yim EK, Peng G, Dai H, et al. Rak functions as a tumor suppressor by regulating PTEN protein stability and function. Cancer Cell. 2009;15:304–14. doi: 10.1016/j.ccr.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blanco-Aparicio C, Renner O, Leal JF, Carnero A. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28:1379–86. doi: 10.1093/carcin/bgm052. [DOI] [PubMed] [Google Scholar]
- 49.Chakravarti A, Delaney MA, Noll E, et al. Prognostic and pathologic significance of quantitative protein expression profiling in human gliomas. Clin Cancer Res. 2001;7:2387–95. [PubMed] [Google Scholar]
- 50.Chakravarti A, Noll E, Black PM, et al. Quantitatively determined survivin expression levels are of prognostic value in human gliomas. J Clin Oncol. 2002;20:1063–8. doi: 10.1200/JCO.2002.20.4.1063. [DOI] [PubMed] [Google Scholar]