

Abstract
Studies have demonstrated that several schizophrenia candidate genes are especially susceptible to changes in transcriptional activity as a result of histone modifications and DNA methylation. Increased expression of epigenetic enzymes which generally reduce transcription have been reported in schizophrenia postmortem brain samples. An abnormal chromatin state leading to reduced candidate gene expression can be explained by aberrant coordination of epigenetic mechanisms in schizophrenia. Dynamic epigenetic processes are difficult to study using static measures such as postmortem brain samples. Therefore, we have developed a model using cultured peripheral blood mononuclear cells (PBMC) capable of pharmacologically probing these processes in human subjects. This approach has revealed several promising findings indicating that schizophrenia subject PBMC chromatin may be less capable of responding to agents which normally ‘open’ chromatin. We suggest that the ability to appropriately modify chromatin structure may be a factor in treatment response. Several pharmacological approaches for targeting epigenetic processes are reviewed.
Keywords: lymphocyte, DNA methylation, histone, HDAC, DNMT, GAD67, reelin, bipolar, demethylase, methyltransferase, chromatin, psychosis
1. Background
For the past 40 years schizophrenia research has directed an intense and exciting effort to identify causative genes. However, peculiarities such as the noncomplete concordance between monozygotic twins, fluctuating disease course, sexual dimorphism, peaks of susceptibility, symptom changes coinciding with major hormonal rearrangements, and parent-of-origin effects are difficult to explain with a purely genetic approach (Petronis, 2004; Harlap et al., 2009).
The study of epigenetics has provided a platform whereby the impact of environmental factors (e.g., hormones, drugs of abuse, medications, infections, toxins, diet, etc.), both within an individual and as passed through generations, can be measured and understood. Also, because epigenetic factors are pharmacologically alterable the potential for therapeutic interventions is profound.
2. Introduction to epigenetics
The nucleosome comprises one of the major components of epigenetic gene regulation. The nucleosome is composed of an octamer of histone proteins (a pair of H2a, H2b, H3 and H4 proteins) as well as approximately 146 base pairs of DNA. Examples of alterations to this unit include methylation of the DNA cytosine base and site specific amino acid histone protein modifications including methylation, acetylation, phosphorylation, ubiquitination, and sumolyation (Akbarian and Huang, 2009). These dynamic and often heritable changes have profound effects on the spatial and temporal regulation of gene expression by altering regulatory region availability to transcription factors (Egger et al., 2004).
One example of a covalent histone modification that increases promoter accessibility is the histone acetyltransferase (HAT) catalyzed acetylation of lysine amino acids on the N-terminal tails of histone 3 or 4 proteins (Turner, 2002). These histone modifications neutralize the proteins’ positively charged N-terminal tails, making the negatively charged DNA molecule more available to the machinery of transcription (Hong et al., 1993). Conversely, the histone deacetylase (HDAC) catalyzed removal of acetyl groups condenses chromatin around gene promoters generally resulting in decreased gene expression (Peterson and Laniel, 2004).
Histone methylation can induce either a transcriptionally facilitative or repressive state depending on the amino acid residue being methylated. For example, while the methylation of the fourth lysine on histone 3 (H3K4) generally increases gene expression, methylation of lysines 9 (H3K9) or 27 (H3K27) of histone 3 generally produces the opposite effect (Peterson and Laniel, 2004).
DNA methylation provides another example of an epigenetic process that affects gene expression. The DNA methylatransferase (DNMT) family of enzymes methylate CpG dinucleotide using S-adenosylmethionine (SAM) as the methyl donor (Abdolmaleky et al., 2004). Generally, increased CpG methylation results in decreased gene expression (Szyf, 1996). Therefore, a CpG island (defined as a >500bp region of DNA with >55% GC content (Takai and Jones, 2002)) within a gene’s regulatory region makes it especially prone to epigenetic gene regulation.
The repressive processes of histone deacetylation, H3K9 or H3K27 methylation and DNA methylation have been found to cooperate in reducing gene expression. This occurs both through direct and indirect means. The direct recruitment of both HDACs and HMTs by DNMTs (Bachman et al., 2001; Fuks et al., 2000, 2001, 2003a; Robertson et al., 2000; Rountree et al., 2000) and DNMTs by HMTs (Epsztejn-Litman et al., 2008) have been reported. In addition, indirect interactions, whereby methyl-CpG binding domain proteins (MBDs), such as MeCP2, lead to increased histone methylation (Fuks et al., 2003b) and histone deacetylation have been documented (Jones et al., 1998). HP1, a repressor protein which is associated with H3K9 methylation (Jacobs et al., 2002; Wallace and Orr-Weaver, 2005), is able to both independently condense chromatin and recruit DNMTs (Lehnertz et al., 2003; Fuks et al., 2003a) (Fig. 1A).
Figure 1. Hypothetical mechanism of action for medications which target restrictive chromatin in schizophrenia.
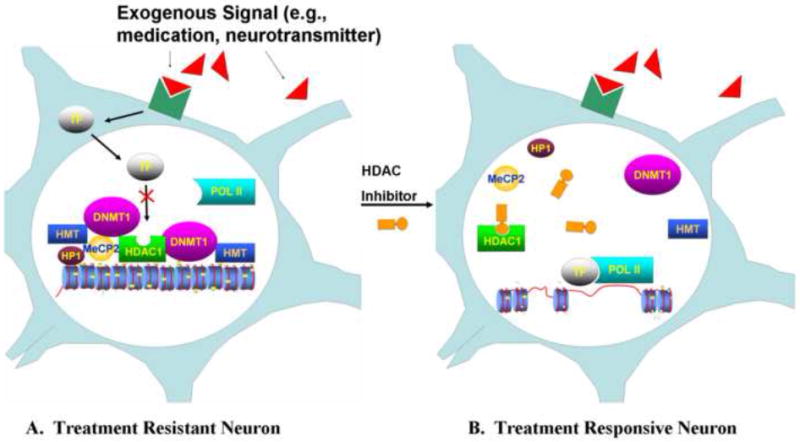
(A) Treatment Resistant Neuron. An exogenous signal (red) (e.g., neurotransmitter, medication) binds to its receptor (dark green) causing a transcription factor (TF) to translocate to the nucleus where it is prevented from serving as a platform for RNA polymerase II (POL II) by a restrictive chromatin state. Histone deacetylases, such as HDAC1, histone methyltransferases (HMT), DNA methyltransferases, such as DNMT1, and the repressor protein, HP1, cooperate both directly and indirectly through methyl-CpG domain binding proteins such as MeCP2, to induce this state. (B) Treatment Responsive Neuron. In this hypothetical model a histone deacetylase inhibitor (HDACi) prevents the deacetylation of histone proteins. This sets off a chain of events whereby the components of the restrictive chromatin state dissociate from the gene promoter region, chromatin structure is remodeled, and the promoter region in now more available for transcription factor binding. The transcription factor then serves as a platform for RNA polymerase II (POL II) binding. In this way, ‘softening’ chromatin around a gene promoter region makes expression levels more readily alterable by exogenous forces such as medications.
3. Epigenetics and Schizophrenia
3.1 GABAergic system, schizophrenia, and epigenetics
The evidence for epigenetic abnormalities being operant in schizophrenia in part comes from the study of schizophrenia candidate gene regulation, including GAD67 and reelin. A decrease in GAD67, an enzyme that catalyzes decarboxylation of glutamate to form GABA in chandelier type GABA interneurons, is thought to lead to asynchronized cortical activity and the working memory deficits observed in schizophrenia (Lewis et al., 2005). Reelin most likely plays a role in synaptic plasticity learning and memory formation. A deficit in reelin likely accounts for the decreased numbers of dendritic spines in postmortem brains of schizophrenia subjects (Guidotti et al., 2005).
Among the most consistent findings in all of schizophrenia research is the downregulation of these genes (Akbarian et al., 1995; Guidotti et al., 2000; Fatemi et al., 2005; Akbarian and Huang, 2006; Hashimoto et al., 2008). The promoters of both reelin and GAD67 reside within CpG islands making them especially prone to regulation through DNA methylation. In cultured NT2 neuronal precursor cells decreased expression of the reelin gene has been associated with increased promoter CpG island methylation (Chen et al., 2002), and decreased acetylated histone levels (Mitchell et al., 2005). In addition, cultured cell and animal model experiments have shown that DNMT inhibitors (Kundakovic et al., 2007), knocking down the expression of DNMT1 (Noh et al., 2005), and HDAC inhibitors result in increased reelin and GAD67 expression (Chen et al., 2002; Kundakovic et al., 2009).
3.2 One-carbon metabolism abnormalities
In the 1960’s and 1970’s clinical researchers performed studies in which schizophrenia patients were treated with L-methionine. Their hypothesis was that methionine, which gets converted to SAM in the body, would serve as a methyl donor to dopamine, thereby hastening its inactivation (Antun et al., 1971; Berlet et al., 1965). Surprisingly, 79% of patients treated with methionine experienced a worsening of symptoms (Wyatt et al., 1971).
Costa and colleagues hypothesized that the methionine-induced worsening of symptoms was because methionine was being converted into SAM, leading to increased DNA methylation and decreased transcription of schizophrenia GABAergic candidate genes, such as reelin and GAD67 (Costa et al., 2002). Since then data from experiments in which mice were administered L-methionine have supported this hypothesis (Tremolizzo et al., 2002; Dong et al., 2005, 2007). Interestingly, L-methionine treated mice exhibited many of the endophenotypes associated with schizophrenia, including: being less socially interactive, less habituation to an intruder, faster ppi, and impaired attention (Tremolizzo et al., 2002, 2005). Administration of the HDAC inhibitor and mood stabilizer, valproic acid (VPA), following L-methionine treatment, resulted in gene expression normalization and corrected several of the behavioral manifestations caused by L-methionine treatment (Tremolizzo et al., 2005).
Subsequent findings from several postmortem experiments have supported the increased DNA methylation hypothesis. Increased levels of SAM (Guidotti et al., 2007), and increased DNA methylation at the reelin promoter (Abdolmaleky et al., 2005; Grayson et al., 2005), SOX10 promoter (Iwamoto et al., 2005), and in female subjects at the MARLIN-1, Pbx1/Meis1, DTNBP1, and HCG9 promoters (Mill et al., 2008) have been reported in schizophrenia patient samples. As expected promoter hypermethylation of the reelin (Abdolmaleky et al., 2005; Grayson et al., 2005) and SOX10 promoters (Iwamoto et al., 2005) were associated with decreased transcription. However, others have not been able to replicate the reelin studies (Mill et al., 2008; Tochigi et al., 2008).
In further support of abnormal DNA methylation contributing to schizophrenia, higher DNMT1 (Veldic et al., 2005) and DNMT3a (Zhubi et al., 2009) mRNA expression have been reported in schizophrenia postmortem cortical GABAergic neurons. Further, increased DNMT1 expression was negatively correlated with decreased expression of GAD67 mRNA (Veldic et al., 2005), and elevated in areas of the brain where reelin is decreased (Veldic et al., 2004). DNMT3a has also been found to colocalize with GAD67 in postmortem brain samples, and increased in similar brain regions as DNMT1 (Zhubi et al., 2009).
Based on animal models it may be possible to surmise the effects of increased DNMT expression on the brain. Following brain ischemic injury DNA methylation increases. By contrast, transgenic DNMT1 deficient mice have been found to be resistant to mild ischemic damage (Endres et al., 2000). In other experiments, dorsal forebrain DNMT1 conditional knockouts introduced in differentiated excitatory cells resulted in increased expression of 1047 genes (6.1%) and decreased expression of 444 genes (2.6%) with no effects on cell survival (Golshani et al., 2005; Hutnick et al., 2009). These animals exhibited aberrant LTP and failed to develop somatosensory projection to the sensory cortex. Based on these studies it may be possible to hypothesize that increased DNMT1 and DNMT3a expression might result in pathology observed in schizophrenia such as reduced counts of several types of neurons, altered expression of relatively few genes, and abnormal synaptic plasticity and memory formation (Reif et al., 2006; Costa et al., 2002). Experiments clarifying the regulation of DNMT genes in the brain and animal overexpression models would greatly help clarify the role of DNMTs in the development of schizophrenia.
3.3 Histone modification abnormalities
Abnormalities in histone modifications have also been identified. Two recent postmortem studies have found elevated levels of HDAC1 in schizophrenia postmortem brain samples (Benes et al., 2008; Sharma et al., 2008). A correlation between higher levels of HDAC1 expression and reduced GAD67 expression has been reported (Sharma et al., 2008). In addition, in female schizophrenia postmortem brain samples lower H3K4 methylation levels at the GAD67 promoter with resultant decreased gene expression has been described (Huang H et al., 2007).
In animal models, non-specific HDAC inhibition has been shown to ameliorate neurodegenerative and cognitive phenotypes (Fischer et al., 2007), reduce ischemic damage (Kim et al., 2009), and enhance learning and memory tasks in even wild type mice. HDAC2 overexpression has been found to decrease dendritic spine density, synapse number, synaptic plasticity and memory formation (Guan et al., 2009). By contrast, si-RNA mediated HDAC1 knockdowns and pharmacological HDAC1 inhibition has been found to impair learning and increase cell death. HDAC1 expression is induced by neurotoxic stimuli and serves to protect against ischemic cell death and DNA damage (Kim et al., 2008). This may indicate that increased HDAC1 expression is a consequence of pathophysiological processes in schizophrenia. On the other hand, pharmacological inhibition of HDAC1 in cellular models results in increased GAD67 expression (Kundakovic et al., 2009), and its expression in postmortem brain samples is negatively correlated with GAD67 expression (Sharma et al., 2008). Taken together increased HDAC1 expression may be both a consequence of and contributor to the underlying pathology in schizophrenia. Parsing the roles and regulation of each of the HDACs could serve to shed additional light on the reported increased expression of HDAC1 in schizophrenia.
Based on the observations of reduced expression of several prominent schizophrenia candidate genes, increased DNA methylation at many of these genes, increased expression of several DNMTs and HDAC1, and reduced H3K4 methylation it would appear that a restrictive chromatin state leading to decreased expression of schizophrenia relevant genes is a parsimonious explanation for disease risk. However, there are indications that the primary abnormality is more complex. For example, decreased COMT promoter (Abdolmaleky et al., 2006) and in males decreased WDR18 promoter methylation have also been reported (Mill et al., 2008). Akbarian et al. (2005) found that high levels of methylated H3R17 and histone 3 phosporylated at serine 10 and acetylated at lysine 14, two signs indicative of ‘open’ chromatin, were associated with significantly less gene expression of several metabolic genes in schizophrenia relative to nonpsychiatric control postmortem brain samples. The same group also reported a lack of association between the restrictive modifications of methylated H3K27 and DNA methylation at the GAD67 promoter in schizophrenia. This was not found in nonpsychiatric controls (Huang and Akbarian, 2007). These findings may indicate schizophrenia subjects are afflicted with an overall dysfunction and lack of coordination in epigenetic machinery, rather than simply overly restrictive chromatin.
3.4 Measuring Epigenetic Parameters in Real Clinical Time
3.4.1 Baseline abnormalities in chromatin structure
Because in psychiatry the organ of interest from living subjects is largely inaccessible the search for peripheral biomarkers has become increasingly appealing. Fraga et al. (2005) found that peripheral blood mononuclear cells (PBMC) provide a reliable means of demonstrating differences on global levels of DNA methylation, acetylated histone 3, and acetylated histone 4 between individuals over a 12 week period. Other studies have shown that peripheral markers are able to discern chromatin structure differences in twins discordant for mental illness (Kuratomi et al., 2008; Tsujita et al., 1998), as well as demonstrate similarities in epigenetic parameters among individuals affected by the same illness (Kuratomi et al., 2008). Petronis et al. (2003) found in two sets of twins, one concordant for schizophrenia and the other discordant, that the affected twin from the discordant pair had a dopamine 2 receptor promoter DNA methylation pattern more similar to the affected concordant twins than to his unaffected twin. Others have reported increased affinity of S-adenosyltransferase (MAT), the enzyme that converts methionine into SAM, for methionine in both red blood cells and frontal cortical postmortem brain samples from schizophrenia subjects (Gomes-Trolin, et al., 1998). S-adenosylhomocyteine (SAH), the SAM metabolite, has also been found to be increased in lymphocytes from schizophrenia subjects (Bromberg et al., 2008).
More recent studies have also indicated peripheral abnormalities in epigenetic mechanisms. We have found reduced levels of the ‘open’ chromatin mark, acetylated histone 3, in PBMC from schizophrenia subjects compared to nonpsychiatric controls (Gavin et al., 2008) and bipolar subjects (Gavin et al., 2009a), as well as elevated levels of the ‘closed’ chromatin mark, dimethylated H3K9 (Gavin et al., 2009b). Recently, Zhubi et al. (2009) reported elevated DNMT1 and DNMT3a expression in PBMC from schizophrenia subjects compared to nonpsychiatric controls.
3.4.2 Abnormal epigenetic mechanisms in schizophrenia
As previously stated, several studies have demonstrated abnormalities in the coordination of epigenetic processes in schizophrenia. Based on the hypothesis that these dynamic processes may not be demonstrable through use of static measures in postmortem brain samples it has become necessary to develop models capable of measuring changes in real clinical time. Increasing evidence suggests that nucleated blood cells may serve as a viable approach for revealing epigenetic processes in a prospective, hypothesis-driven manner.
Issidorides and colleagues (1975) reported abnormally condensed nucleohistone staining patterns in schizophrenia patient neutrophils, normalized using the antipsychotic pimozide. Later studies by Kosower et al. (1995) found lymphocytes treated with a DNMT inhibitor exhibited a significantly smaller extension of the 1q heterochromatic C-band in schizophrenia subjects compared to nonpsychiatric controls.
More recently, we conducted a study in which schizophrenia and bipolar subjects were clinically treated for four weeks with VPA. It was found that this treatment induced significantly less histone 3 and histone 4 acetylation in schizophrenia subjects’ PBMCs (Sharma et al., 2006).
As an extension of these studies we developed an in vitro PBMC assay in order to perform similar studies unencumbered by the limitations of a clinical study. Namely, by developing an in vitro assay we were protected from the unforeseeable or undisclosed changes in a patient’s physiology such as medication noncompliance, substance use, environmental exposures such as infections and diet, hormonal fluctuations, and differences in metabolism of HDAC inhibitors. Further, in vivo studies are limited by the fact that few known chromatin altering agents are approved for use in psychiatric patients. Finally, in vivo comparisons to nonpsychiatric controls would not be ethical (Gavin et al., In Press).
In PBMC cultures, treated for 24 hours with the HDAC inhibitor Trichostatin A (TSA), there were more modest changes in acetylated histone 3 and dimethylated H3K9 levels in cells from schizophrenia compared with nonpsychiatric control subjects (Gavin et al., 2008, 2009b). Additionally, there was a significant correlation between TSA-induced increases in GAD67 expression and acetylated histone 3 levels among nonpsychiatric subjects, but not among schizophrenia subjects (Gavin et al., 2008). This may imply, as the results of other studies suggest (Huang and Akbarian, 2007), that in schizophrenia the abnormality exists in the coordination of epigenetic processes.
4. Therapeutics
Because epigenetic mechanisms are amenable to pharmacological intervention the potential for developing agents which target chromatin remodeling and DNA methylation is profound. Preliminary data suggests that subjects with higher baseline PBMC levels of acetylated histone 3 are more likely to respond to VPA treatment both in terms of mania and psychotic symptoms (Gavin et al., 2009a). Therefore, one might predict that a high acetylated histone 3 level, as was found in bipolar subjects (Gavin et al., 2009a), could be used as a predictor of HDAC inhibitor therapeutic response. This would conform to clinical observation where VPA is a known treatment of bipolar disorder.
On the other hand, there are several indicators that some schizophrenia patients are characterized by lower levels of acetylated histone 3 (Gavin et al., 2008), higher dimethylated H3K9 (Gavin et al., 2009b), and chromatin that is less responsive to HDAC inhibitors (Sharma et al., 2006; Gavin et al., 2008, 2009a, 2009b). Therefore, a normalization of restrictive chromatin, based on these types of measures, might be beneficial. This is based on the hypothesis that restrictive chromatin in schizophrenia results in less access of candidate gene promoters to other stimuli (Fig. 1A). Perhaps by making gene promoters more available using HDAC inhibitors, HMT inhibitors, histone demethylase inhibitors, DNA methyltransferase inhibitors, or DNA demethylase inducers it may be possible to ‘soften’ chromatin surrounding candidate gene promoters, thereby facilitating the gene expression altering effects of antipsychotic medications (Fig. 1B) (Sharma, 2005).
As a caveat to these possibilities it must be noted that schizophrenia is a life-long illness with associated cytoarchitectural differences. Therefore the possibility of completely reversing the illness in the fully developed brain seems somewhat remote. On the other hand, agents that manipulate epigenetic factors may be unique in their abilities to reverse what appear to be entrenched deficits. For example, Weaver et al. (2004) reported that rat pups raised by low nurturing mothers (low LG-ABN) exhibited exaggerated fear responses, had greater HPA axis responses to stress, and decreased expression of the glucocorticoid receptor exon 17 as adults. TSA was able to reverse all of these effects (Weaver et al., 2004). It has also been shown in animal models that following an ischemic event, HDAC inhibitors can minimize damage by decreasing brain infarct volume (Kim et al., 2007), and even increase neurogenesis (Kim et al., 2009). Therefore, while these agents will likely not be curative they may potentially slow disease progression and perhaps reverse some of the cytoarchitectural abnormalities present in schizophrenia.
4.1 HDAC inhibitors
Considering both reelin and GAD67 expression have been shown to be upregulated using HDAC inhibitors, it is possible that a disproportionate number of schizophrenia candidate genes are affected by abnormalities in histone acetylation. There is evidence to support this hypothesis based on elevated levels of HDAC1 in schizophrenia postmortem brain samples (Benes et al., 2008; Sharma et al., 2008), global reductions in acetylated histones at baseline in schizophrenia subjects’ PBMCs (Gavin et al., 2008, 2009a), and a decreased ability to increase these levels with HDAC inhibitors (Sharma et al., 2006; Gavin et al., 2008). The therapeutic selectivity of these drugs is further increased by the fact that repression of transcription by deacetylated histones is specific to cell types and tissues and perhaps even within different brain regions (Simonini et al., 2006). Also, individual HDAC inhibitors have been shown to preferentially inhibit particular classes of HDACs (Hu et al., 2003)
There are five types of HDAC inhibitors based on structure: short chain fatty acids (e.g., sodium butyrate, phenylbutyrate, and VPA); the hydroxamic acids (e.g., TSA, suberoylanilide hydroxamic acid (SAHA), panobinostat (LBH589), belinostat (PXD101)); the epoxyketones (e.g., trapoxin); the cyclic peptide (e.g., romidepsin (FK228)); and the benzamides (e.g., MS-275 and MGCD0103). All four classes inhibit class I (HDAC1, 2, 3, and 8) and II (HDAC4, 5, 6, 7, 9, and 10) HDACs, although the benzamides preferentially inhibit class I HDACs (Acharya et al., 2005; Szyf, 2009; Atadja, 2009). Several of the hydroxamic acids (TSA and SAHA (Hockly et al., 2003; Faraco et al., 2006; Romieu et al., 2008)), the short chain fatty acids, and the benzamides are known to cross the blood brain barrier (BBB) in animal models (Kumar et al., 2005; Ryu et al., 2005; Simonini et al., 2006). Only VPA is known to efficiently cross the BBB in humans (Gopaul et al., 2003). Phenylbutyrate, SAHA, VPA, MS-275, MGCD0103 have been shown to be relatively well-tolerated in clinical populations (Hogarth et al., 2007; Luu et al., 2008; Hauschild et al., 2008). VPA and SAHA are currently FDA approved, VPA for bipolar disorder and epilepsy and SAHA for T-cell lymphoma. Phenylbutyrate, MS-275, and MGCD0103 are currently undergoing clinical trials for Huntington’s disease and various cancers.
4.2 HMT Inhibitors/Histone demethylase Inhibitors
Abnormalities in the repressive histone marks dimethylated H3K9 (Gavin et al., 2009b) and methylated H3K27 (Huang H et al., 2007) have been found in schizophrenia, as has an abnormality in the level of the open chromatin mark methylated H3K4 (Huang and Akbarian, 2007). Recently, two HMT inhibitors have been described (Greiner et al., 2005; Kubicek et al., 2007). However, very little is known about their in vivo effects.
Histone demethylase inhibitors which specifically increase the levels of the open chromatin mark methylated H3K4 have been described. A recent study discovered that novel biguanide and bisguanidine polyamine analogues are capable of inhibiting the H3K4 demethylating enzyme (Huang Y et al., 2007). Interestingly, the antidepressants tranylcypromine and phenelzine have been shown to have H3K4 histone demethylase inhibitory properties (Lee et al., 2006). In the literature there are studies showing beneficial effects of MAOIs in schizophrenia (Siris et al., 1978). In one study, subjects treated with tranylcypromine plus a typical antipsychotic showed particular improvement in negative symptoms (Bucci et al., 1987). The idea that chromatin altering medications may serve as a treatment for primary negative symptoms as defined by the deficit syndrome, which generally have been shown to be refractory to traditional antipsychotic treatment is an attractive one (Kirkpatrick et al., 2001).
4.3 DNMT inhibitors
DNMT inhibitors include agents such as doxorubicin, 5-aza-2’-deoxycytidine (AZA), zebularine (ZEB), and procainamide. These drugs have been shown to increase the expression of GAD67 and reelin in cultured cell lines while decreasing the expression of DNMT1 (Kundakovic et al., 2007). Therefore, there is the potential for DNMT inhibitors to normalize these abnormalities. Unfortunately, these agents have several disadvantages. AZA and ZEB are the most potent DNMT1 inhibitors, but do not cross the BBB and are active only during the S phase of the cell cycle, making them ineffective for post-mitotic cells (Yoo et al., 2004).
Recently, Satta et al. (2009) reported that nicotine decreases DNMT1 expression in GABAergic mouse neurons leading to decreased methylation at the GAD67 promoter and increased GAD67 protein expression. This effect was found to occur as a result of nicotinic receptor agonism. These improve cognitive functioning in schizophrenia (Olincy et al., 2006; Barr et al., 2008; Jubelt et al., 2008), and may suggest in part why 80% of schizophrenia patients use tobacco (de Leon, 1996). The specific nicotinic receptors that mediate this improved cognition have yet to be established. However, an alpha7-nicotine receptor agonist has been shown in small studies to improve cognition in schizophrenia subjects (Olincy et al., 2006).
4.4 DNA Demethylase Inducers
The means of active mammalian DNA demethylation (as opposed to the passive loss of methyl groups as occurs during DNA replication) has been a subject of much controversy over the last 20 years. The paucity of replicable results between studies and methodological errors has resulted in slow progress in characterizing the components which mediate active DNA demethylation.
Although the mechanism of active DNA demethylation remains elusive there have been several reports that it is possible to pharmacologically induce this process. Several HDAC inhibitors have been suggested to be capable of doing this (Weaver et al., 2004; Detich et al., 2003; Dong et al., 2008), perhaps by allowing greater access of the putative DNA demethylase to gene promoters (Detich et al., 2003; Dong et al., 2008). One recent article found clozapine and sulpiride but not haloperidol or olanzapine to be able to induce demethylation at the reelin promoter (Dong et al., 2008). The mechanisms of these effects have yet to be elucidated.
5. Conclusion
In summary, epigenetic mechanisms are becoming increasingly understood as are their relevance to disease. Cell lines, animal models, postmortem brain studies, and clinical studies all indicate the existence of epigenetic abnormalities in schizophrenia. Finally, as these abnormalities continue to be characterized, and those schizophrenia subjects most afflicted by these abnormalities are identified, there is the potential to develop new medications that target this system.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdolmaleky H, Smith C, et al. Methylomics in psychiatry: Modulation of gene-environment interactions may be through DNA methylation. American Journal of Medical Genetics. 2004;127B:51–9. doi: 10.1002/ajmg.b.20142. [DOI] [PubMed] [Google Scholar]
- Abdolmaleky H, Cheng K, et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. American Journal of Medical Genetics. 2005;134B:60–6. doi: 10.1002/ajmg.b.30140. [DOI] [PubMed] [Google Scholar]
- Abdolmaleky H, Cheng K, et al. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Human Molecular Genetics. 2006;15:3132–45. doi: 10.1093/hmg/ddl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya M, Sparreboom A, et al. Rational development of histone deacetylase inhibitors as anticancer agents: a review. Molecular Pharmacology. 2005;68:917–32. doi: 10.1124/mol.105.014167. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Kim J, et al. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Archives of General Psychiatry. 1995;52:258–66. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Ruehl M, et al. Chromatin alterations associated with down-regulated metabolic gene expression in the prefrontal cortex of subjects with schizophrenia. Archives of General Psychiatry. 2005;62:829–40. doi: 10.1001/archpsyc.62.8.829. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Huang H. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Research Reviews. 2006;52:293–304. doi: 10.1016/j.brainresrev.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Huang H. Epigenetic regulation in human brain-focus on histone lysine methylation. Biological Psychiatry. 2009;65:198–203. doi: 10.1016/j.biopsych.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antun F, Burnett G, et al. The effects of L-methionine (without MAOI) in schizophrenia. Journal of Psychiatric Research. 1971;8:63–71. doi: 10.1016/0022-3956(71)90009-4. [DOI] [PubMed] [Google Scholar]
- Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Letters. 2009;280:233–41. doi: 10.1016/j.canlet.2009.02.019. [DOI] [PubMed] [Google Scholar]
- Bachman K, Rountree M, et al. Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. Journal of Biological Chemistry. 2001;276:32282–7. doi: 10.1074/jbc.M104661200. [DOI] [PubMed] [Google Scholar]
- Barr R, Culhane M, et al. The effects of transdermal nicotine on cognition in nonsmokers with schizophrenia and nonpsychiatric controls. Neuropsychopharmacology. 2008;33:480–90. doi: 10.1038/sj.npp.1301423. [DOI] [PubMed] [Google Scholar]
- Benes F, Lim B, et al. Circuitry-based gene expression profiles in GABA cells of the trisynaptic pathway in schizophrenics versus bipolars. Proceedings of the National Academy of Sciences U S A. 2008;105:20935–40. doi: 10.1073/pnas.0810153105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlet H, Matsumoto K, et al. Biochemical correlates of behavior in schizophrenic patients. Schizophrenic patients receiving tryptophan and methionine or methionine together with a monoamine oxidase inhibitor. Archives of General Psychiatry. 1965;13:521–31. doi: 10.1001/archpsyc.1965.01730060039006. [DOI] [PubMed] [Google Scholar]
- Bromberg A, Levine J, et al. No association between global leukocyte DNA methylation and homocysteine levels in schizophrenia patients. Schizophrenia Research. 2008;101:50–7. doi: 10.1016/j.schres.2008.01.009. [DOI] [PubMed] [Google Scholar]
- Bucci L. The negative symptoms of schizophrenia and the monoamine oxidase inhibitors. Psychopharmacology (Berl) 1987;91:104–8. doi: 10.1007/BF00690936. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sharma R, et al. On the epigenetic regulation of the human reelin promoter. Nucleic Acids Research. 2002;30:2930–9. doi: 10.1093/nar/gkf401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa E, Davis J, et al. The heterozygote reeler mouse as a model for the development of a new generation of antipsychotics. Current Opinion in Pharmacology. 2002;2:56–62. doi: 10.1016/s1471-4892(01)00121-7. [DOI] [PubMed] [Google Scholar]
- de Leon J. Smoking and vulnerability for schizophrenia. Schizophrenia Bulletin. 1996;22:405–9. doi: 10.1093/schbul/22.3.405. [DOI] [PubMed] [Google Scholar]
- Detich N, Bovenzi V, et al. Valproate induces replication-independent active DNA demethylation. Journal of Biological Chemistry. 2003;278:27586–92. doi: 10.1074/jbc.M303740200. [DOI] [PubMed] [Google Scholar]
- Dong E, Agis-Balboa R, et al. Reelin and glutamic acid decarboxylase67 promoter remodeling in an epigenetic methionine-induced mouse model of schizophrenia. Proceedings of the National Academy of Sciences U S A. 2005;102:12578–83. doi: 10.1073/pnas.0505394102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong E, Guidotti A, et al. Histone hyperacetylation induces demethylation of reelin and 67-kDa glutamic acid decarboxylase promoters. Proceedings of the National Academy of Sciences U S A. 2007;104:4676–81. doi: 10.1073/pnas.0700529104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong E, Nelson M, et al. Clozapine and sulpiride but not haloperidol or olanzapine activate brain DNA demethylation. Proceedings of the National Academy of Sciences U S A. 2008;105:13614–9. doi: 10.1073/pnas.0805493105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger G, Liang G, et al. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–63. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- Endres M, Meisel A, et al. DNA methyltransferase contributes to delayed ischemic brain injury. Journal of Neuroscience. 2000;20:3175–81. doi: 10.1523/JNEUROSCI.20-09-03175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epsztejn-Litman S, Feldman N, et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nature Structural and Molecular Biology. 2008;15:1176–83. doi: 10.1038/nsmb.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraco G, Pancani T, et al. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Molecular Pharmacology. 2006;70:1876–84. doi: 10.1124/mol.106.027912. [DOI] [PubMed] [Google Scholar]
- Fatemi S, Stary J, et al. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophrenia Research. 2005;72:109–22. doi: 10.1016/j.schres.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, et al. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–82. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Fraga M, Ballestar E, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences U S A. 2005;102:10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, Burgers W, et al. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nature Genetics. 2000;24:88–91. doi: 10.1038/71750. [DOI] [PubMed] [Google Scholar]
- Fuks F, Burgers W, et al. Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. The EMBO Journal. 2001;20:2536–44. doi: 10.1093/emboj/20.10.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, Hurd P, et al. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Research. 2003a;31:2305–12. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, Hurd P, et al. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. Journal of Biological Chemistry. 2003b;278:4035–40. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- Gavin D, Kartan S, et al. Reduced baseline acetylated histone 3 levels, and a blunted response to HDAC inhibition in lymphocyte cultures from schizophrenia subjects. Schizophrenia Research. 2008;103:330–2. doi: 10.1016/j.schres.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin D, Kartan S, et al. Histone deacetylase inhibitors and candidate gene expression: An in vivo and in vitro approach to studying chromatin remodeling in a clinical population. Journal of Psychiatric Research. 2009a;43:870–6. doi: 10.1016/j.jpsychires.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Gavin D, Rosen C, et al. Dimethylated lysine 9 of histone 3 is elevated in schizophrenia and exhibits a divergent response to histone deacetylase inhibitors in lymphocyte cultures. Journal of Psychiatry and Neuroscience. 2009b;34:232–7. [PMC free article] [PubMed] [Google Scholar]
- Gavin D, Sharma R. Chromatin from Peripheral Blood Mononuclear Cells as Biomarkers for Epigenetic Abnormalities in Schizophrenia. Cardiovascular Psychiatry and Neurology. 2009 doi: 10.1155/2009/409562. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golshani P, Hutnick L, et al. Conditional Dnmt1 deletion in dorsal forebrain disrupts development of somatosensory barrel cortex and thalamocortical long-term potentiation. Thalamus & Related Systems. 2005;3:227–233. doi: 10.1017/S1472928807000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes-Trolin C, Yassin M, et al. Erythrocyte and brain methionine adenosyltransferase activities in patients with schizophrenia. Journal of Neural Transmission. 1998;105:1293–305. doi: 10.1007/s007020050132. [DOI] [PubMed] [Google Scholar]
- Gopaul V, Tang W, et al. Amino acid conjugates: metabolites of 2- propylpentanoic acid (valproic acid) in epileptic patients. Drug Metabolism and Disposition. 2003;31:114–21. doi: 10.1124/dmd.31.1.114. [DOI] [PubMed] [Google Scholar]
- Gottesman I. Schizophrenia Genesis: The Origins of Madness. W H Freeman; New York: 1991. [Google Scholar]
- Gottesman I, Shields J. Schizophrenia: The Epigenetic Puzzle. Cambridge University Press; Cambridge, United Kingdom: 1982. [Google Scholar]
- Grayson D, Jia X, et al. Reelin promoter hypermethylation in schizophrenia. Proceedings of the National Academy of Sciences U S A. 2005;102:9341–6. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner D, Bonaldi T, et al. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nature Chemical Biology. 2005;1:143–5. doi: 10.1038/nchembio721. [DOI] [PubMed] [Google Scholar]
- Guan J, Haggarty S, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti A, Auta J, et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Archives of General Psychiatry. 2000;57:1061–9. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- Guidotti A, Auta J, et al. GABAergic dysfunction in schizophrenia: new treatment strategies on the horizon. Psychopharmacology (Berl) 2005;180:191–205. doi: 10.1007/s00213-005-2212-8. [DOI] [PubMed] [Google Scholar]
- Guidotti A, Ruzicka W, et al. S-adenosyl methionine and DNA methyltransferase-1 mRNA overexpression in psychosis. Neuroreport. 2007;18:57–60. doi: 10.1097/WNR.0b013e32800fefd7. [DOI] [PubMed] [Google Scholar]
- Harlap S, Perrin M, et al. Schizophrenia and birthplace of paternal and maternal grandfather in the Jerusalem perinatal cohort prospective study. Schizophrenia Research. 2009;111:23–31. doi: 10.1016/j.schres.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Arion D, et al. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Molecular Psychiatry. 2008;13:147–61. doi: 10.1038/sj.mp.4002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauschild A, Trefzer U, et al. Multicenter phase II trial of the histone deacetylase inhibitor pyridylmethyl-N-{4-[(2-aminophenyl)-carbamoyl]-benzyl}-carbamate in pretreated metastatic melanoma. Melanoma Research. 2008;18:274–8. doi: 10.1097/CMR.0b013e328307c248. [DOI] [PubMed] [Google Scholar]
- Hockly E, Richon V, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proceedings of the National Academy of Sciences U S A. 2003;100:2041–6. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogarth P, Lovrecic L, et al. Sodium phenylbutyrate in Huntington’s disease: a dose-finding study. Movement Disorders. 2007;22:1962–4. doi: 10.1002/mds.21632. [DOI] [PubMed] [Google Scholar]
- Hong L, Schroth G, et al. Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 “tail” to DNA. Journal of Biological Chemistry. 1993;268:305–14. [PubMed] [Google Scholar]
- Hu E, Dul E, et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. Journal of Pharmacology and Experimental Therapeutics. 2003;307:720–8. doi: 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- Huang H, Akbarian S. GAD1 mRNA expression and DNA methylation in prefrontal cortex of subjects with schizophrenia. PLoS ONE. 2007;2:e809. doi: 10.1371/journal.pone.0000809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Matevossian A, et al. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. Journal of Neuroscience. 2007;27:11254–62. doi: 10.1523/JNEUROSCI.3272-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Greene E, et al. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proceedings of the National Academy of Sciences U S A. 2007;104:8023–8. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutnick L, Golshani P, et al. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Human Molecular Genetics. 2009;18:2875–88. doi: 10.1093/hmg/ddp222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issidorides M, Stefanis C, et al. Altered chromatin ultrastructure in neutrophils of schizophrenics. Nature. 1975;258:612–4. doi: 10.1038/258612a0. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Bundo M, et al. DNA methylation status of SOX10 correlates with its downregulation and oligodendrocyte dysfunction in schizophrenia. Journal of Neuroscience. 2005;25:5376–81. doi: 10.1523/JNEUROSCI.0766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs S, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science. 2002;295:2080–3. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- Jones P, Veenstra G, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nature Genetics. 1998;19:187–91. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Jubelt L, Barr R, et al. Effects of transdermal nicotine on episodic memory in non-smokers with and without schizophrenia. Psychopharmacology (Berl) 2008;199:89–98. doi: 10.1007/s00213-008-1133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Rowe M, et al. Histone deacetylase inhibitors exhibit anti- inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. Journal of Pharmacology and Experimental Therapeutics. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- Kim D, Frank C, et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron. 2008;60:803–17. doi: 10.1016/j.neuron.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Leeds P, et al. The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. Journal of Neurochemistry. 2009;110:1226–40. doi: 10.1111/j.1471-4159.2009.06212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick B, Buchanan R, et al. A separate disease within the syndrome of schizophrenia. Archives of General Psychiatry. 2001;58:165–71. doi: 10.1001/archpsyc.58.2.165. [DOI] [PubMed] [Google Scholar]
- Kosower N, Gerad L, et al. Constitutive heterochromatin of chromosome 1 and Duffy blood group alleles in schizophrenia. American Journal of Medical Genetics. 1995;60:133–8. doi: 10.1002/ajmg.1320600209. [DOI] [PubMed] [Google Scholar]
- Kubicek S, O’Sullivan R, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Molecular Cell. 2007;25:473–81. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Kumar A, Choi K, et al. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–14. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Kundakovic M, Chen Y, et al. DNA methyltransferase inhibitors coordinately induce expression of the human reelin and glutamic acid decarboxylase 67 genes. Molecular Pharmacology. 2007;71:644–53. doi: 10.1124/mol.106.030635. [DOI] [PubMed] [Google Scholar]
- Kundakovic M, Chen Y, et al. The reelin and GAD67 promoters are activated by epigenetic drugs that facilitate the disruption of local repressor complexes. Molecular Pharmacology. 2009;75:342–54. doi: 10.1124/mol.108.051763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuratomi G, Iwamoto K, et al. Aberrant DNA methylation associated with bipolar disorder identified from discordant monozygotic twins. Molecular Psychiatry. 2008;13:429–41. doi: 10.1038/sj.mp.4002001. [DOI] [PubMed] [Google Scholar]
- Lachner M, O’Carroll D, et al. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–20. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- Lee M, Wynder C, et al. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chemical Biology. 2006;13:563–7. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Lehnertz B, Ueda Y, et al. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Current Biology. 2003;13:1192–200. doi: 10.1016/s0960-9822(03)00432-9. [DOI] [PubMed] [Google Scholar]
- Lewis D, Hashimoto T, et al. Cortical inhibitory neurons and schizophrenia. Nature Reviews Neuroscience. 2005;6:312–24. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Luu T, Morgan R, et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: a California Cancer Consortium study. Clinical Cancer Research. 2008;14:7138–42. doi: 10.1158/1078-0432.CCR-08-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mill J, Tang T, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. American Journal of Human Genetics. 2008;82:696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell C, Chen Y, et al. Histone deacetylase inhibitors decrease reelin promoter methylation in vitro. Journal of Neurochemistry. 2005;93:483–92. doi: 10.1111/j.1471-4159.2005.03040.x. [DOI] [PubMed] [Google Scholar]
- Noh J, Sharma R, et al. DNA methyltransferase 1 regulates reelin mRNA expression in mouse primary cortical cultures. Proceedings of the National Academy of Sciences U S A. 2005;102:1749–54. doi: 10.1073/pnas.0409648102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olincy A, Harris J, et al. Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Archives of General Psychiatry. 2006;63:630–8. doi: 10.1001/archpsyc.63.6.630. [DOI] [PubMed] [Google Scholar]
- Peterson C, Laniel M. Histones and histone modifications. Current Biology. 2004;14:R546–51. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Petronis A, Gottesman I, et al. Monozygotic twins exhibit numerous epigenetic differences: clues to twin discordance? Schizophrenia Bulletin. 2003;29:169–78. doi: 10.1093/oxfordjournals.schbul.a006988. [DOI] [PubMed] [Google Scholar]
- Petronis A. The origin of schizophrenia: genetic thesis, epigenetic antithesis, and resolving synthesis. Biological Psychiatry. 2004;55:965–70. doi: 10.1016/j.biopsych.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Reif A, Fritzen S, et al. Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Molecular Psychiatry. 2006;11:514–22. doi: 10.1038/sj.mp.4001791. [DOI] [PubMed] [Google Scholar]
- Robertson K, Ait-Si-Ali S, et al. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nature Genetics. 2000;25:338–42. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- Romieu P, Host L, et al. Histone deacetylase inhibitors decrease cocaine but not sucrose self-administration in rats. Journal of Neuroscience. 2008;28:9342–8. doi: 10.1523/JNEUROSCI.0379-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rountree M, Bachman K, et al. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nature Genetics. 2000;25:269–77. doi: 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- Ryu H, Smith K, et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. Journal of Neurochemistry. 2005;93:1087–98. doi: 10.1111/j.1471-4159.2005.03077.x. [DOI] [PubMed] [Google Scholar]
- Satta R, Maloku E, et al. Nicotine decreases DNA methyltransferase 1 expression and glutamic acid decarboxylase 67 promoter methylation in GABAergic interneurons. Proceedings of the National Academy of Sciences U S A. 2008;105:16356–61. doi: 10.1073/pnas.0808699105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R. Schizophrenia, epigenetics and ligand-activated nuclear receptors: a framework for chromatin therapeutics. Schizophrenia Research. 2005;72:79–90. doi: 10.1016/j.schres.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Sharma R, Rosen C, et al. Valproic acid and chromatin remodeling in schizophrenia and bipolar disorder: preliminary results from a clinical population. Schizophrenia Research. 2006;88:227–31. doi: 10.1016/j.schres.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Sharma R, Grayson D, et al. Histone deactylase 1 expression is increased in the prefrontal cortex of schizophrenia subjects: analysis of the National Brain Databank microarray collection. Schizophrenia Research. 2008;98:111–7. doi: 10.1016/j.schres.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonini M, Camargo L, et al. The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases. Proceedings of the National Academy of Sciences U S A. 2006;103:1587–92. doi: 10.1073/pnas.0510341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siris S, van Kammen D, et al. Use of antidepressant drugs in schizophrenia. Archives of General Psychiatry. 1978;35:1368–77. doi: 10.1001/archpsyc.1978.01770350094009. [DOI] [PubMed] [Google Scholar]
- Szyf M. The DNA methylation machinery as a target for anticancer therapy. Pharmacology and Therapeutics. 1996;70:1–37. doi: 10.1016/0163-7258(96)00002-2. [DOI] [PubMed] [Google Scholar]
- Szyf M. Epigenetics, DNA methylation, and chromatin modifying drugs. Annual Review of Pharmacology and Toxicology. 2009;49:243–63. doi: 10.1146/annurev-pharmtox-061008-103102. [DOI] [PubMed] [Google Scholar]
- Takai D, Jones P. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proceedings of the National Academy of Sciences U S A. 2002;99:3740–5. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tochigi M, Iwamoto K, et al. Methylation status of the reelin promoter region in the brain of schizophrenic patients. Biological Psychiatry. 2008;63:530–3. doi: 10.1016/j.biopsych.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Tremolizzo L, Carboni G, et al. An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proceedings of the National Academy of Sciences U S A. 2002;99:17095–100. doi: 10.1073/pnas.262658999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremolizzo L, Doueiri M, et al. Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biological Psychiatry. 2005;57:500–9. doi: 10.1016/j.biopsych.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Tsujita T, Niikawa N, et al. Genomic discordance between monozygotic twins discordant for schizophrenia. American Journal of Psychiatry. 1998;155:422–4. doi: 10.1176/ajp.155.3.422. [DOI] [PubMed] [Google Scholar]
- Turner B. Cellular memory and the histone code. Cell. 2002;111:285–91. doi: 10.1016/s0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- Veldic M, Caruncho H, et al. DNA-methyltransferase 1 mRNA is selectively overexpressed in telencephalic GABAergic interneurons of schizophrenia brains. Proceedings of the National Academy of Sciences U S A. 2004;101:348–53. doi: 10.1073/pnas.2637013100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldic M, Guidotti A, et al. In psychosis, cortical interneurons overexpress DNA-methyltransferase 1. Proceedings of the National Academy of Sciences U S A. 2005;102:2152–7. doi: 10.1073/pnas.0409665102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace J, Orr-Weaver T. Replication of heterochromatin: insights into mechanisms of epigenetic inheritance. Chromosoma. 2005;114:389–402. doi: 10.1007/s00412-005-0024-6. [DOI] [PubMed] [Google Scholar]
- Warren K, McCully C, et al. Plasma and cerebrospinal fluid pharmacokinetics of the histone deacetylase inhibitor, belinostat (PXD101), in non-human primates. Cancer Chemotherapy and Pharmacology. 2008;62:433–7. doi: 10.1007/s00280-007-0622-5. [DOI] [PubMed] [Google Scholar]
- Weaver I, Cervoni N, et al. Epigenetic programming by maternal behavior. Nature Neuroscience. 2004;7:847–54. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Wyatt R, Termini B, et al. Part II. Sleep Studies. Schizophrenia Bulletin. 1971;1:45–66. [Google Scholar]
- Yoo C, Cheng J, et al. Zebularine: a new drug for epigenetic therapy. Biochemistry Society Transactions. 2004;32:910–2. doi: 10.1042/BST0320910. [DOI] [PubMed] [Google Scholar]
- Zhubi A, Veldic M, et al. An upregulation of DNA-methyltransferase 1 and 3a expressed in telencephalic GABAergic neurons of schizophrenia patients is also detected in peripheral blood lymphocytes. Schizophrenia Research. 2009;111:115–22. doi: 10.1016/j.schres.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]