

Abstract
Research on depression and antidepressant drugs is necessary, as many patients display poor response to therapy. Different symptomatic and pathophysiological features have been proposed as end points of the depressive phenotype and of the antidepressant action, including anhedonia, depressed mood, alterations in morphology and activity of some brain areas (amygdala, nucleus accumbens, hippocampus, prefrontal cortex and cingulate cortex), modifications in the connectivity between brain structures, changes in neurotransmitters (serotonin, noradrenaline, glutamate and neuropeptides), brain plasticity (neurogenesis, neurotrophins) and abnormal function of the hypothalamic-pituitary adrenal axis. However, few models have been proposed to describe how these end points could induce the depressive phenotype and are involved in the mechanism of action of antidepressants. Here we propose a connectionist-inspired network of depression and antidepressant action, in which the different aetiological factors participating in the release of a depressive episode are represented by input nodes, the different symptomatic as well as pathophysiological end points are represented by an intermediate layer, and the onset of depression or of comorbid disease is represented by the output node. The occurrence of depression and the mechanism of the antidepressant action thus depend upon the weight of the interactions between the different end points, none of them being per se crucial to the onset of a depressive phenotype or to the antidepressant action. This model is heuristic to draw future lines of research concerning new antidepressant therapies, designing new animal models of depression and for a better understanding of the depressive pathology and of its comorbid pathology such as anxiety disorders.
Keywords: antidepressant, hypothalamic-pituitary adrenal axis, neuroplasticity, glutamate, serotonin, hippocampus, prefrontal cortex, amygdala, cingulate cortex, depression
Introduction
The lifetime prevalence for major depression is estimated as high as 16.2% in the USA (Greenberg et al., 2003; Kessler et al., 2003) and according to the World Health Organization, it will be the second most prevalent cause of illness-induced disability by 2020 (Murray and Lopez, 1997). The nosography of this disease encompasses various symptoms including anhedonia, depressed mood, fatigue, increased stress sensitivity, thoughts of worthlessness, inappropriate guilt, helplessness, apathy, shift towards negative emotions (sadness, emotional blunting, irritability and anxiety), cognitive alterations (impaired working memory, bias towards negative stimuli), body weight and sleep pattern abnormalities (see Table 1). Anhedonia and depressed mood are considered as core symptoms present in all patients, while other alterations vary among patients, so that the disease displays high symptomatic heterogeneity. This symptomatic variability sometimes raises the question of the construct validity of depression as a relevant entity, related to a monolithic pattern of biological alterations. To further complicate the picture, some of these symptoms also occur in other diagnostic categories, raising the question whether these symptoms are specific to each disease. For example, depressive episodes are also found in disorders from the ‘bipolar spectrum’, characterized by a cycling between depressive periods and mania or hypomania. Additionally, some disorders not only share symptoms found in major depression, but are also comorbid with the depressive pathology. For example, some symptoms of major depression overlap with those of generalized anxiety disorder (GAD), and high comorbidity among both pathologies is well established (Moller, 2002), suggesting common aetiological factors. Factors conferring a high vulnerability to major depression and/or anxiety disorders can be either genetic or environmental (i.e. poor maternal care or perinatal stress) in origin (Figure 1). Interestingly, twin studies (Kendler et al., 1992; 2007; Roy et al., 1995; Kendler, 1996) found consistent evidence that major depression and GAD share genetic risk factors such as polymorphism of the serotonin (5-HT: 5-hydroxytryptamine) transporter (serotonin transporter: SERT). Indeed, this polymorphism is associated to predisposition traits favouring depression (Schinka et al., 2004; Sen et al., 2004) and GAD (Wray et al., 2009). Such relationship is also found for polymorphism of the cathecol-O-methyl transferase gene, of the brain-derived neurotrophic factor (BDNF) gene and of the tryptophan hydroxylase-1 (TPH1) gene (see Hettema, 2008 for a review).
Table 1.
Diagnostic criteria for major depression
| Depressed mood |
| Decreased interest in pleasurable activities and ability to experience pleasure (anhedonia) |
| Weight loss or weight gain, increased or decreased appetite |
| Insomnia or hypersomnia |
| Psychomotor agitation or retardation |
| Fatigue or loss of energy |
| Feelings of hopelessness, worthlessness and guilt |
| Decreased ability to think or concentrate |
| Recurrent thoughts of death and suicide |
| Decreased ability to perform daily tasks efficiently |
Diagnostic for major depression is made according to the criteria defined by the Diagnostic Statistical Manual of Mental Disorders if a minimum of five symptoms (including at least one of the bolded symptoms) from the above list have been present during the same 2 week period and disrupt normal occupational and social functioning. (Bolded symptoms are considered cardinal and more specific signs of depression.)
Figure 1.
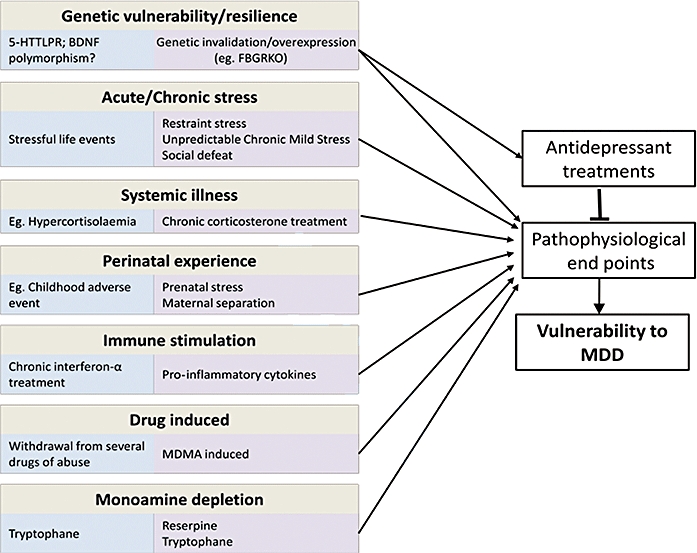
Representation of aetiological factors involved in depression. Examples of triggers in humans are given in blue boxes. Examples of experimental models used in animals to reproduce these triggers are given in purple boxes. These aetiological factors are thought to induce changes in pathophysiological end points precipitating a depression. Antidepressant treatments may act by reversing these changes. BDNF, brain-derived neurotrophic factor; MDD, major depressive disorder; MDMA, methylenedioxymethamphetamine.
Major depression can be treated using antidepressant (AD) pharmacotherapy, particularly by manipulating monoaminergic targets. For example, selective serotonin reuptake inhibitors (SSRIs) block the SERT thus increasing 5-HT, and monoamine oxydase inhibitors target monoamine oxydase, the enzyme metabolizing monoamines, also increasing 5-HT and/or noradrenergic neurotransmission. It is generally admitted that the aetiological factors involved in the release of a depressive episode might cause changes in specific end points. An end point is defined here as alterations that are targeted by the different therapies. These alterations are almost pathophysiological changes, but neuropsychological alterations can also be considered as end points as they can be targeted by behavioural therapies such as emotional regulation or cognitive and behavioural therapy. The different treatments act to reverse the alterations of these different end points (Figure 1), but the relevance of that for depression is unclear due to the heterogeneity of this disease and its high comorbidity. Indeed, do the therapies act by relieving the pathophysiological expression of a crucial end point recapitulating major depression or do they target several end points each related to particular symptoms, some of them also being present in comorbid pathologies? This paper is aimed at challenging these issues, describing the different proposed end points and trying to construct a theoretical model of their involvement in the pathophysiology of depression and in the AD's action. Data from the clinic will be discussed, but in some cases research is rather based upon data from preclinical studies, using animal models. Indeed, such models enable to elucidate the causal involvement of a given target by using invasive techniques (lesion of a brain structure, invalidation of a given gene and so forth) and permit a description of the end point pointing to cellular or molecular processes that cannot be studied in humans. Therefore we will briefly describe the animal models that are sometimes used to resolve these issues before describing the end points and presenting our model.
Animal models
An animal model of a psychiatric pathology has to meet several validity criteria in order to be relevant to the clinic. It first has to satisfy face validity that is to provoke changes (behavioural as well as pathophysiological) considered as equivalent to those observed in the human pathology. These alterations might be measured via species-specific methodologies (questionnaires, behaviour, imaging, biochemical dosage and so forth). For example, in humans, anhedonia can be assessed via anhedonia scales (Chapman et al., 1976) while in rodents it will be evaluated via reward-based tests. Some end points are not measurable in preclinical models, as they involve cognitive processes that are present only in higher primates (Belzung and Philippot, 2007) such as the excessive culpability seen in depression. On the other side, some end points cannot be assessed in humans, as for example the ones that can only be detected via immunoshistochemistry. A list of the different end points, proposed by the clinical as well as the preclinical literature is presented in Table 2. The animal model has also to fulfil a second validity criterion: the construct validity that is similarity in causation and in the theoretical construct. Indeed, in animals, the depressive-like phenotype has to be elicited by experimental manipulations considered as isomorphic to the factors involved in the etiopathogeny of the human disease (Figure 1). As depression is related to a complex aetiology, including developmental as well as triggering factors (stress), animal models are based upon induction of vulnerability during the developmental period (genetic overexpression or invalidation, maternal separation), on learning-related changes of coping (learned helplessness) or on application of mild stressors in adult subjects [social stress, unpredictable chronic mild stress (UCMS), Figure 1]. A third issue an animal model has to address is the predictive validity criterion. This means that treatments that counteract the human pathology should also reverse the alterations observed in the animal model: for example, chronic (but not acute) ADs or electroconvulsive therapy (ECT) should be effective in the animal model.
Table 2.
Examples of potential end points for antidepressant treatments based on clinical or preclinical data
| Altered cortico-limbic processing |
| Cg25 |
| Hippocampus |
| Amygdala |
| Prefrontal cortex |
| Nucleus accumbens/ventral tegmental area |
| Neuroplasticity |
| Neurotrophins |
| BDNF, VEGF and associated intracellular pathways |
| LTP, LTD |
| Neurogenesis |
| Glial pathology |
| Neurotransmitter changes |
| Serotonin |
| Dopamine |
| Glutamate |
| GABA |
| Noradrenaline |
| HPA axis dys-regulations |
| Hypercortisolemia |
| Decreased glucocorticoids-induced negative feedback |
| Altered glucocorticoids receptors function/expression |
| Neuropeptides |
| Corticotrophin-releasing factor |
| Vasopressin |
| Neuropeptide Y |
| Hypocretin |
| Immune system |
| Pro-inflammatory cytokines |
| Biological rhythms dys-regulations |
| Sleep/wake cycle |
| Others |
| Melatonin |
| Histone deacetylation |
| Cognitive traits and behavioural adaptation |
| Stress coping |
| Emotional regulation |
| Executive function |
| Motivation/apathy (e.g. self-care behaviour/grooming behaviour) |
BDNF, brain-derived neurotrophic factor; GABA, gamma amino butyric acid; HPA, hypothalamic-pituitary adrenal; LTD, long-term depression; LTP, long-term potentiation; VEGF, vascular endothelial growth factor.
End points of the antidepressant action: do they target a final common pathway?
If ADs target pathophysiological end points, the question is whether some pathways underlying the therapeutic effects can be considered as crucial, explaining per se the AD effect, or whether the ADs rather target a combination of end points to achieve recovery. The first hypothesis is based on the assumption that a unique final common pathway underlies the therapeutic effects. In regard to the symptomatic heterogeneity of the disease, this appears as illusory. However, it can be that a given treatment acts on a core symptom of the disease, so that it might induce remission via this end point, the other associated symptoms remaining unchanged. As to the second option, explanations should be provided on the mechanisms explaining how an effect on this combination of end points may induce remission. In order to better understand this issue, it seems necessary to use some examples from the literature. It is beyond the scope of this paper to provide an exhaustive picture of all the end points that have been studied (see Figure 1 and Table 2) but we might rather illustrate our argument using some specific examples, such as brain areas, brain connectivity, neurotransmission, neuroplasticity and hypothalamic-pituitary adrenal (HPA) axis.
Brain circuitry and connectivity
A first well-described end point concerns brain circuitry. Here, we will focus on the macroscopic alterations observed in depression such as modifications in the volume of an area measured by magnetic resonance imaging or in the activity of a region studied via functional magnetic resonance imaging or positron emission tomography. As to the functional aspect, the experimental approach used is often to try to investigate brain area activation during a particular situation, thus establishing a relationship between this task and a brain structure. For example, anhedonia can be related to altered functioning of the nucleus accumbens (Nestler and Carlezon, 2006), depressed mood with altered activity of the anterior cingulate cortex (Drevets et al., 2008), cognitive dysfunction to altered processing in the hippocampus (Spedding et al., 2003) and in the prefrontal cortex (PFC) (Rogers et al., 2004). If ADs target some brain alterations underlying particular symptoms, rather than depression per se, it is possible that it may also relieve alterations present in the comorbid pathologies. This is the reason why we will also discuss the presence of the changes in comorbid disorders, focusing on the example of GAD.
In major depression, several morphological alterations have been reported in cortical areas such as the PFC and the subgenual cingulate cortex (Cg25) as well as in some subcortical areas, including the hippocampus and the amygdala (Ressler and Mayberg, 2007). For example, hippocampal volume decrease is found in patients who suffered multiple depression episodes (MacQueen et al., 2003) while amygdala is enlarged in first episode patients and reduced in recurrent depression (Frodl et al., 2002; Frodl et al., 2003). As to functional alterations, depression is associated with decreased baseline activity in the temporal cortex and in the insula, increased activity in the cerebellum (Fitzgerald et al., 2008), in the ventromedial PFC (see Koenigs and Grafman, 2009, for a review) and in the anterior cingulate cortex (Drevets, 2000). Most of the depression-related changes in brain activity are abolished in remittent patients, indicating that these changes are state markers. For example, paroxetine treatment reverses the frontal, the cingulate and the hippocampal changes seen in depressed patients (Goldapple et al., 2004). Convergent data can be found from animal studies. For example, using c-fos immunostaining, it has been shown that chronic fluoxetine reverses the pattern of activation induced by the novelty suppression of feeding test in the cingulate cortex and the hippocampus but also in the bed nucleus of the stria terminalis, the nucleus accumbens and the piriform cortex (Bechtholt et al., 2008).
However, are these alterations crucial for the AD's action? This seems true in the case of some cerebral end points underlying core symptoms of depression, such as the anterior cingulate cortex activity. Indeed, theta activity increase (Pizzagalli et al., 2001; Mulert et al., 2007) as well as activation detected by neuroimaging at baseline (Mayberg et al., 1997; Saxena et al., 2003) or during precise tasks (Davidson et al., 2003) in this area predict the outcome to monoaminergic ADs but also to putative ADs such as the N-methyl-D-aspartate (NMDA) antagonist ketamine (Salvadore et al., 2009) or to non-pharmacological therapies including sleep deprivation (Wu et al., 1999) or cognitive behavioural therapy (Fu et al., 2008). All these findings support the idea of cingulate cortex activity as a crucial end point of the depressive pathology and the AD's action. However, the picture is more complex as greater pretreatment activity of this area is also found in those GAD patients better responding to venlafaxine (Whalen et al., 2008). In the case of GAD, other end points seem crucial as amygdala activity is also a marker of the outcome to fluoxetine therapy (Whalen et al., 2008). Other pathophysiological AD-sensitive features seem crucial to particular symptoms, rather than to the disease. For example, altered activity of the cortico-limbic network when confronted to facial expressions of joy (Fu et al., 2007), altered activity in the anterior cingulate cortex and the insula when faced with facial expression of sadness (Chen et al., 2007), modified activity pattern in the PFC following painful stimulus (Schweinhardt et al., 2008) and attenuated nucleus accumbens activation in response to positive stimuli (Epstein et al., 2006) have all been described in depressed patients. These specific changes are reversed by ADs: for example, the cortico-limbic changes observed when depressed subjects are confronted to facial expression of joy are reversed by fluoxetine (Fu et al., 2007).
Some of the above-mentioned cerebral alterations could be related not to a modified activity within the targeted brain area per se, but to altered excitatory or inhibitory input to this structure. For example, perception of a threatening stimulus is associated with increased activation of the amygdala (Davis and Whalen, 2001). In normal subjects, an inhibitory feedback from cortical areas limits this increased amygdala activity, and this might be dysfunctional in subjects vulnerable to depression such as carriers of the short variant of the SERT gene (Pezawas et al., 2005). Similar results are found in GAD patients as weaker negative connectivity between amygdala and ventrolateral PFC has been described in these patients independently from the comorbid diagnosis of depression (Monk et al., 2008). Therefore, connectivity between amygdala and PFC cannot be considered as an end point crucial for depression, but rather as an end point of a particular symptom seen in depressed as well as in comorbid patients. Further, depressed subjects display increased activity in the extended amygdala when confronted to emotional facial expressions; this is correlated to activity in the anterior cingulate cortex and this correlation is decreased in depressed patients (Anand et al., 2005). Again, rodent models might help in describing the synaptic process underlying such modifications, as changes in brain connectivity can be assessed via long-term potentiation (LTP) and long-term depression (LTD). Indeed, LTP and LTD correspond to semi-permanent changes in connectivity between brain regions (as between the hippocampus and the PFC) or within subareas of a region (as between CA3 and CA1 within the hippocampus). Interestingly, in rodents, exposure to stress impairs LTP in the hippocampal–medial PFC pathway (Rocher et al., 2004; Cerqueira et al., 2007), an effect reversed by ADs (Rocher et al., 2004). Stress also blocks the induction of LTP at the projection from the amygdala to the PFC (Maroun and Richter-Levin, 2003), and at the reverse projection, from PFC to amygdala, stress induces the promotion of LTP and inhibition of LTD (Maroun, 2006). Similar changes have also been observed within the hippocampus. Indeed, stress deteriorates hippocampal LTP while AD from different pharmacological classes such as SSRIs or tianeptine but also ECT increase LTP (Shakesby et al., 2002; Pittenger and Duman, 2008). AD-related alterations in LTD have also been observed in the hippocampus as chronic stress facilitates LTD, an effect prevented by chronic fluvoxamine (Holderbach et al., 2007). These data indicate that it is rather difficult to isolate one particular end point. It would rather be relevant to elucidate the function of these connectivity changes. Indeed, Airan et al. (2007) found that dentate gyrus activity is reduced in chronically stressed animals, whereas CA1 activity is increased, suggesting elevated hippocampal output, and reduced hippocampal activity in depressed-like rodents. This is reversed after AD. Interestingly, the activity propagation in the dentate gyrus relative to CA1 provides a reliable indicator of the behavioural phenotype. Authors suggest that depression-like behaviour could thus be associated with altered dentate gyrus associative/predictive activity or increased error signals from CA1, resulting in the failure to adapt to environmental changes.
Neurotransmission changes
Inappropriate functioning and connectivity among key cortico-limbic systems might be related to impaired neurotransmission among brain structures. Several neurotransmitter systems have been involved in depression and in AD response, including the monoaminergic, and more recently the glutamatergic systems.
As all used ADs target monoaminergic neurotransmission, particularly 5-HT and noradrenaline, these systems have been proposed to be the crucial end point underlying recovery. Indeed, brain 5-HT availability is increased by most ADs and associated with modulation of 5-HT receptors, particularly desensitization of the 5-HT1A autoreceptor (Chaput et al., 1991; Blier and de Montigny, 1998). Is this crucial for AD effects? This seems to be the case as depletion of 5-HT completely blocks the effects of SSRIs (Redrobe et al., 2005; O'Leary et al., 2007). Further, a single nucleotide polymorphism (SNP) for the SERT, the 5-HT1A, the 5-HT2A or the 5-HT3A receptors, or for enzymes involved in the biosynthesis of 5-HT (TPH) are all associated with altered response to monoaminergic ADs (Kato and Serretti, 2008), supporting the view of the 5-HT system as the crucial target of the AD response. However, this is a simplification. Indeed, while rapid depletion of 5-HT can induce a relapse in patients responsive to fluoxetine, no effect is found in patients successfully treated with an AD having a strong noradrenergic component (Delgado et al., 1999). Moreover, mice having targeted invalidation of the 5-HT1A receptors gene become insensitive to the effects of fluoxetine in the novelty suppression of feeding test, while still responding to imipramine, again a compound whose action is not only affecting 5-HT transmission (Santarelli et al., 2003). Therefore, 5-HT seems causally involved in the effects of 5-HT-acting ADs, but is not crucial for the effects of other ADs. Further, 5-HT is not critical in the pathophysiology of depression as most studies investigating 5-HT function in depression provide equivocal results. For example, unaltered platelet 5-HT concentration has been repeatedly reported in depressed patients (Muck-Seler et al., 1991; Muck-Seler et al., 1996; Jakovljevic et al., 1997; Pivac et al., 1997). However, other studies report 5-HT-related changes. For example, decreased plasma tryptophan levels (Coppen et al., 1973; Cowen et al., 1989), reduced cerebrospinal fluid 5-hydroxyindoleacetic acid (metabolite of 5-HT) (Asberg et al., 1976), reduced 5-HT1A receptor binding potential in the raphe, the cingulate cortex and the insula (Drevets et al., 1999; Sargent et al., 2000; Meltzer et al., 2004; Drevets et al., 2007) have been reported. Finally, some of these pathophysiological alterations are not crucial for the AD's effects. For example, the change in 5-HT1A receptors is not reversed by AD (Drevets et al., 1999; Sargent et al., 2000).
Cryan et al. (2004) highlighted the role of noradrenaline in the effects of several ADs as mice unable to synthesize noradrenaline due to targeted disruption of the dopamine beta-hydroxylase gene (Dbh−/−) fail to respond to several noradrenergic-acting ADs in the tail suspension test but also to the ones of a monoamine oxydase inhibitor and of several SSRIs, suggesting a major role of noradrenaline in AD effects.
Recently research pointed to glutamatergic targets, as this neurotransmitter is the major brain excitatory amino-acid, involved in connectivity among brain areas and LTP. Interestingly, altered levels of glutamate have been seen in cortico-limbic structures found to be dys-regulated in depression (Sanacora et al., 2008). Conversely, ADs might work by stabilizing glutamatergic transmission (Bonanno et al., 2005; Hashimoto, 2009). At the receptor level, alterations are mainly described in the expression of NMDA receptors (reduction in NMDA receptor binding in the frontal cortex and in NR1 subunit in the temporal cortex) and/or the α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptors. Further, glutamatergic agents elicit AD properties (Sanacora et al., 2008). Indeed, ketamine, a NMDA antagonist, elicits AD effects in depressed patients (Zarate et al., 2006). In animal studies, mGlu1/mGlu5 and mGlu2/mGlu3 antagonists have AD-like properties. However, these glutamatergic targets are not causally involved in the effects of monoaminergic ADs. Indeed, deletion of the gene encoding the mGlu5 protein alters the AD effects of the mGlu5 antagonist 6-methyl-2-(phenylethynyl)pyridine (MPEP) in the forced swim test, but not the ones of imipramine (Li et al., 2006). Similarly, mGlu7 receptor knockout mice are insensitive to the AD effects of an mGlu7 ligand in the tail suspension test while the effects of imipramine are still present (Palucha et al., 2007).
All together, these data indicate that changes in neurotransmission are observed in depressed patients and are targeted by ADs, but generally the therapeutic effects are specific to the targeted system: 5-HT-related end points are targeted by 5-HT ADs, glutamatergic targets by glutamatergic ADs and so on. One can assume that the neurotransmission alterations provoke the dysfunction of particular brain areas, and the reversal of this pattern can be achieved targeting the neurotransmission via system-specific tools.
Neuroplasticity
When a subject is faced with environmental challenges, network construction and reorganization occurs leading to neuronal remodelling, formation of novel synapses and birth of new neurons: all these modifications can be grouped under the term neuroplasticity. Recently, the idea that neuroplasticity might be considered as an end point of the AD's action has prompted much enthusiasm. Indeed, altered plasticity may provoke dysfunction of brain structure activity as well as in communication among brain areas. Two neuroplasticity-related targets have received major interest: neurogenesis and neurotrophic factors.
Concerning neurotrophic factors, a particular focus has been on BDNF, not only because it stimulates LTP but also because most monoaminergic ADs but also putative ADs (such as NMDA antagonist memantine), as well as non-pharmacological therapy (ECT and transcranial magnetic stimulation) all promote expression of BDNF in the hippocampus (Duman and Monteggia, 2006). Infusions of BDNF into the hippocampus can mimic AD-like effects (reviewed in Duman and Monteggia, 2006), and overexpression of the TrkB receptor (the primary receptor of BDNF) in mice induces AD-like effects in the forced swim test (Koponen et al., 2005). Moreover, social defeat stress, an animal model of depression, decreases BDNF in the hippocampus (Tsankova et al., 2006). Can BDNF be considered as a crucial end point in the depressive-like phenotype and in AD action? In any case, such formulation is an oversimplification, as the reverse pattern is observed in another brain region: indeed, social defeat leads to a BDNF increase in the nucleus accumbens that is prevented by AD treatment (Berton et al., 2006). A relevant formulation might be that plasticity per se requires BDNF, and that plasticity is a medium to remission or to recovery of some depressive-related symptoms in specific areas. For example, BDNF polymorphism seems associated with particular forms of depression or symptoms, such as depression associated to fatigue (Utge et al., 2009) or poorer working memory, slowed response speed, neuroticism, elevations in autonomic arousal and higher anxiety (Gatt et al., 2009). Studies also pointed to involvement of BDNF polymorphism in other pathologies, such as some markers of psychosis (Golimbet et al., 2008; Rybakowski, 2008), substance dependence (Jiang et al., 2009; Wojnar et al., 2009) and Alzheimer disease (Huang et al., 2007). Further, it is also associated with some non-psychiatric/neurologic disease that can increase vulnerability to depression, such as type 2 diabetes (Krabbe et al., 2007).
Another plasticity-related process that received much attention is adult hippocampal neurogenesis. It is now well established that chronic monoaminergic ADs induce an increase in the number of new hippocampal neurons (Malberg et al., 2000; Manev et al., 2001). Similar effects are reported with putative ADs acting via other pathways such as glutamatergic ligands (Yoshimizu and Chaki, 2004), a synthetic cannabinoid (Jiang et al., 2005), tianeptine (Czeh et al., 2001; McEwen et al., 2002), corticotrophin-releasing factor 1 (CRF1) or vasopressin 1b (V1b) receptor antagonists (Alonso et al., 2004) or a melanin-concentrating hormone antagonist (David et al., 2007) as well as with non-pharmacological therapy, such as ECT (Malberg et al., 2000). In some cases, neurogenic effects of ADs are observed in normal animals (Malberg et al., 2000) and in other cases AD restore a decrease in neurogenesis induced by stress (Alonso et al., 2004). Enthusiasm for this target became higher with the observation that AD's effects are prevented by X-ray hippocampal irradiation, which abolishes neurogenesis (Santarelli et al., 2003) suggesting that hippocampal neurogenesis might represent the crucial end point necessary to the AD's action. This involves neurotrophic action as Li et al. (2008) showed that genetic ablation of the TrkB receptor in neural progenitor cells in the dentate gyrus impairs the neurogenic and behavioural effects of ADs. There are however limitations to this hypothesis. Indeed, it was observed that the neurogenesis dependence of the AD's effects vary depending on the species and genetic background of animals (Miller et al., 2008), the magnitude of the neurogenesis ablation [partial ablation of neurogenesis by antimitotic drugs does not elicit suppression of the AD's effects (Bessa et al., 2009)], the pharmacological class of ADs and the type of behavioural paradigms used (Zhao et al., 2008). For example, neurogenesis, if required for the response to monoaminergic-acting ADs or to synthetic cannabinoid HU210, is not necessary for the effects of a melanin-concentrating hormone antagonist (David et al., 2007) and of a CRF1 antagonist or a V1b antagonist (Surget et al., 2008). It was also suggested that the involvement of neurogenesis in the AD's effects is necessary to some but not all depression-related phenotypes. Indeed, hippocampal irradiation suppresses the effects of fluoxetine in the novelty suppression of feeding test, considered as a test measuring the anxiolytic effects of ADs, but not in the open field and in the forced swim test (David et al., 2009). Similarly, irradiation prevents the AD-like effects of a CRF1 antagonist in the novelty suppression of feeding test, but not the effects of this compound on the coat state and in the splash test (Surget et al., 2008). So, it seems that neurogenesis is crucial for the effects of ADs on some specific symptoms of the depressive-like state, rather than on depression per se. Further, neural cell proliferation is not reduced in depressed patients (Reif et al., 2006), which again is a limit to the hypothesis of neurogenesis as the crucial end point. It is now hypothesized that neurogenesis, rather than being a necessary mechanism by which all ADs exert their action, contributes to an optimal functioning of the hippocampus. When neurogenesis is decreased, hippocampal activity is altered (Airan et al., 2007), thus rendering appropriate processing of the context more difficult. As the hippocampus projects towards brain regions involved in emotion (amygdala and PFC), hedonicity (nucleus accumbens) and stress (hypothalamus), abolition of neurogenesis may induce an overall change in the cortico-limbic network activity, thus increasing vulnerability to depression.
Neuroendocrine stress axis
Hippocampus, amygdala and PFC are known to contribute to the central regulation of the HPA axis (Ulrich-Lai and Herman, 2009), and hippocampal neurogenesis is directly involved in this phenomenon (Schloesser et al., 2009). The HPA axis has also been proposed as a crucial element of the pathophysiology of depression and the AD response. Indeed, depressive patients display higher salivary cortisol, particularly at awakening (Vreeburg et al., 2009). This is associated with increased concentrations of CRF in the cerebrospinal fluid (Nemeroff et al., 1984), increased adrenocorticotropin (ACTH) pulse frequency (Mortola et al., 1987), blunted ACTH response following CRF administration (Gold et al., 1984; Holsboer et al., 1984), lower number of CRF receptors in the PFC (Nemeroff et al., 1988), low cortisol suppression after dexamethasone (Carroll et al., 1981) and alterations in the cortisol and ACTH concentrations induced by CRF after suppression by dexamethasone, all indicating a reduced HPA feedback (Holsboer, 2000; Ising et al., 2007). Further, normalization of HPA function has been associated with sustained remission (Ribeiro et al., 1993; Holsboer and Barden, 1996; O'Toole et al., 1997; Zobel et al., 1999; Zobel et al., 2001) suggesting that aberrant HPA function might trigger depression, ADs normalizing these changes (Pariante, 2003). This is confirmed in animal models. For example, after chronic stress, hypersecretion of CRF and vasopressin, functional decrease in glucocorticoid receptor (GR) activity, increased adrenal sensitivity to ACTH and diminished negative feedback of the HPA axis have been observed, which all lead to HPA hyperactivity. Chronic imipramine reverses these changes (Raone et al., 2007) while fluoxetine or tianeptine counteract the increased corticosterone levels occurring after prenatal stress (Szymanska et al., 2009). Is HPA normalization correlated to the effects of ADs or does normalization represent a prerequisite for stable remission? Some arguments indicate that HPA normalization might be causal in the AD response. For instance, HPA axis dysfunction is associated with reduced SSRI efficacy (Young et al., 2004), and patients not exhibiting cortisol suppression to a dexamethasone/CRH challenge after 2–3 weeks of treatment are not likely to respond to the therapy (Ising et al., 2007). Regarding the receptors, several SNPs of the GRs influence HPA reactivity and negative feedback (Derijk and de Kloet, 2008) and some of these variants affect response to ADs (Binder et al., 2004; Brouwer et al., 2006). For instance, Binder et al. (2004) found that SNPs in FKBP5, a GR-regulating cochaperone of hsp-90 important for fine-tuning of the HPA axis, are associated with AD response and with recurrence of depressive episodes. Further, CRF1 antagonists have shown relative success in attenuating depressive-like behaviours in animal models and in clinical trials (Alonso et al., 2004; Holsboer and Ising, 2008) supporting the idea of HPA function as a crucial end point involved in the pathophysiology as well as in ADs effects. However, other data rather support the idea that the HPA-related changes are not as central. First, these changes are not present in all depressed patients, as hypercortisolemia is found only in 40–60% of depressed patients (Parker et al., 2003) and dexamethasone suppression is not found in all studies (Vreeburg et al., 2009). This might be explained by the fact that the outcome of the dexamethasone suppression test might be influenced by psychiatric comorbidity and depression subtypes (Nemeroff, 1996; Amsterdam, 1998; Veen et al., 2009a). Second, therapeutic improvement can occur independently from an action on the HPA dysfunction. Indeed, paralleling some clinical data (Watson et al., 2002; Gervasoni et al., 2004), UCMS-induced behavioural changes in certain strains of mice can occur without corticosterone alterations and are reversed by chronic AD (Ibarguen-Vargas et al., 2008). Using mice with an acquired forebrain-specific disruption of GR (FBGRKO) Boyle et al. (2005) showed that AD induced a therapeutic effect, but failed to reverse the loss of negative feedback induced by the GR deletion. Further, most remittent patients have higher salivary cortisol, indicating that hypercortisolemia might rather be a trait marker (Vreeburg et al., 2009) of the depressive personality, not reversed by ADs. Third, HPA changes might be associated with some specific symptoms/comorbidity. Indeed, HPA alteration is not associated with severity, chronicity or symptom profile of the depressed patients, except for comorbid anxiety (Veen et al., 2009b). Fourth, some stress-related targets can also be involved independently from the HPA. For example, preclinical data indicate that CRF and vasopressine may target depressive behaviour as well as anxious behaviour, in a way sometimes independent from hypothalamic function. Indeed, in rodents, CRF1 and V1b antagonists are effective both in models of depression and in models of pathological anxiety (Griebel et al., 2002; Ducottet et al., 2003; Louis et al., 2006; Salome et al., 2006). Effectiveness of CRF1 antagonists has also been described in some clinical studies (Holsboer and Ising, 2008) even if others failed to replicate the same findings (Binneman et al., 2008). The AD effects of the V1b antagonist occur via extra-hypothalamic mechanisms such as the lateral septum (Stemmelin et al., 2005) and different amygdala nuclei (Salome et al., 2006), while anxiolytic action of the same compound occurs via the basolateral amygdala (Salome et al., 2006). Other data show an HPA-independent action of CRF as Muller et al. (2003) showed that postnatal inactivation of CRF1 in limbic regions (amygdala, hippocampus and neocortex) lead to a decrease in anxiety behaviours, without affecting CRF1 expression in the pituitary.
All these data converge on the idea that the AD action can be achieved either via some particularly crucial end points underlying core symptoms, or via a combination of end points each involved in a particular symptoms. The question than is whether these end points participate in the induction of the depressive-phenotype or whether their role is limited to the AD action.
Have the proposed end points a causal role in the depressive pathology?
If an end point is altered in depressive subjects and involved in the AD mechanism does this mean that it is causally involved in the disease, triggering the depressive episode? Recently, Banasr and Duman (2008) showed that glial ablation in the PFC is sufficient to induce depressive-like behaviours in adult rats, illustrating that an end point might be causally involved in a depressive phenotype. However, the picture is rarely as straightforward as most factors important for the AD's action are not causally involved in the pathology. For instance, several polymorphisms have been described that alter AD's response, including polymorphism of the SERT, the 5-HT1A, the 5-HT2A and the 5-HT3A receptors, of the noradrenaline transporter, the BDNF gene, the genes altering substance P levels (angiotensin-converting enzyme gene: ACE), genes modifying the HPA axis functioning (CRF1 or FKBP5 gene) and glutamatergic neurotransmission (dystrobrevin-binding protein 1 gene) (Kato and Serretti, 2008 for review). However, no evidence indicates that these abnormalities are associated with increased occurrence of depression. Further, 5-HT depletion only induced depression episodes in those patients having a high vulnerability (Ruhe et al., 2007). In mice, no depressive-like effect of 5-HT depletion is observed in vulnerable subjects such as stressed BALB/c mice (Yalcin et al., 2008). Dbh−/− mice behave normally (Cryan et al., 2001) as do mice with an inducible knockout of BDNF in the forebrain (Monteggia et al., 2004). Abolition of hippocampal neurogenesis does not provoke depression-like phenotype in mice (Santarelli et al., 2003; Surget et al., 2008), and neurogenesis level in unstressed mice does not predict vulnerability to UCMS (Mineur et al., 2007). Further, as mentioned above, several of the putative end points are associated not only to major depression, but also to other disease. For example 5-HT alterations and modification in connectivity between the amydgala and the PFC also appear in GAD patients, and BDNF polymorphism is associated with depression as well as with psychosis, substance abuse or Alzheimer disease. The same reasoning applies for the symptomatic features of depression. For example, difficulty in concentration is present in most affective disorders as well as in several neurological diseases, so that this cannot be considered as a cause of depression. For this reason, most of these end points cannot be considered as a single critical target, causing the depressive phenotype in a monolithic way.
An alternative model as to the relationship between end points and depressive feature
We showed that the hypothesis that depression and/or AD effects might be related to a single end point, explaining the whole phenotype of the pathology, causally involved in the disease and recapitulating all the mechanisms underlying the AD's efficacy is questionable. We also presented data showing that the depressive phenotype might rather be related to an ensemble of independent pathophysiological end points, each important for a particular aspect of the symptomatology. But how do these end points produce the disorder and how can ADs achieve remission? Here we present a theoretical model to resolve this issue, using a connectionist-inspired network (Figure 2). Connectionist models are based upon a connected network of nodes where information is treated in a parallel and distributed fashion. These networks include several layers such as an input layer, one or more intermediate layers performing internal processing and an output layer. The different nodes from a same layer can also be inter-connected. The output represents the activity pattern of different nodes from the network. Each node has several inputs, a threshold (or input–output) function and an output. The strength of the link between nodes can be modified by experience. In Figure 2, each pathophysiological end point (e.g. 5-HT neurotransmission, hippocampal neurogenesis, HPA function, nucleus accumbens and anterior cingulate activity) is represented by a node of the first intermediate layer and each behavioural/cognitive feature is represented by a node of the second intermediate layer, both being inter-connected. In order to simplify the figure, we focused on five neurobiological-related and four symptomatic-related end points. The input layer corresponds to aetiological factors either present during the developmental period and involved in the vulnerability to the disease (genetic factors as well as early environmental features) or factors precipitating the depressive episode during adulthood (life event for example). The vulnerability/resilience will change the threshold sufficient for a particular triggering event to elicit a pathophysiological end point. The output layer corresponds to the state of the subject (normal, depressed, or with a comorbid pathology or symptomatology such as GAD). Several important aspects have to be considered: (i) the pathophysiological end points have causal relationship with symptomatic end points; (ii) among the pathophysiological end points, some are crucial because they might induce a core symptom (anhedonia, depressed mood) and others are secondary; (iii) some non-crucial pathophysiologial end points can induce a depressive episode via connections with other nodes, thus inducing overall changes of the network; (iv) in reason of the above-mentioned arguments, different pattern of node activation can underlie depression/remission/resilience; (v) symptomatic end points can also have interrelations. For example, stress coping can regulate executive functions; (vi) back-propagation might enable a node of the second layer to act on nodes of the first layer. For example, relevant stress coping can change the way a triggering event is perceived, thus altering the effect of the life event on the pathophysiological end points; and (vii) the different patterns of node activation may explain the high heterogeneity of the disease as well as its comorbidity.
Figure 2.

Theoretical model of depression and antidepressant effects based on a connectionist inspired network. Blue arrows represent normal interactions between nodes. Red arrows represent pathological interactions. Antidepressant effects are indicated by green arrows. Increased weight/interaction between nodes is represented by bold arrows. Filled red nodes indicate a pathological change of state, while filled green nodes represent an antidepressant-induced reversal from a pathological state to a normal state. Core behavioural/cognitive traits relevant to the depression diagnostic (hedonic behaviour and mood) are indicated by plain nodes while dashed nodes represent secondary traits (stress coping or cognitive processing). Different colours of the output correspond to the state of the subject (green: normal, red; depressed, purple: comorbid pathology). (A) Normal state, (B) depressive episode induced by dys-regulation of a single end point leading to a core behavioural impairment (e.g. mood), (C) depressive disorder induced by dys-regulation of multiple end points leading to impairments in multiple psychological functions together mediating symptoms of depression, (D) comorbid pathological state induced by dys-regulation of a single secondary end point, (E) hypothetical effects of an antidepressant treatment in scenario 2B, (F) hypothetical effects of an antidepressant in scenario 2B, (G) hypothetical effects of a cognitive behavioural therapy or an emotional regulation therapy on a subject displaying profile 2C and (H) hypothetical view proposing that depression can be recapitulated by a crucial process, neuroplasticity.
Different configurations of this theoretical network are illustrated that may help in the discussion of such model. Figure 2A represents the state of the network in a normal subject. During development, he has been subjected to factors providing either vulnerability or resilience to depression. No particular event triggering depression has occurred, so that the different pathophysiological end points have a normal expression, which is related to normal behaviour. In Figure 2B, the subject has been subjected to a triggering event that, combined to a particular vulnerability state, has induced abnormal expression of the crucial end point E, which is able to induce a core symptom of the disease (depressed mood). In Figure 2C, the triggering event occurring in a vulnerable subject induces pathological expression of an end point (end point A) that is not crucial per se. However, as this end point has strong relationships with end point D, itself related to end point C, it might elicit alterations in stress coping, executive function and anhedonia, all together causing a depressive episode. The example of Figure 2D illustrates occurrence of a comorbid disease. Here, the subject is resilient to depression, but he is faced with an event triggering abnormal expression of biological marker B related to stress coping and executive function. However, resilience of this subject might protect him from cognitive dysfunction, so that the subject will display a symptom of a comorbid disease. Figure 2E and F illustrates the effects of AD therapy on such network. In Figure 2E, the treatment targets a crucial end point, and thus a core symptom, inducing a therapeutic effect by counteracting the abnormalities displayed in Figure 2B. The treatment thus acts to reverse the pathophysiological alterations underlying the depressive state. The scheme displayed in Figure 2F is rather interesting, as here the AD effects are achieved via a pathway different from the one underlying the depressive state (the one of Figure 2B) showing that recovery can occur without reversing the pathophysiological alterations. This could be relevant for recurrent depression, as the presence of this pathophysiological end point in the remittent patient might be a key factor in precipitating a new depressive episode. However, this remains to be tested. Figure 2G illustrates the effects of a cognitive behavioural therapy or of emotion regulation therapy applied to a patient in the state described by Figure 2C. According to this scheme, these therapies target emotional/cognitive processes, which may in turn alter the coping of the subject with the triggering event, thus modifying its aptitude to induce pathophysiological end points. However, this is purely speculative. Figure 2H is a theoretical case, showing that a connectionist network can also be used to model a case where depression can be explained by a common and unique critical process. Indeed, here all pathophysiological end points (neurotransmission, HPA axis activity and so on) influence a unique process (neuroplasticity) occurring in different brain areas. Depending of the regional brain pattern in which the plasticity occurs, a different clinical picture will appear. This might however be an oversimplification, as mentioned above. Many other schemes and theoretical views can be constructed. For example, the participation of brain connectivity/LTP in depressive behaviour and AD's effects might be illustrated by changes in the weight between two brain regions (two end points).
Conclusion
This model is a theoretical view that can help in discussing the role of the different pathophysiological end points described in depression as well as the different mechanisms leading to recovery, via pharmacotherapy or via other approaches. It is coherent with data from the literature we reviewed, such as for example the observation that a drug can induce recovery without reversing pathophysiological alterations, the data showing that ADs can act via different mechanisms, the fact that AD can treat depression as well as comorbid pathologies sharing common pathophysiological end points, the heterogeneity of the symptomatic features of the disease as well as the observation that not all pathophysiological alterations are present in all patients. It represents an interesting framework needing to be tested and refined via experimental manipulations. For example, the weight of the connections between the different nodes has to be established, as well as the level of the different thresholds that render an end point relevant to the pathology and to the action of the therapy. Here we present only a simplified version of the theoretical model, and much other end points have to be included in the network, such as for example cytokines (see Table 2 for a more exhaustive list). This model will enable to progress as to the mechanism of action of the commonly used ADs but also to predict new pathways of AD action, thus permitting to design new treatments of the future. In reason of the complexity of this network, and of the weight of the different relations between the candidate nodes, it might be that the state of the network may vary among patients, so that the future ADs have to target the pathophysiological abnormalities seen in particular subjects, thus shifting to personalized medicine. Animal models should be refined and take such models into account. In any case, this model clearly shows that the research for a unique and crucial end point, explaining all symptoms in all patients is rather a myth. The crucial end point may indeed be the state of this network, rather than one of its components.
Acknowledgments
We are very thankful to Michael Spedding and Etienne Billette de Villemeur, for attentive reading of the manuscript and helpful comments.
Glossary
Abbreviations:
- 5-HT
5-hydroxytryptamine
- ACTH
adrenocorticotropin
- AD
antidepressant
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazole propionate
- Cg25
subgenual cingulate cortex
- CRF
corticotrophin-releasing factor
- Dbh
dopamine beta-hydroxylase gene
- ECT
electroconvulsive therapy
- GABA
gamma amino butyric acid
- GAD
generalized anxiety disorder
- GR
glucocorticoid receptor
- HPA
hypothalamic-pituitary adrenal
- LTD
long-term depression
- LTP
long-term potentiation
- NMDA
N-methyl-D-aspartate
- PFC
prefrontal cortex
- SNP
single nucleotide polymorphism
- SSRIs
selective serotonin reuptake inhibitors
- TPH1
tryptophan hydroxylase-1
- UCMS
unpredictable chronic mild stress
Conflict of interest
Catherine Belzung is a consultant for Takeda, Japan.
References
- Airan RD, Meltzer LA, Roy M, Gong Y, Chen H, Deisseroth K. High-speed imaging reveals neurophysiological links to behavior in an animal model of depression. Science. 2007;317:819–823. doi: 10.1126/science.1144400. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso R, Griebel G, Pavone G, Stemmelin J, Le FG, Soubrie P. Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry. 2004;9:278–286. doi: 10.1038/sj.mp.4001464. [DOI] [PubMed] [Google Scholar]
- Amsterdam JD. Selective serotonin reuptake inhibitor efficacy in severe and melancholic depression. J Psychopharmacol. 1998;12:S99–S111. doi: 10.1177/0269881198012003061. [DOI] [PubMed] [Google Scholar]
- Anand A, Li Y, Wang Y, Wu J, Gao S, Bukhari L, et al. Activity and connectivity of brain mood regulating circuit in depression: a functional magnetic resonance study. Biol Psychiatry. 2005;57:1079–1088. doi: 10.1016/j.biopsych.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Asberg M, Thoren P, Traskman L, Bertilsson L, Ringberger V. ‘Serotonin depression’– a biochemical subgroup within the affective disorders? Science. 1976;191:478–480. doi: 10.1126/science.1246632. [DOI] [PubMed] [Google Scholar]
- Banasr M, Duman RS. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry. 2008;64:863–870. doi: 10.1016/j.biopsych.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtholt AJ, Valentino RJ, Lucki I. Overlapping and distinct brain regions associated with the anxiolytic effects of chlordiazepoxide and chronic fluoxetine. Neuropsychopharmacology. 2008;33:2117–2130. doi: 10.1038/sj.npp.1301616. [DOI] [PubMed] [Google Scholar]
- Belzung C, Philippot P. Anxiety from a phylogenetic perspective: is there a qualitative difference between human and animal anxiety? Neural Plast. 2007;2007:59676. doi: 10.1155/2007/59676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA, et al. The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol Psychiatry. 2009;14:764–773. doi: 10.1038/mp.2008.119. [DOI] [PubMed] [Google Scholar]
- Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, Putz B, et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet. 2004;36:1319–1325. doi: 10.1038/ng1479. [DOI] [PubMed] [Google Scholar]
- Binneman B, Feltner D, Kolluri S, Shi Y, Qiu R, Stiger T. A 6-week randomized, placebo-controlled trial of CP-316,311 (a selective CRH1 antagonist) in the treatment of major depression. Am J Psychiatry. 2008;165:617–620. doi: 10.1176/appi.ajp.2008.07071199. [DOI] [PubMed] [Google Scholar]
- Blier P, de Montigny C. Possible serotonergic mechanisms underlying the antidepressant and anti-obsessive-compulsive disorder responses. Biol Psychiatry. 1998;44:313–323. doi: 10.1016/s0006-3223(98)00114-0. [DOI] [PubMed] [Google Scholar]
- Bonanno G, Giambelli R, Raiteri L, Tiraboschi E, Zappettini S, Musazzi L, et al. Chronic antidepressants reduce depolarization-evoked glutamate release and protein interactions favoring formation of SNARE complex in hippocampus. J Neurosci. 2005;25:3270–3279. doi: 10.1523/JNEUROSCI.5033-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle MP, Brewer JA, Funatsu M, Wozniak DF, Tsien JZ, Izumi Y, et al. Acquired deficit of forebrain glucocorticoid receptor produces depression-like changes in adrenal axis regulation and behavior. Proc Natl Acad Sci USA. 2005;102:473–478. doi: 10.1073/pnas.0406458102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer JP, Appelhof BC, van Rossum EF, Koper JW, Fliers E, Huyser J, et al. Prediction of treatment response by HPA-axis and glucocorticoid receptor polymorphisms in major depression. Psychoneuroendocrinology. 2006;31:1154–1163. doi: 10.1016/j.psyneuen.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Carroll BJ, Feinberg M, Greden JF, Tarika J, Albala AA, Haskett RF, et al. A specific laboratory test for the diagnosis of melancholia. Standardization, validation, and clinical utility. Arch Gen Psychiatry. 1981;38:15–22. doi: 10.1001/archpsyc.1981.01780260017001. [DOI] [PubMed] [Google Scholar]
- Cerqueira JJ, Mailliet F, Almeida OF, Jay TM, Sousa N. The prefrontal cortex as a key target of the maladaptive response to stress. J Neurosci. 2007;27:2781–2787. doi: 10.1523/JNEUROSCI.4372-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman LJ, Chapman JP, Raulin ML. Scales for physical and social anhedonia. J Abnorm Psychol. 1976;85:374–382. doi: 10.1037//0021-843x.85.4.374. [DOI] [PubMed] [Google Scholar]
- Chaput Y, de MC, Blier P. Presynaptic and postsynaptic modifications of the serotonin system by long-term administration of antidepressant treatments. An in vivo electrophysiologic study in the rat. Neuropsychopharmacology. 1991;5:219–229. [PubMed] [Google Scholar]
- Chen CH, Ridler K, Suckling J, Williams S, Fu CH, Merlo-Pich E, et al. Brain imaging correlates of depressive symptom severity and predictors of symptom improvement after antidepressant treatment. Biol Psychiatry. 2007;62:407–414. doi: 10.1016/j.biopsych.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Coppen A, Eccleston EG, Peet M. Total and free tryptophan concentration in the plasma of depressive patients. Lancet. 1973;2:60–63. doi: 10.1016/s0140-6736(73)93259-5. [DOI] [PubMed] [Google Scholar]
- Cowen PJ, Parry-Billings M, Newsholme EA. Decreased plasma tryptophan levels in major depression. J Affect Disord. 1989;16:27–31. doi: 10.1016/0165-0327(89)90051-7. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Dalvi A, Jin SH, Hirsch BR, Lucki I, Thomas SA. Use of dopamine-beta-hydroxylase-deficient mice to determine the role of norepinephrine in the mechanism of action of antidepressant drugs. J Pharmacol Exp Ther. 2001;298:651–657. [PubMed] [Google Scholar]
- Cryan JF, O'Leary OF, Jin SH, Friedland JC, Ouyang M, Hirsch BR, et al. Norepinephrine-deficient mice lack responses to antidepressant drugs, including selective serotonin reuptake inhibitors. Proc Natl Acad Sci USA. 2004;101:8186–8191. doi: 10.1073/pnas.0401080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeh B, Michaelis T, Watanabe T, Frahm J, de BG, van KM, et al. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc Natl Acad Sci USA. 2001;98:12796–12801. doi: 10.1073/pnas.211427898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DJ, Klemenhagen KC, Holick KA, Saxe MD, Mendez I, Santarelli L, et al. Efficacy of the MCHR1 antagonist N-[3-(1-{[4-(3,4-difluorophenoxy)phenyl]methyl}(4-piperidyl))-4-methylphenyl]-2-methylpropanamide (SNAP 94847) in mouse models of anxiety and depression following acute and chronic administration is independent of hippocampal neurogenesis. J Pharmacol Exp Ther. 2007;321:237–248. doi: 10.1124/jpet.106.109678. [DOI] [PubMed] [Google Scholar]
- David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, et al. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 2009;62:479–493. doi: 10.1016/j.neuron.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson RJ, Irwin W, Anderle MJ, Kalin NH. The neural substrates of affective processing in depressed patients treated with venlafaxine. Am J Psychiatry. 2003;160:64–75. doi: 10.1176/appi.ajp.160.1.64. [DOI] [PubMed] [Google Scholar]
- Davis M, Whalen PJ. The amygdala: vigilance and emotion. Mol Psychiatry. 2001;6:13–34. doi: 10.1038/sj.mp.4000812. [DOI] [PubMed] [Google Scholar]
- Delgado PL, Miller HL, Salomon RM, Licinio J, Krystal JH, Moreno FA, et al. Tryptophan-depletion challenge in depressed patients treated with desipramine or fluoxetine: implications for the role of serotonin in the mechanism of antidepressant action. Biol Psychiatry. 1999;46:212–220. doi: 10.1016/s0006-3223(99)00014-1. [DOI] [PubMed] [Google Scholar]
- Derijk RH, de Kloet ER. Corticosteroid receptor polymorphisms: determinants of vulnerability and resilience. Eur J Pharmacol. 2008;583:303–311. doi: 10.1016/j.ejphar.2007.11.072. [DOI] [PubMed] [Google Scholar]
- Drevets WC. Neuroimaging studies of mood disorders. Biol Psychiatry. 2000;48:813–829. doi: 10.1016/s0006-3223(00)01020-9. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Frank E, Price JC, Kupfer DJ, Holt D, Greer PJ, et al. PET imaging of serotonin 1A receptor binding in depression. Biol Psychiatry. 1999;46:1375–1387. doi: 10.1016/s0006-3223(99)00189-4. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Thase ME, Moses-Kolko EL, Price J, Frank E, Kupfer DJ, et al. Serotonin-1A receptor imaging in recurrent depression: replication and literature review. Nucl Med Biol. 2007;34:865–877. doi: 10.1016/j.nucmedbio.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct. 2008;213:93–118. doi: 10.1007/s00429-008-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducottet C, Griebel G, Belzung C. Effects of the selective nonpeptide corticotropin-releasing factor receptor 1 antagonist antalarmin in the chronic mild stress model of depression in mice. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:625–631. doi: 10.1016/S0278-5846(03)00051-4. [DOI] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Epstein J, Pan H, Kocsis JH, Yang Y, Butler T, Chusid J, et al. Lack of ventral striatal response to positive stimuli in depressed versus normal subjects. Am J Psychiatry. 2006;163:1784–1790. doi: 10.1176/ajp.2006.163.10.1784. [DOI] [PubMed] [Google Scholar]
- Fitzgerald PB, Laird AR, Maller J, Daskalakis ZJ. A meta-analytic study of changes in brain activation in depression. Hum Brain Mapp. 2008;29:683–695. doi: 10.1002/hbm.20426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodl T, Meisenzahl E, Zetzsche T, Bottlender R, Born C, Groll C, et al. Enlargement of the amygdala in patients with a first episode of major depression. Biol Psychiatry. 2002;51:708–714. doi: 10.1016/s0006-3223(01)01359-2. [DOI] [PubMed] [Google Scholar]
- Frodl T, Meisenzahl EM, Zetzsche T, Born C, Jager M, Groll C, et al. Larger amygdala volumes in first depressive episode as compared to recurrent major depression and healthy control subjects. Biol Psychiatry. 2003;53:338–344. doi: 10.1016/s0006-3223(02)01474-9. [DOI] [PubMed] [Google Scholar]
- Fu CH, Williams SC, Brammer MJ, Suckling J, Kim J, Cleare AJ, et al. Neural responses to happy facial expressions in major depression following antidepressant treatment. Am J Psychiatry. 2007;164:599–607. doi: 10.1176/ajp.2007.164.4.599. [DOI] [PubMed] [Google Scholar]
- Fu CH, Williams SC, Cleare AJ, Scott J, Mitterschiffthaler MT, Walsh ND, et al. Neural responses to sad facial expressions in major depression following cognitive behavioral therapy. Biol Psychiatry. 2008;64:505–512. doi: 10.1016/j.biopsych.2008.04.033. [DOI] [PubMed] [Google Scholar]
- Gatt JM, Nemeroff CB, Dobson-Stone C, Paul RH, Bryant RA, Schofield PR, et al. Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol Psychiatry. 2009;14:681–695. doi: 10.1038/mp.2008.143. [DOI] [PubMed] [Google Scholar]
- Gervasoni N, Bertschy G, Osiek C, Perret G, Denis R, Golaz J, et al. Cortisol responses to combined dexamethasone/CRH test in outpatients with a major depressive episode. J Psychiatr Res. 2004;38:553–557. doi: 10.1016/j.jpsychires.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Gold PW, Chrousos G, Kellner C, Post R, Roy A, Augerinos P, et al. Psychiatric implications of basic and clinical studies with corticotropin-releasing factor. Am J Psychiatry. 1984;141:619–627. doi: 10.1176/ajp.141.5.619. [DOI] [PubMed] [Google Scholar]
- Goldapple K, Segal Z, Garson C, Lau M, Bieling P, Kennedy S, et al. Modulation of cortical-limbic pathways in major depression: treatment-specific effects of cognitive behavior therapy. Arch Gen Psychiatry. 2004;61:34–41. doi: 10.1001/archpsyc.61.1.34. [DOI] [PubMed] [Google Scholar]
- Golimbet VE, Lebedeva IS, Korovaitseva GI, Lezheiko TV, Yumatova PE. Association of 5-HTTLPR serotonin transporter gene polymorphism and Val66Met brain-derived neurotrophic factor gene polymorphism with auditory N100 evoked potential amplitude in patients with endogenous psychoses. Bull Exp Biol Med. 2008;146:605–608. doi: 10.1007/s10517-009-0348-y. [DOI] [PubMed] [Google Scholar]
- Greenberg PE, Kessler RC, Birnbaum HG, Leong SA, Lowe SW, Berglund PA, et al. The economic burden of depression in the United States: how did it change between 1990 and 2000? J Clin Psychiatry. 2003;64:1465–1475. doi: 10.4088/jcp.v64n1211. [DOI] [PubMed] [Google Scholar]
- Griebel G, Simiand J, Serradeil-Le GC, Wagnon J, Pascal M, Scatton B, et al. Anxiolytic- and antidepressant-like effects of the non-peptide vasopressin V1b receptor antagonist, SSR149415, suggest an innovative approach for the treatment of stress-related disorders. Proc Natl Acad Sci USA. 2002;99:6370–6375. doi: 10.1073/pnas.092012099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K. Emerging role of glutamate in the pathophysiology of major depressive disorder. Brain Res Rev. 2009;61:105–123. doi: 10.1016/j.brainresrev.2009.05.005. [DOI] [PubMed] [Google Scholar]
- Hettema JM. What is the genetic relationship between anxiety and depression? Am J Med Genet C Semin Med Genet. 2008;148C:140–146. doi: 10.1002/ajmg.c.30171. [DOI] [PubMed] [Google Scholar]
- Holderbach R, Clark K, Moreau JL, Bischofberger J, Normann C. Enhanced long-term synaptic depression in an animal model of depression. Biol Psychiatry. 2007;62:92–100. doi: 10.1016/j.biopsych.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- Holsboer F, Barden N. Antidepressants and hypothalamic-pituitary-adrenocortical regulation. Endocr Rev. 1996;17:187–205. doi: 10.1210/edrv-17-2-187. [DOI] [PubMed] [Google Scholar]
- Holsboer F, Ising M. Central CRH system in depression and anxiety – evidence from clinical studies with CRH1 receptor antagonists. Eur J Pharmacol. 2008;583:350–357. doi: 10.1016/j.ejphar.2007.12.032. [DOI] [PubMed] [Google Scholar]
- Holsboer F, Von BU, Gerken A, Stalla GK, Muller OA. Blunted corticotropin and normal cortisol response to human corticotropin-releasing factor in depression. N Engl J Med. 1984;311:1127. doi: 10.1056/NEJM198410253111718. [DOI] [PubMed] [Google Scholar]
- Huang R, Huang J, Cathcart H, Smith S, Poduslo SE. Genetic variants in brain-derived neurotrophic factor associated with Alzheimer's disease. J Med Genet. 2007;44:e66. doi: 10.1136/jmg.2006.044883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarguen-Vargas Y, Surget A, Touma C, Palme R, Belzung C. Multifaceted strain-specific effects in a mouse model of depression and of antidepressant reversal. Psychoneuroendocrinology. 2008;33:1357–1368. doi: 10.1016/j.psyneuen.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Ising M, Horstmann S, Kloiber S, Lucae S, Binder EB, Kern N, et al. Combined dexamethasone/corticotropin releasing hormone test predicts treatment response in major depression – a potential biomarker? Biol Psychiatry. 2007;62:47–54. doi: 10.1016/j.biopsych.2006.07.039. [DOI] [PubMed] [Google Scholar]
- Jakovljevic M, Muck-Seler D, Pivac N, Ljubicic D, Bujas M, Dodig G. Seasonal influence on platelet 5-HT levels in patients with recurrent major depression and schizophrenia. Biol Psychiatry. 1997;41:1028–1034. doi: 10.1016/s0006-3223(96)00212-0. [DOI] [PubMed] [Google Scholar]
- Jiang W, Zhang Y, Xiao L, Van CJ, Ji SP, Bai G, et al. Cannabinoids promote embryonic and adult hippocampus neurogenesis and produce anxiolytic- and antidepressant-like effects. J Clin Invest. 2005;115:3104–3116. doi: 10.1172/JCI25509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Zhou J, Mash DC, Marini AM, Lipsky RH. Human BDNF isoforms are differentially expressed in cocaine addicts and are sorted to the regulated secretory pathway independent of the Met66 substitution. Neuromolecular Med. 2009;11:1–12. doi: 10.1007/s12017-008-8051-0. [DOI] [PubMed] [Google Scholar]
- Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry. 2008 doi: 10.1038/mp.2008.116. in press. [DOI] [PubMed] [Google Scholar]
- Kendler KS. Major depression and generalised anxiety disorder. Same genes, (partly)different environments – revisited. Br J Psychiatry Suppl. 1996;30:68–75. [PubMed] [Google Scholar]
- Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ. Major depression and generalized anxiety disorder. Same genes, (partly) different environments? Arch Gen Psychiatry. 1992;49:716–722. doi: 10.1001/archpsyc.1992.01820090044008. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Gardner CO, Gatz M, Pedersen NL. The sources of co-morbidity between major depression and generalized anxiety disorder in a Swedish national twin sample. Psychol Med. 2007;37:453–462. doi: 10.1017/S0033291706009135. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R) JAMA. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- Koenigs M, Grafman J. The functional neuroanatomy of depression: distinct roles for ventromedial and dorsolateral prefrontal cortex. Behav Brain Res. 2009;201:239–243. doi: 10.1016/j.bbr.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koponen E, Rantamaki T, Voikar V, Saarelainen T, MacDonald E, Castren E. Enhanced BDNF signaling is associated with an antidepressant-like behavioral response and changes in brain monoamines. Cell Mol Neurobiol. 2005;25:973–980. doi: 10.1007/s10571-005-8468-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krabbe KS, Nielsen AR, Krogh-Madsen R, Plomgaard P, Rasmussen P, Erikstrup C, et al. Brain-derived neurotrophic factor (BDNF) and type 2 diabetes. Diabetologia. 2007;50:431–438. doi: 10.1007/s00125-006-0537-4. [DOI] [PubMed] [Google Scholar]
- Li X, Need AB, Baez M, Witkin JM. Metabotropic glutamate 5 receptor antagonism is associated with antidepressant-like effects in mice. J Pharmacol Exp Ther. 2006;319:254–259. doi: 10.1124/jpet.106.103143. [DOI] [PubMed] [Google Scholar]
- Li Y, Luikart BW, Birnbaum S, Chen J, Kwon CH, Kernie SG, et al. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron. 2008;59:399–412. doi: 10.1016/j.neuron.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis C, Cohen C, Depoortere R, Griebel G. Antidepressant-like effects of the corticotropin-releasing factor 1 receptor antagonist, SSR125543, and the vasopressin 1b receptor antagonist, SSR149415, in a DRL-72 s schedule in the rat. Neuropsychopharmacology. 2006;31:2180–2187. doi: 10.1038/sj.npp.1301036. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Magarinos AM, Reagan LP. Structural plasticity and tianeptine: cellular and molecular targets. Eur Psychiatry. 2002;17(Suppl. 3):318–330. doi: 10.1016/s0924-9338(02)00650-8. [DOI] [PubMed] [Google Scholar]
- MacQueen GM, Campbell S, McEwen BS, Macdonald K, Amano S, Joffe RT, et al. Course of illness, hippocampal function, and hippocampal volume in major depression. Proc Natl Acad Sci USA. 2003;100:1387–1392. doi: 10.1073/pnas.0337481100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manev R, Uz T, Manev H. Fluoxetine increases the content of neurotrophic protein S100beta in the rat hippocampus. Eur J Pharmacol. 2001;420:R1–R2. doi: 10.1016/s0014-2999(01)00989-x. [DOI] [PubMed] [Google Scholar]
- Maroun M. Stress reverses plasticity in the pathway projecting from the ventromedial prefrontal cortex to the basolateral amygdala. Eur J Neurosci. 2006;24:2917–2922. doi: 10.1111/j.1460-9568.2006.05169.x. [DOI] [PubMed] [Google Scholar]
- Maroun M, Richter-Levin G. Exposure to acute stress blocks the induction of long-term potentiation of the amygdala-prefrontal cortex pathway in vivo. J Neurosci. 2003;23:4406–4409. doi: 10.1523/JNEUROSCI.23-11-04406.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayberg HS, Brannan SK, Mahurin RK, Jerabek PA, Brickman JS, Tekell JL, et al. Cingulate function in depression: a potential predictor of treatment response. Neuroreport. 1997;8:1057–1061. doi: 10.1097/00001756-199703030-00048. [DOI] [PubMed] [Google Scholar]
- Meltzer CC, Price JC, Mathis CA, Butters MA, Ziolko SK, Moses-Kolko E, et al. Serotonin 1A receptor binding and treatment response in late-life depression. Neuropsychopharmacology. 2004;29:2258–2265. doi: 10.1038/sj.npp.1300556. [DOI] [PubMed] [Google Scholar]
- Miller BH, Schultz LE, Gulati A, Cameron MD, Pletcher MT. Genetic regulation of behavioral and neuronal responses to fluoxetine. Neuropsychopharmacology. 2008;33:1312–1322. doi: 10.1038/sj.npp.1301497. [DOI] [PubMed] [Google Scholar]
- Mineur YS, Belzung C, Crusio WE. Functional implications of decreases in neurogenesis following chronic mild stress in mice. Neuroscience. 2007;150:251–259. doi: 10.1016/j.neuroscience.2007.09.045. [DOI] [PubMed] [Google Scholar]
- Moller HJ. Anxiety associated with comorbid depression. J Clin Psychiatry. 2002;63(Suppl. 14):22–26. [PubMed] [Google Scholar]
- Monk CS, Telzer EH, Mogg K, Bradley BP, Mai X, Louro HM, et al. Amygdala and ventrolateral prefrontal cortex activation to masked angry faces in children and adolescents with generalized anxiety disorder. Arch Gen Psychiatry. 2008;65:568–576. doi: 10.1001/archpsyc.65.5.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, et al. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci USA. 2004;101:10827–10832. doi: 10.1073/pnas.0402141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortola JF, Liu JH, Gillin JC, Rasmussen DD, Yen SS. Pulsatile rhythms of adrenocorticotropin (ACTH) and cortisol in women with endogenous depression: evidence for increased ACTH pulse frequency. J Clin Endocrinol Metab. 1987;65:962–968. doi: 10.1210/jcem-65-5-962. [DOI] [PubMed] [Google Scholar]
- Muck-Seler D, Jakovljevic M, Deanovic Z. Effect of antidepressant treatment on platelet 5-HT content and relation to therapeutic outcome in unipolar depressive patients. J Affect Disord. 1991;23:157–164. doi: 10.1016/0165-0327(91)90028-q. [DOI] [PubMed] [Google Scholar]
- Muck-Seler D, Jakovljevic M, Pivac N. Platelet 5-HT concentrations and suicidal behaviour in recurrent major depression. J Affect Disord. 1996;39:73–80. doi: 10.1016/0165-0327(96)00024-9. [DOI] [PubMed] [Google Scholar]
- Mulert C, Juckel G, Brunnmeier M, Karch S, Leicht G, Mergl R, et al. Rostral anterior cingulate cortex activity in the theta band predicts response to antidepressive medication. Clin EEG Neurosci. 2007;38:78–81. doi: 10.1177/155005940703800209. [DOI] [PubMed] [Google Scholar]
- Muller MB, Zimmermann S, Sillaber I, Hagemeyer TP, Deussing JM, Timpl P, et al. Limbic corticotropin-releasing hormone receptor 1 mediates anxiety-related behavior and hormonal adaptation to stress. Nat Neurosci. 2003;6:1100–1107. doi: 10.1038/nn1123. [DOI] [PubMed] [Google Scholar]
- Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet. 1997;349:1498–1504. doi: 10.1016/S0140-6736(96)07492-2. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB. The corticotropin-releasing factor (CRF) hypothesis of depression: new findings and new directions. Mol Psychiatry. 1996;1:336–342. [PubMed] [Google Scholar]
- Nemeroff CB, Widerlov E, Bissette G, Walleus H, Karlsson I, Eklund K, et al. Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science. 1984;226:1342–1344. doi: 10.1126/science.6334362. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB, Owens MJ, Bissette G, Andorn AC, Stanley M. Reduced corticotropin releasing factor binding sites in the frontal cortex of suicide victims. Arch Gen Psychiatry. 1988;45:577–579. doi: 10.1001/archpsyc.1988.01800300075009. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- O'Leary OF, Bechtholt AJ, Crowley JJ, Hill TE, Page ME, Lucki I. Depletion of serotonin and catecholamines block the acute behavioral response to different classes of antidepressant drugs in the mouse tail suspension test. Psychopharmacology (Berl) 2007;192:357–371. doi: 10.1007/s00213-007-0728-9. [DOI] [PubMed] [Google Scholar]
- O'Toole SM, Sekula LK, Rubin RT. Pituitary-adrenal cortical axis measures as predictors of sustained remission in major depression. Biol Psychiatry. 1997;42:85–89. doi: 10.1016/S0006-3223(96)00293-4. [DOI] [PubMed] [Google Scholar]
- Palucha A, Klak K, Branski P, van der PH, Flor PJ, Pilc A. Activation of the mGlu7 receptor elicits antidepressant-like effects in mice. Psychopharmacology (Berl) 2007;194:555–562. doi: 10.1007/s00213-007-0856-2. [DOI] [PubMed] [Google Scholar]
- Pariante CM. Depression, stress and the adrenal axis. J Neuroendocrinol. 2003;15:811–812. doi: 10.1046/j.1365-2826.2003.01058.x. [DOI] [PubMed] [Google Scholar]
- Parker KJ, Schatzberg AF, Lyons DM. Neuroendocrine aspects of hypercortisolism in major depression. Horm Behav. 2003;43:60–66. doi: 10.1016/s0018-506x(02)00016-8. [DOI] [PubMed] [Google Scholar]
- Pezawas L, Meyer-Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS, et al. 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci. 2005;8:828–834. doi: 10.1038/nn1463. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. 2008;33:88–109. doi: 10.1038/sj.npp.1301574. [DOI] [PubMed] [Google Scholar]
- Pivac N, Muck-Seler D, Jakovljevic M. Platelet 5-HT levels and hypothalamic-pituitary-adrenal axis activity in schizophrenic patients with positive and negative symptoms. Neuropsychobiology. 1997;36:19–21. doi: 10.1159/000119354. [DOI] [PubMed] [Google Scholar]
- Pizzagalli D, Pascual-Marqui RD, Nitschke JB, Oakes TR, Larson CL, Abercrombie HC, et al. Anterior cingulate activity as a predictor of degree of treatment response in major depression: evidence from brain electrical tomography analysis. Am J Psychiatry. 2001;158:405–415. doi: 10.1176/appi.ajp.158.3.405. [DOI] [PubMed] [Google Scholar]
- Raone A, Cassanelli A, Scheggi S, Rauggi R, Danielli B, De Montis MG. Hypothalamus-pituitary-adrenal modifications consequent to chronic stress exposure in an experimental model of depression in rats. Neuroscience. 2007;146:1734–1742. doi: 10.1016/j.neuroscience.2007.03.027. [DOI] [PubMed] [Google Scholar]
- Redrobe JP, Dumont Y, Fournier A, Baker GB, Quirion R. Role of serotonin (5-HT) in the antidepressant-like properties of neuropeptide Y (NPY) in the mouse forced swim test. Peptides. 2005;26:1394–1400. doi: 10.1016/j.peptides.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Reif A, Fritzen S, Finger M, Strobel A, Lauer M, Schmitt A, et al. Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol Psychiatry. 2006;11:514–522. doi: 10.1038/sj.mp.4001791. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Mayberg HS. Targeting abnormal neural circuits in mood and anxiety disorders: from the laboratory to the clinic. Nat Neurosci. 2007;10:1116–1124. doi: 10.1038/nn1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro SC, Tandon R, Grunhaus L, Greden JF. The DST as a predictor of outcome in depression: a meta-analysis. Am J Psychiatry. 1993;150:1618–1629. doi: 10.1176/ajp.150.11.1618. [DOI] [PubMed] [Google Scholar]
- Rocher C, Spedding M, Munoz C, Jay TM. Acute stress-induced changes in hippocampal/prefrontal circuits in rats: effects of antidepressants. Cereb Cortex. 2004;14:224–229. doi: 10.1093/cercor/bhg122. [DOI] [PubMed] [Google Scholar]
- Rogers MA, Kasai K, Koji M, Fukuda R, Iwanami A, Nakagome K, et al. Executive and prefrontal dysfunction in unipolar depression: a review of neuropsychological and imaging evidence. Neurosci Res. 2004;50:1–11. doi: 10.1016/j.neures.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Roy MA, Neale MC, Pedersen NL, Mathe AA, Kendler KS. A twin study of generalized anxiety disorder and major depression. Psychol Med. 1995;25:1037–1049. doi: 10.1017/s0033291700037533. [DOI] [PubMed] [Google Scholar]
- Ruhe HG, Mason NS, Schene AH. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: a meta-analysis of monoamine depletion studies. Mol Psychiatry. 2007;12:331–359. doi: 10.1038/sj.mp.4001949. [DOI] [PubMed] [Google Scholar]
- Rybakowski JK. BDNF gene: functional Val66Met polymorphism in mood disorders and schizophrenia. Pharmacogenomics. 2008;9:1589–1593. doi: 10.2217/14622416.9.11.1589. [DOI] [PubMed] [Google Scholar]
- Salome N, Stemmelin J, Cohen C, Griebel G. Differential roles of amygdaloid nuclei in the anxiolytic- and antidepressant-like effects of the V1b receptor antagonist, SSR149415, in rats. Psychopharmacology (Berl) 2006;187:237–244. doi: 10.1007/s00213-006-0424-1. [DOI] [PubMed] [Google Scholar]
- Salvadore G, Cornwell BR, Colon-Rosario V, Coppola R, Grillon C, Zarate CA, Jr, et al. Increased anterior cingulate cortical activity in response to fearful faces: a neurophysiological biomarker that predicts rapid antidepressant response to ketamine. Biol Psychiatry. 2009;65:289–295. doi: 10.1016/j.biopsych.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7:426–437. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- Sargent PA, Kjaer KH, Bench CJ, Rabiner EA, Messa C, Meyer J, et al. Brain serotonin1A receptor binding measured by positron emission tomography with [11C]WAY-100635: effects of depression and antidepressant treatment. Arch Gen Psychiatry. 2000;57:174–180. doi: 10.1001/archpsyc.57.2.174. [DOI] [PubMed] [Google Scholar]
- Saxena S, Brody AL, Ho ML, Zohrabi N, Maidment KM, Baxter LR., Jr Differential brain metabolic predictors of response to paroxetine in obsessive-compulsive disorder versus major depression. Am J Psychiatry. 2003;160:522–532. doi: 10.1176/appi.ajp.160.3.522. [DOI] [PubMed] [Google Scholar]
- Schinka JA, Busch RM, Robichaux-Keene N. A meta-analysis of the association between the serotonin transporter gene polymorphism (5-HTTLPR) and trait anxiety. Mol Psychiatry. 2004;9:197–202. doi: 10.1038/sj.mp.4001405. [DOI] [PubMed] [Google Scholar]
- Schloesser RJ, Manji HK, Martinowich K. Suppression of adult neurogenesis leads to an increased hypothalamo-pituitary-adrenal axis response. Neuroreport. 2009;20:553–557. doi: 10.1097/WNR.0b013e3283293e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweinhardt P, Kalk N, Wartolowska K, Chessell I, Wordsworth P, Tracey I. Investigation into the neural correlates of emotional augmentation of clinical pain. Neuroimage. 2008;40:759–766. doi: 10.1016/j.neuroimage.2007.12.016. [DOI] [PubMed] [Google Scholar]
- Sen S, Villafuerte S, Nesse R, Stoltenberg SF, Hopcian J, Gleiberman L, et al. Serotonin transporter and GABAA alpha 6 receptor variants are associated with neuroticism. Biol Psychiatry. 2004;55:244–249. doi: 10.1016/j.biopsych.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Shakesby AC, Anwyl R, Rowan MJ. Overcoming the effects of stress on synaptic plasticity in the intact hippocampus: rapid actions of serotonergic and antidepressant agents. J Neurosci. 2002;22:3638–3644. doi: 10.1523/JNEUROSCI.22-09-03638.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spedding M, Neau I, Harsing L. Brain plasticity and pathology in psychiatric disease: sites of action for potential therapy. Curr Opin Pharmacol. 2003;3:33–40. doi: 10.1016/s1471-4892(02)00008-5. [DOI] [PubMed] [Google Scholar]
- Stemmelin J, Lukovic L, Salome N, Griebel G. Evidence that the lateral septum is involved in the antidepressant-like effects of the vasopressin V1b receptor antagonist, SSR149415. Neuropsychopharmacology. 2005;30:35–42. doi: 10.1038/sj.npp.1300562. [DOI] [PubMed] [Google Scholar]
- Surget A, Saxe M, Leman S, Ibarguen-Vargas Y, Chalon S, Griebel G, et al. Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol Psychiatry. 2008;64:293–301. doi: 10.1016/j.biopsych.2008.02.022. [DOI] [PubMed] [Google Scholar]
- Szymanska M, Budziszewska B, Jaworska-Feil L, Basta-Kaim A, Kubera M, Leskiewicz M, et al. The effect of antidepressant drugs on the HPA axis activity, glucocorticoid receptor level and FKBP51 concentration in prenatally stressed rats. Psychoneuroendocrinology. 2009;34:822–832. doi: 10.1016/j.psyneuen.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. 2009;10:397–409. doi: 10.1038/nrn2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utge S, Soronen P, Partonen T, Loukola A, Kronholm E, Pirkola S, et al. A population-based association study of candidate genes for depression and sleep disturbance. Am J Med Genet B Neuropsychiatr Genet. 2009 doi: 10.1002/ajmg.b.31002. in press. [DOI] [PubMed] [Google Scholar]
- Veen G, Derijk RH, Giltay EJ, van VI, van PJ, Zitman FG. The influence of psychiatric comorbidity on the dexamethasone/CRH test in major depression. Eur Neuropsychopharmacol. 2009a;19:409–415. doi: 10.1016/j.euroneuro.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Veen G, Giltay EJ, Derijk RH, van VI, van PJ, Zitman FG. Salivary cortisol, serum lipids, and adiposity in patients with depressive and anxiety disorders. Metabolism. 2009b;58:821–827. doi: 10.1016/j.metabol.2009.02.009. [DOI] [PubMed] [Google Scholar]
- Vreeburg SA, Hoogendijk WJ, van PJ, Derijk RH, Verhagen JC, Van DR, et al. Major depressive disorder and hypothalamic-pituitary-adrenal axis activity: results from a large cohort study. Arch Gen Psychiatry. 2009;66:617–626. doi: 10.1001/archgenpsychiatry.2009.50. [DOI] [PubMed] [Google Scholar]
- Watson S, Gallagher P, Del-Estal D, Hearn A, Ferrier IN, Young AH. Hypothalamic-pituitary-adrenal axis function in patients with chronic depression. Psychol Med. 2002;32:1021–1028. doi: 10.1017/s0033291702005998. [DOI] [PubMed] [Google Scholar]
- Whalen PJ, Johnstone T, Somerville LH, Nitschke JB, Polis S, Alexander AL, et al. A functional magnetic resonance imaging predictor of treatment response to venlafaxine in generalized anxiety disorder. Biol Psychiatry. 2008;63:858–863. doi: 10.1016/j.biopsych.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojnar M, Brower KJ, Strobbe S, Ilgen M, Matsumoto H, Nowosad I, et al. Association between Val66Met brain-derived neurotrophic factor (BDNF) gene polymorphism and post-treatment relapse in alcohol dependence. Alcohol Clin Exp Res. 2009;33:693–702. doi: 10.1111/j.1530-0277.2008.00886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray NR, James MR, Gordon SD, Dumenil T, Ryan L, Coventry WL, et al. Accurate, large-scale genotyping of 5HTTLPR and flanking single nucleotide polymorphisms in an association study of depression, anxiety and personality measures. Biol Psychiatry. 2009;66:468–476. doi: 10.1016/j.biopsych.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Buchsbaum MS, Gillin JC, Tang C, Cadwell S, Wiegand M, et al. Prediction of antidepressant effects of sleep deprivation by metabolic rates in the ventral anterior cingulate and medial prefrontal cortex. Am J Psychiatry. 1999;156:1149–1158. doi: 10.1176/ajp.156.8.1149. [DOI] [PubMed] [Google Scholar]
- Yalcin I, Belzung C, Surget A. Mouse strain differences in the unpredictable chronic mild stress: a four-antidepressant survey. Behav Brain Res. 2008;193:140–143. doi: 10.1016/j.bbr.2008.04.021. [DOI] [PubMed] [Google Scholar]
- Yoshimizu T, Chaki S. Increased cell proliferation in the adult mouse hippocampus following chronic administration of group II metabotropic glutamate receptor antagonist, MGS0039. Biochem Biophys Res Commun. 2004;315:493–496. doi: 10.1016/j.bbrc.2004.01.073. [DOI] [PubMed] [Google Scholar]
- Young EA, Altemus M, Lopez JF, Kocsis JH, Schatzberg AF, DeBattista C, et al. HPA axis activation in major depression and response to fluoxetine: a pilot study. Psychoneuroendocrinology. 2004;29:1198–1204. doi: 10.1016/j.psyneuen.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]
- Zobel AW, Yassouridis A, Frieboes RM, Holsboer F. Prediction of medium-term outcome by cortisol response to the combined dexamethasone-CRH test in patients with remitted depression. Am J Psychiatry. 1999;156:949–951. doi: 10.1176/ajp.156.6.949. [DOI] [PubMed] [Google Scholar]
- Zobel AW, Nickel T, Sonntag A, Uhr M, Holsboer F, Ising M. Cortisol response in the combined dexamethasone/CRH test as predictor of relapse in patients with remitted depression. a prospective study. J Psychiatr Res. 2001;35:83–94. doi: 10.1016/s0022-3956(01)00013-9. [DOI] [PubMed] [Google Scholar]