

Abstract
Background and purpose:
Excitation–transcriptional coupling involves communication between plasma membrane ion channels and gene expression in the nucleus. Calcium influx through L-type Ca2+ channels induces phosphorylation of the transcription factor, cyclic-AMP response element binding protein (CREB) and downstream activation of the cyclic-AMP response element (CRE) promoter regions. Tyrosine nitration of Ca2+ channels attenuates interactions with c-Src kinase, decreasing Ca2+ channel currents and smooth muscle contraction during colonic inflammation. In this study we examined the effect of tyrosine nitration and colonic inflammation on Ca2+ channel mediated phosphorylation of CREB and CRE activation.
Experimental approach:
CREB and phospho-CREB were detected by Western blots and CRE activation measured by dual luciferase assay. Chinese hamster ovary (CHO) cells were transfected with hCav1.2b and hCav1.2b c-terminal mutants. Colonic inflammation was induced by intracolonic instillation of 2,4,6 trinitrobenzene sulphonic acid in mouse colon.
Key results:
In hCav1.2b transfected CHO cells and in native colonic smooth muscle, depolarization with 80 mM KCl induced CREB phosphorylation (pCREB). Treatment with peroxynitrite inhibited KCl-induced pCREB. Following experimental colitis, KCl-induced CREB phosphorylation was abolished in smooth muscle, concomitant with tyrosine nitration of Ca2+ channels. Depolarization increased CRE activation in hCav1.2b CHO cells by 2.35 fold which was blocked by nifedipine and by protein nitration of Ca2+ channels with peroxynitrite. The Src-kinase inhibitor, PP2, blocked depolarization-induced CRE activation. Mutation of the C-terminus tyrosine residue, Y2134F, but not Y1861F, blocked CRE activation.
Conclusions and implications:
Post-translational modification of Ca2+ channels due to tyrosine nitration modified excitation–transcriptional coupling in colonic inflammation.
Keywords: nitration, tyrosine kinase, Ca2+ channel, colonic inflammation, gene expression
Introduction
Voltage-gated Ca2+ channels play an essential role in many processes including those related to smooth muscle contraction (excitation–contraction coupling), neurotransmitter release (excitation–secretion coupling) and gene expression (excitation–transcription coupling). Excitation–transcription coupling has been demonstrated in several cell types, including smooth muscle, to involve Ca2+ channel dependent activation of transcription factors (Barlow et al., 2006; Wamhoff et al., 2006). One of the transcription factors that is strongly coupled to L-type Ca2+ channel activation is the cyclic AMP response element binding protein (CREB) (Dolmetsch et al., 2001). Nuclear CREB is phosphorylated at Ser133 upon cell depolarization. In hippocampal neurons, translocation of calmodulin to the nucleus acts as a primary mechanism leading to activation of calmodulin dependent kinase(s) and phosphorylation of CREB (Deisseroth et al., 1998). The issue of how voltage changes may result in excitation–transcription coupling was recently addressed by Wheeler et al. (2008), who demonstrated the dependence of nuclear CREB phosphorylation on the open probability of the L-type Ca2+ channel rather than bulk increases in intracellular Ca2+ thus defining the gating of the Ca2+ channel as an important step in Ca2+-mediated gene transcription.
Ca2+ currents are significantly decreased during colonic inflammation leading to impaired motility (Akbarali et al., 2000; Liu et al., 2001; Kinoshita et al., 2003). This contributes to the morbidity of colitis associated with ischemia, infection and inflammatory bowel disease. In addition to the critical role in muscle contraction, the L-type Ca2+ channels are necessary for gene expression of smooth muscle differentiation markers (Wamhoff et al., 2004) and colonic inflammation results in expression of several pro- and anti-inflammatory mediators whose gene expression may be regulated in smooth muscle cells (Shi and Sarna, 2005). Thus down-regulation of the Ca2+ channel during inflammation is likely to have profound effects, both towards changes in motility as well as gene expression. We have recently addressed the potential mechanisms by which colonic inflammation may alter Ca2+ currents. Our studies show that attenuation of the smooth muscle Ca2+ currents may occur as a result of altered regulation of Ca2+ channel phosphorylation rather than decreased protein expression in mouse colonic inflammation. In mouse colon, inflammation induced by either dextran sulphate sodium (DSS) (Kang et al., 2004) or TNBS (Ross et al., 2007) did not alter protein or mRNA expression of the L-type Ca2+ channel, Cav1.2b (channel nomenclature follows Alexander et al., 2008)but the regulation of the channel by the tyrosine kinase, c-Src kinase, was altered. Similar results have also been shown in atrial cells from patients with chronic atrial fibrillation (AF). Ca2+ currents are reduced by more than 50% in atrial cells from AF patients, however, neither protein expression of the α or the auxillary β subunit or mRNA expression of the Ca2+ channel were altered (Christ et al., 2004). We further showed that nitration by peroxynitrite of tyrosine residues within the carboxy terminus of the smooth muscle isoform of the Ca2+ channel, Cav1.2b, prevents phosphorylation by c-Src kinase resulting in decreased Ca2+ currents (Kang et al., 2007; Ross et al., 2007). Colonic inflammation induces neutrophil and macrophage recruitment in the colonic wall resulting in enhanced expression of the inducible nitric oxide synthase (iNOS). Peroxynitrite (ONOO-), formed from the reaction of nitric oxide (NO) with superoxide (O2-) is thought to be the major nitrating agent in vivo and has been shown to nitrate tyrosine residues in intact cells and in vitro chemical studies (Ischiropoulos et al., 1992; Estevez et al., 1998), including nitrotyrosine expression in epithelia from patients with ulcerative colitis (Singer et al., 1996). Our previous studies demonstrated that the Ca2+ channels from mouse colonic smooth muscle were nitrated following induction of inflammation and we have further established that residues Y2134 and Y1837 within the C-terminus are potential tyrosine phosphorylation sites (Kang et al., 2007),which, when nitrated, result in decreased Ca2+ currents.
The objective of this study was to determine whether tyrosine nitration of the Ca2+ channel affects downstream activation of the transcription factor CREB and the activation of its DNA binding to CRE.
Methods
Cell culture and transient transfection
Chinese hamster ovary (CHO) cells were maintained and grown to ∼80% confluence in DMEM/F12 medium supplemented with 10% fetal bovine serum, 100 units·mL−1 penicillin, and 100 µg·mL−1 streptomycin (Invitrogen, Carlsbad, CA, USA) in a humidified atmosphere of 5% CO2 and 95% O2 at 37°C. Transient transfections were performed using Genejammer reagent (Stratagene, La Jolla, CA, USA) as described by the manufacturer, and experiments were performed 24–72 h after transfection.
Induction of colitis
All animal care and experimental protocols followed the guidelines and approval by the Virginia Commonwealth University Institutional Animal Care and Use Committee. Colonic inflammation was induced by intracolonic instillation of trinitrobenzene sulphonic acid (TNBS; 2.5% in 50% ethanol) in male Balb/C mice (25–30 g) through a 3.5-French catheter carefully inserted into the rectum. Under isoflurane anaesthesia, a catheter tip was inserted 4 cm proximal to the anal verge. Mice were carefully held in a vertical position (head down) for 60 s after TNBS instillation to ensure distribution of the TNBS within the entire colon. Control animals received the same volume (100 µL) of vehicle solution. The mice were weighed daily and inspected for diarrhea and rectal bleeding for assessing the disease activity index (DAI) as previously reported (Akbarali et al., 2000; Jin et al., 2004). Briefly, scores were assigned ranging from 0 to 4, with 0 being no weight loss, normal stool consistency and no rectal bleeding, and 4 assigned when the weight loss exceeded 20% of initial weight with gross bleeding. All mice used in this study had a DAI of >3.5.
Real-time PCR
Quantitative real-time PCR assay for interleukin (IL)-1β was performed to confirm inflammatory changes on a MiniOpticon Real-Time PCR System (Bio-Rad, Hercules, CA, USA). Ribosomal 18S RNA was used as the internal control. Colons were isolated from normal and inflamed mice and total RNA was extracted by the Total RNA Purification System (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. cDNA synthesis and subsequent polymerization was performed in a single step using the SensiMix One-Step Kit (Quantace, Taunton, MA, USA). The reaction mixture (50 µL) contained 200 nM forward primer, 200 nM reverse primer, 1X SensiMix One-Step buffer, 1X SYBR Green I solution, 10 Units RNase inhibitor, and 200 ng total RNA. With the use of the one-step SensiMix kit, reverse transcription was performed for 30 min at 42°C with an enzyme activation step for 10 min at 95°C. The PCR protocol consisted of 40 cycles of denaturation (15 s at 95°C), annealing (30 s at 60°C) and extension (30 s at 72°C). The cycle threshold value, Ct count, for each sample was normalized by subtracting it from the Ct value of the reference gene 18S to establish ΔCt (ΔCt = Il-1β Ct – 18S Ct) for each sample. The fold change was calculated as 2-ΔΔCt. All primers were designed using the Vector NTI software (Invitrogen, Carlsbad, CA, USA), and the sequences in this study used were: 5′-CCTGAACTCAACTGTGAAATGC-3′ (forward) and 5′-CGAGATTTGAAGCTGGATGC-3′ (reverse) for Il-1β (NM_008361), 5′-TCAAGAACGAAAGTCGGAGG-3′ (forward) and 5′-GGACATCTAAGGGCATCAC-3′ (reverse) for 18S (X00686).
CRE luciferase assays
For estimation of CRE activity, CHO cells were co-transfected using Genejammer transfection reagents with the Ca2+ channel β2 subunit that aids in the membrane expression of the α subunit and depending on the experimental protocol, one or more of the α subunit plasmids, hCav1.2b, single or triple tyrosine Ca2+ channel mutants. The inducible CRE-responsive firefly luciferase construct, negative and positive controls from the CRE reporter kit (SABiosciences, Frederick MD, USA) were also co-transfected in these cells. The negative control consists of non-inducible firefly luciferase construct while the positive control has a constitutively expressing firefly luciferase and a GFP construct used for determining transfection efficiency. The CRE reporter kit is premixed with Renilla luciferase that is under the control of a CMV promoter and serves as the internal control. After 48- to 72-h incubation, cells (2 × 104 cells·mL−1) in a 48-well plate were exposed to 80 mM KCl HEPES buffer (60.4 mM NaCl, 80 mM KCl, 0.33 mM NaH2PO4, 5 mM HEPES, 1 mM MgCl2, 2 mM CaCl2, 5 mM D-glucose, pH 7.4), and treated with either forskolin (10 µM), peroxynitrite (150 µM), nifedipine (1 µM), ionomycin (1 µM) or PP2 (5 µM) for the desired durations. After treatment, cells were incubated with 65 µL of passive lysis buffer on a rocking platform at room temperature for 15 min and cell lysates were then transferred to a new tube prior to reporter analyses. A dual-luciferase assay was performed using the Promega Dual-Luciferase assay kit according to the manufacturer's protocols and firefly and Renilla luciferase activities were determined using a Modulus Microplate Luminometer (Turner BioSystems, Sunnyvale, CA, USA). To calculate the relative CRE activity, the firefly luciferase activities were normalized against the corresponding Renilla luciferase and the ratio of CRE: Renilla determined. All experiments were performed in triplicate.
Immunoblotting analysis of nuclear phosphorylated CREB proteins
CHO cells transfected with human jejunal voltage-gated Ca2+ channel, hCav1.2b, and β2 subunit were treated with KCl, nifedipine, and/or BayK 8644 in HEPES buffer for the indicated times. Distal colons were excised from normal and inflamed mice, stripped of mucosa, and treated with 80 mM KCl in Krebs solution for the indicated times in an organ bath, and bubbled continuously with carbogen (95% O2 and 5% CO2). Nuclear extractions from Ca2+channel-transfected CHO cells and mouse colons were prepared using Promega Nuclear extraction kit and protein concentration was measured by BCA assay kit. Samples were separated on 10–12% SDS PAGE gels and transferred onto Immobilon membranes (Bio-Rad, Hercules, CA, USA). After transfer, the membranes were blocked for 1 h at 4°C with TBS-T (0.1% Tween 20 and 20 mM Tris buffered saline, pH 7.6) containing 5% BSA and incubated overnight at 4oC with anti-rabbit pCREB (1:500) and anti-mouse CREB (1:1000) primary antibodies diluted in TBS-T containing 5% non-fat milk. The membranes were then incubated for 1 h at room temperature with secondary antibodies, each of anti-mouse IRDye 800 and anti-rabbit IRDye 680, diluted (1:5000) in 5% non-fat milk blocking buffer with 0.1% Tween 20. After rinsing with TBS-T buffer three times, the membranes were visualized using a LiCor Odyssey Infrared imaging system (LiCor Biosciences, Lincoln, NE, USA).
Immunoprecipitation of nitrosylated Ca2+ channels from mouse Colons
Equal amount of membrane extracts from distal colons of normal and inflamed mice prepared using Pierce Mem-PER Eukaryotic Membrane Protein Extraction Reagent kit, were preincubated with 0.5 µg of the appropriate normal control IgG, corresponding to the host species of the primary antibody, together with 20 µL of Protein A/G PLUS-agarose at 4°C for 30 min. After removal of the beads by centrifugation, the supernatants were incubated on ice with 10 µg of primary antibodies. After an hour, 20 µL of Protein A/G PLUS-agarose was added and incubated at 4°C on a rocker platform for 1 h to overnight. The beads were washed three times in PBS and were then resuspended in 40 µL of 2X electrophoresis sample buffer. Samples were boiled for 3 min and used for electrophoresis and immunoblotting as described in the previous section.
Immunohistochemical staining of Ca2+ channels and pCREB proteins
Immunohistochemical staining was performed by standard protocols. CHO cells transfected with wild-type Ca2+ channels or triple mutant with β2 subunit, were depolarized with 80 mM KCl in the HEPES buffer for 30–60 min, fixed in 4% formaldehyde in PBS for 15 min at room temperature and then washed in PBS three times for 5 min each. After formaldehyde fixation, cells were incubated in ice-cold 100% methanol for 10 min at −20°C, rinsed in PBS for 5 min and then blocked for 60 min with PBS/0.3% Triton X-100 containing 10% non-immune goat serum. Subsequently, samples were incubated with either anti-rabbit Cav1.2 antibody (1:50) or anti-pCREB antibody (1:50) in PBS/0.3% Triton X-100 for overnight at 4°C. Cells were washed in PBS three times. Cells were then treated with the Alexa Fluor® 568 conjugated rabbit polyclonal secondary antibody at 1:1000 dilution in PBS/0.3% Triton X-100 for 1 h at room temperature in the dark. Images were collected using an Olympus FV300 confocal microscope (Olympus America Inc, Center Valley, PA, USA) at the appropriate excitation wavelength.
Whole cell electrophysiology
Changes in membrane potential and calcium currents were measured in transfected CHO cells by whole cell current clamp and voltage clamp techniques respectively. All experiments were performed at room temperature using EPC10 (Heka, Germany) patch clamp amplifier. Patch pipettes were pulled from thin-walled borosilicate glass, and the resistance was 3–5 MΩ when filled with internal solution. For current clamp experiments, the internal solution consisted of 100 mM K+-aspartate, 30 mM KCl, 2 mM MgCl2, 5 mM HEPES, 5 mM EGTA, 5 mM ATP (disodium salt). The cells were continuously perfused with HEPES-buffered solution (135 mM NaCl, 5.4 mM KCl, 0.33 mM NaH2PO4, 5 mM HEPES, 1 mM Mg2Cl, 2 mM CaCl2, and 5.5 mM glucose: pH 7.4 with NaOH). For 80 mM K+ HEPES buffer, 75 mM NaCl in HEPES buffer was substituted by equimolar concentrations of KCl. Calcium currents were measured in voltage-clamp mode with the internal solution containing 100 mM Cs-aspartate, 30 mM CsCl, 2 mM MgCl2, 5 mM HEPES, 5 mM EGTA, 5 mM ATP. Steady-state inactivation was measured using two-step voltage protocols whereas voltage-dependence of activation was calculated from peak current-voltage relationships assuming a reversal potential of +60 mV. Peak conductance was normalized and plotted against the test potential. The sigmoidal curves were fitted by Boltzmann functions as described previously (Akbarali and Giles, 1993).
Data analysis
The data are expressed as means ± SE or mean ± SD as indicated from three to five individual experiments, each run in duplicate. Statistical analysis was performed by ANOVA or by paired Student's t-test, where appropriate, followed by Bonferroni's post hoc test. P values <0.05 were considered significant.
Materials
Protease inhibitors were purchased from Roche Diagnostics (Indianapolis, IN, USA), BCA protein assay kit and nuclear extraction kit from Pierce (Rockford, IL, USA), 3-morpholinosydnonimine (SIN-1) was purchased from TOCRIS Bioscience (Ellisville, MO, USA), peroxynitrite (ONOO-) was obtained from Cayman Chemical (Ann Arbor, MI, USA), nifedipine, BayK 8644, ionomycin, forskolin, and IBMX were purchased from Sigma-Aldrich (St. Louis, MO, USA), the tyrosine kinase inhibitor PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine) was purchased from Calbiochem (San Diego, CA, USA). Isoflurane was supplied by Baxter (Deerfield, IL, USA). The CRE reporter assay kit was purchased from SuperArray Bioscience Corp (Frederick, MD, USA). The dual-luciferase reporter assay system was purchased from Promega (Madison, WI, USA). The anti-rabbit calcium channel antibody was from Alomone labs (Jerusalem, Israel). The anti-rabbit pCREB and anti-mouse CREB antibodies were purchased from Cell Signaling (Danvers, MA, USA). Non-immune Goat serum and Alexa Fluor® 568 conjugated rabbit polyclonal antibody were from Invitrogen (Carlsbad, CA, USA). The goat anti-rabbit IRDye® 680 and goat anti-mouse IRDye® 800CW antibodies were purchased from LiCor Biosciences (Lincoln, NE, USA).
Results
Under current clamp, perfusion of isolated hCav1.2b CHO cells with 80 mM K+ resulted in membrane depolarization from resting potentials of ∼−50 mV to between −15 mV to −6 mV (n= 2) (Figure 1A). To test whether this depolarization was within the ‘window’ current range for Ca2+channels, the voltage dependence of steady-state activation and inactivation was measured, plotted and fitted with Boltzmann functions. As shown in Figure 1B, the two curves overlap resulting in a window current between −25–0 mV, suggesting the presence of non-inactivating Ca2+ currents. As 80-mM K+-induced depolarization lies within this range, this concentration was used to determine Ca2+-mediated activation of the transcription factor CREB and CRE.
Figure 1.
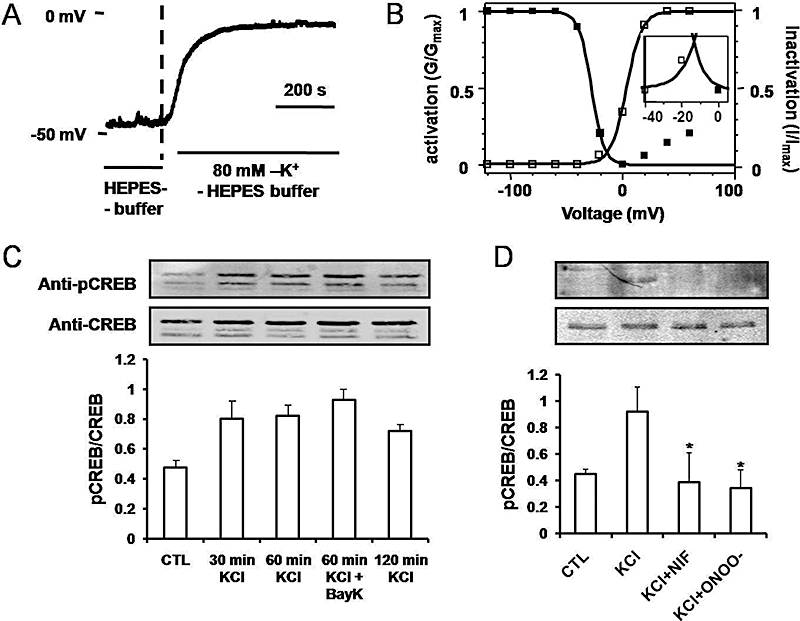
Membrane depolarization and Ca2+ channel window currents in hCav1.2b-transfected CHO cells. (A) Current clamp recording showing depolarization to a steady membrane potential of −6 mV by perfusion with 80 mM KCl-HEPES buffer. (B) Ca2+ channel window currents obtained by overlap of Boltzmann fits for voltage dependent inactivation and activation curves. Inset: Detail of the window current potential range. (C) Time dependent pCREB expression by 80 mM KCl. The hCav1.2b transfected CHO cells were treated with 80 mM KCl for 0 to 120 min and pCREB expression normalized to total CREB. (D) Inhibition of pCREB expression by nifedipine (1 µM) and peroxynitrite (150 µM). CHO cells were incubated with either nifedipine (NIF) or peroxynitrite (ONOO-) for 30 min prior to depolarization for 30 min by KCl. Ratio of phospho-CREB to total CREB are presented in the bottom panels as mean ± SD (n= 3). *P < 0.001 versus KCl. CREB, cyclic AMP response element binding protein.
Nitration of the Ca2+ channel and colonic inflammation reduce depolarization-induced CREB phosphorylation
In hCav1.2b transfected CHO cells, phospho-CREB (pCREB) and total CREB from nuclear extracts were measured by Western blots using anti-pCREB and CREB antibodies. Increase in pCREB could be observed within 30 min and remained elevated, for up to 120 min following 80 mM KCl-induced depolarization (Figure 1C). The combination of KCl with the Ca2+ channel agonist, BayK8644 (1 µM), did not lead to a further increase in pCREB. KCl-induced pCREB was abolished in the presence of nifedipine (10 µM) (Figure 1D). Our previous studies have shown that treatment with peroxynitrite results in the tyrosine nitration of the Ca2+ channel (Ross et al., 2007) and inhibits Ca2+ channel currents. We therefore examined whether CREB phosphorylation induced by membrane depolarization is also affected by nitration. Pretreatment with peroxynitrite (ONOO-) attenuated the KCl-induced increase in pCREB (Figure 1D). Tyrosine nitration of hCav1.2b by peroxynitrite was further confirmed by anti-nitrotyrosine antibody in Western blots of transfected CHO cells (Figure 2A). The expression of pCREB in the nucleus was also determined by immunohistochemistry with anti-pCREB antibody (Figure 2B). Ca2+ channel-transfected CHO cells were treated with 80 mM KCl for 30 min and 60 min, respectively, and then stained with anti-pCREB antibody. KCl-induced depolarization resulted in pCREB expression in the nucleus but not in the absence of depolarization. These findings in hCav1.2b-transfected CHO cells demonstrate depolarization-mediated Ca2+ influx results in pCREB expression and that tyrosine nitration of the Ca2+ channel attenuated pCREB.
Figure 2.
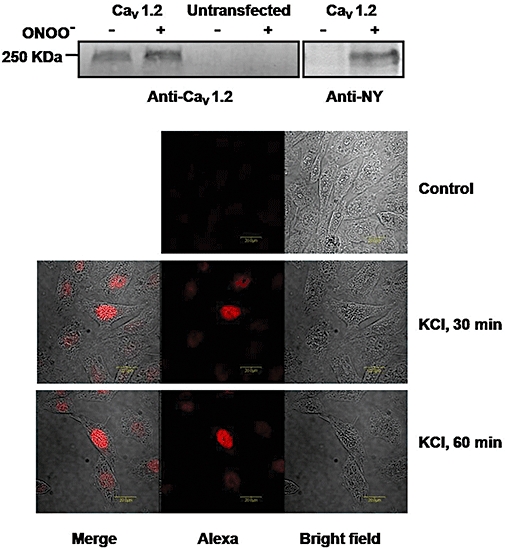
Protein tyrosine nitration of the Ca2+ channel following in vitro treatment with peroxynitrite (ONOO-) in hCav1.2b-transfected CHO cells. (A) Immunoblot with anti-Cav1.2 antibody (left) and anti-nitrotyrosine antibody (right). Protein samples were immunoprecipitated with the Ca2+ channel antibody before immunoblot. Treatment with peroxynitrite enhanced tyrosine nitration of hCav1.2b. (B) Expression of phospho-CREB in hCav1.2b-transfected CHO cells detected by anti-pCREB antibody (1:50 dilution). Staining in the nucleus was observed in hCav1.2b transfected cells treated with 80 mM KCl for either 30 min or 60 min but not in the absence of depolarization (top panel). An Alexa Fluor 568 conjugated antibody (1:1000 dilutions) was used as the secondary antibody. Representative images from three experiments are shown. CREB, cyclic AMP response element binding protein.
In mouse colonic smooth muscle tissues, depolarization with 80 mM KCl resulted in increased expression of pCREB in nuclear extracts that was evident within 15 min with further increases observed at 30 and 60 min (Figure 3A). Treatment with peroxynitrite prevented KCl-induced increases in pCREB expression (Figure 3B). KCl-induced expression of pCREB in smooth muscle was blocked by nifedipine (1 µM) but not by the ATP-sensitive K+ channel blocker, glibenclamide (10 µM) (Figure 3B). These studies suggested that tyrosine nitration results in decreased Ca2+-influx dependent phosphorylation of CREB in mouse colonic smooth muscle. Figure 4A shows that colonic inflammation significantly enhanced tyrosine nitration of several proteins. Cell lysates from smooth muscle tissues were immunoprecipitated with anti-rabbit nitrotyrosine antibodies and immunoblotted with mouse anti-nitrotyrosine antibodies. Several protein bands with enhanced nitrotyrosine were evident from inflamed samples. To determine if the Ca2+ channel was nitrated, control and inflamed colonic muscle was immunoprecipitated with the Ca2+ channel antibody followed by immunoblot with anti-nitrotyrosine. Significant increase in Ca2+ channel nitration occurred following inflammation (Figure 4B). We also tested whether inflammation induced nitration of CREB protein. Figure 4C shows that nuclear CREB was not nitrated in inflamed samples. Colonic inflammation, however, reduced depolarization-induced pCREB expression (Figure 5A). Colonic inflammation was confirmed by increase in mRNA expression for Il-1β (Figure 5B). These findings suggest that nitration of the Ca2+ channel following inflammation may alter activation of the transcription factors.
Figure 3.
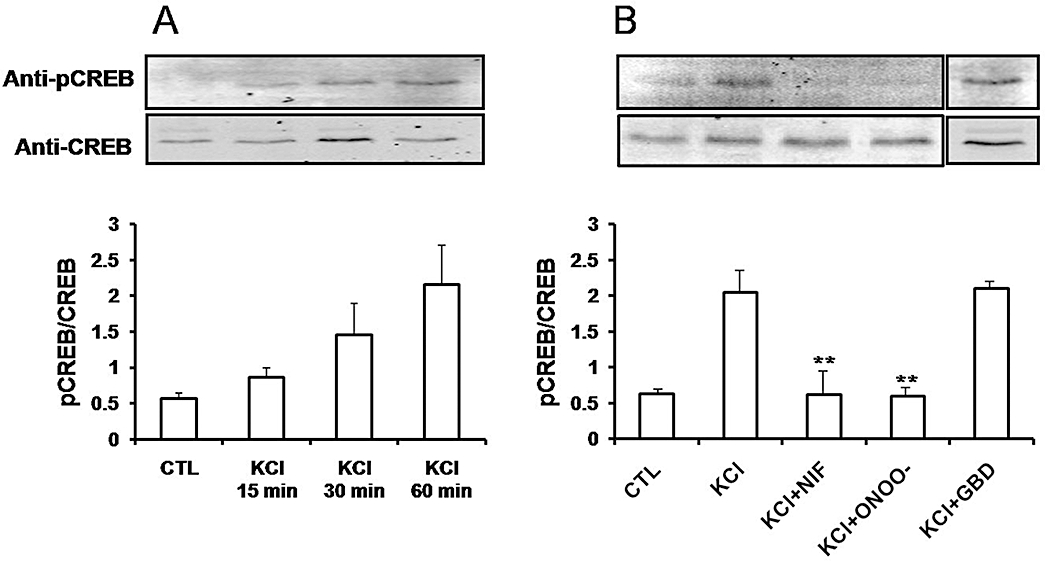
Depolarization-induced phosphorylation of CREB in smooth muscle. Mouse colonic smooth muscle strips were treated with 80 mM KCl for 15, 30 and 60 min (A), and were pretreated with either 1 µM of nifedipine (NIF), 150 µM of peroxynitrite (ONOO-) or 10 µM glibenclamide (GBD) for 30 min prior to depolarization with 80 mM KCl (B). Expression level of phospho-CREB was determined by Western blot using anti-pCREB. Total expression of CREB was detected with anti-CREB antibody. The expression levels are shown as a ratio of pCREB compared with total CREB. The results represent the mean ± SD (n= 3). **P < 0.001 versus KCl. CREB, cyclic AMP response element binding protein.
Figure 4.
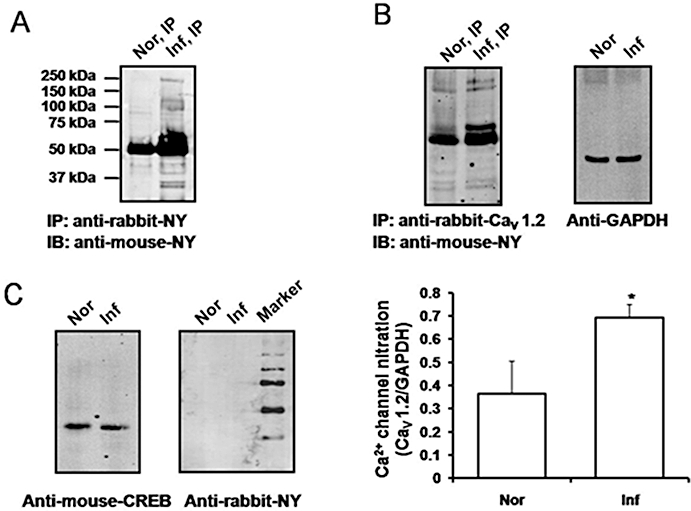
Protein tyrosine nitration in smooth muscle from inflamed colon. Membrane extracts were obtained from distal colonic smooth muscle cells from normal (Nor) and inflamed (Inf) mice, and lysates were subject to immunoprecipitation with rabbit anti-nitrotyrosine (NY) (A) or rabbit anti-Ca2+ channel (Cav1.2) (B) antibodies and immunoblotted using mouse anti-nitrotyrosine (anti-NY) antibodies. Nitration of Ca2+ channels (arrow) was significantly higher in inflamed colon (bottom panel). Equal amount of protein loading was confirmed using anti-GAPDH antibody. *P < 0.05 versus normal (n= 3). Nuclear extracts from distal colons of normal and inflamed mice were subject to immunoblot with either mouse anti-CREB or anti-NY antibodies (C). Protein nitration of CREB was not detected in either normal or inflamed colonic samples. CREB, cyclic AMP response element binding protein.
Figure 5.
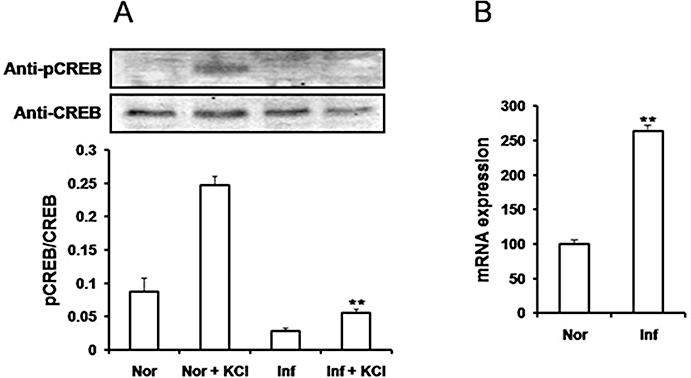
Depolarization-induced pCREB is decreased in inflamed colon. (A) Nuclear extracts were prepared from distal colon of normal (Nor) and inflamed (Inf) mice treated with 80 mM KCl, and expression of pCREB was measured by immunoblots with anti-pCREB and CREB antibodies. Phospho-CREB expression was not enhanced by depolarization in inflamed colon. **P < 0.01 versus Nor + KCl (n= 3). (B) quantitative comparison of gene expression shows increased expression of IL-1β during colonic inflammation. Total RNA was obtained from normal and inflamed distal colon tissues, and fold increase of IL-1β mRNA level was assessed by real-time RT-PCR after being normalized by the mRNA level of 18S. IL-1β mRNA was increased by 2.5-fold due to inflammation. All real-time PCR experiments were carried out in triplicate. Data analysed by unpaired t-test; n= 4. **P < 0.01 versus normal. CREB, cyclic AMP response element binding protein.
Nitration of Ca2+ channel by peroxynitrite (ONOO-) prevents CRE activation by KCl-induced membrane depolarization
CRE activation occurs downstream of CREB phosphorylation. To determine whether KCl-induced membrane depolarization affects CRE activation, CHO cells were co-transfected with the Ca2+ channel hCav1.2b, β2 and CRE reporter genes. CRE activities were determined using the dual luciferase assay. We tested the effects of various treatments on CRE activation (Figure 6). In these experiments, the ratio of firefly (CRE) to Renilla (internal control) for untreated control cells was 0.32 ± 0.008. Depolarization with 80 mM KCl for 30 min significantly increased CRE activation by over 2.35 fold (ratio 0.83 ± 0.001). Forskolin enhanced levels of cAMP and, as expected, increased CRE activation by 2.77 fold (ratio 0.88 ± 0.002). Pretreatment with nifedipine to block Ca2+ entry resulted in the inhibition of CRE activation by KCl. We next examined whether nitration of Ca2+ channels affects depolarization-induced CRE activation. Transfected CHO cells were pretreated with SIN-1/peroxynitrite (150 µM) prior to depolarization with 80 mM KCl. Nitration prevented CRE activation by KCl. Treatment with ionomycin to increase global intracellular Ca2+ did not result in CRE activation. These results suggest that the L-type Ca2+ channel plays an important role in the induction/repression of gene transcription and that nitration of the tyrosine residues by ONOO- can prevent downstream effects of Ca2+ channel activation.
Figure 6.
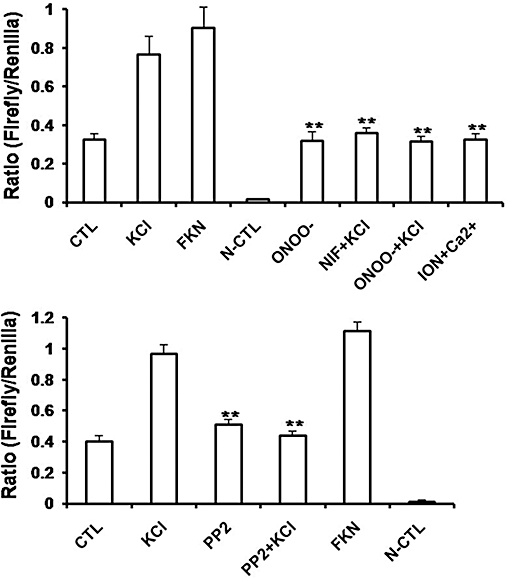
Inhibition of depolarization-induced CRE activation by tyrosine nitration. The dual luciferase assay was performed to measure expression of CRE-promoter-based firefly luciferase. Results are expressed as ratio of firefly to Renilla luciferase (internal control). (A) The hCav1.2b transfected CHO cells were treated with 80 mM KCl, or 10 µM forskolin (FKN) for 30 min. Pretreatment of the cells with 150 µM peroxynitrite (ONOO) or 1 µM nifedipine for 30 min attenuated KCl-induced CRE activation. Ionomycin (1 µM) in the presence of calcium (ION +Ca) did not induce increase in CRE activation. Data represent mean ± SEM. **P < 0.001 versus KCl, unpaired t-test; n= 3–6. (B) Effect of PP2 on depolarization-induced CRE activation. The hCav1.2b transfected CHO cells were treated with 10 µM of PP2, the Src kinase inhibitor for 10 min prior to depolarization with KCl. Experiments were performed in triplicates. Data represent mean ± SEM. **P < 0.001 versus KCl, unpaired t-test; n= 3–6. CRE, cyclic AMP response element.
Src kinase regulation of Ca2+ channel is involved in CRE activation
The smooth muscle channel, Cav1.2b, is regulated by c-Src kinase through phosphorylation of tyrosine residues, Y2134 and Y1837, and PP2, the Src kinase inhibitor, attenuates Ca2+ channel currents (Hu et al., 1998; Kang et al., 2007). We therefore tested the effect of PP2 on depolarization-induced CRE activation. As shown in Figure 6B, PP2 (10 µM) abolished KCl-induced CRE activation. PP2 treatment in the absence of depolarization did not induce CRE activation. Forskolin was used to induce CRE activation as a positive control. No CRE activity was observed in CHO cells transfected with the negative control constructs. We next tested the functional role of C-terminus tyrosine residues of the Ca2+ channel in transcriptional activation. We have previously demonstrated that mutating tyrosine residue Y2134F in the hCav1.2b results in loss of Src-SH2 binding and tyrosine phosphorylation of the Ca2+ channel (Kang et al., 2007). This and mutations of all three tyrosine residues within the terminal 330 amino acids also results in decreased regulation of the Ca2+ currents by Src kinase. CHO cells were transfected with either single or triple tyrosine mutants of the Ca2+ channel. Depolarization of cells containing the triple mutant Y1837F-Y1861F-Y2134F with KCl did not result in CRE activation. In these cells, forskolin-induced CRE activation remained unaffected (Figure 7A). To test for specificity, we also examined CRE activation in cells containing the Y2134F mutant. Mutation of this tyrosine residue prevented KCl-induced CRE activation but not that induced by forskolin (Figure 7B). On the other hand, depolarization-induced CRE activation was not affected in the mutant Y1861F which is not involved in Src regulation (Kang et al., 2007) (Figure 7C). The expression of the mutant Ca2+ channels on the membranes was confirmed by immunohistochemistry (Figure 8).
Figure 7.
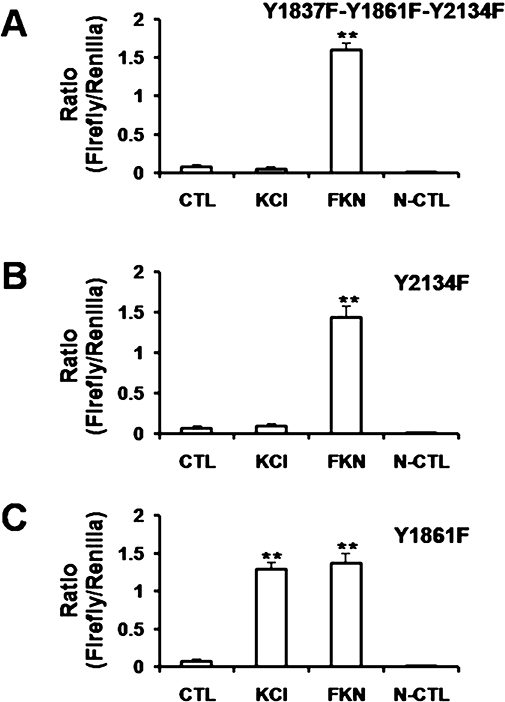
Effect of single or triple tyrosine mutations of hCav1.2b on depolarization-induced CRE activation. The triple mutant, Y1837F-Y1861F-Y2134F (A) or single mutants, Y2134F (B) and Y1861F (C) of the Ca2+ channel were transfected in CHO cells. After 48–72 h, cells were treated with 80 mM KCl or 10 µM forskolin (FKN) for 30 min. CRE luciferase activation was measured as ratio of internal control (Renilla). Depolarization-induced CRE activation was blocked in the triple mutant and in the Y2134F mutant but not in the Y1861F mutant. Experiments were in triplicate. Data are mean ± SEM. **P < 0.001 versus KCl, unpaired t-test; n= 3–6. CRE, cyclic AMP response element.
Figure 8.
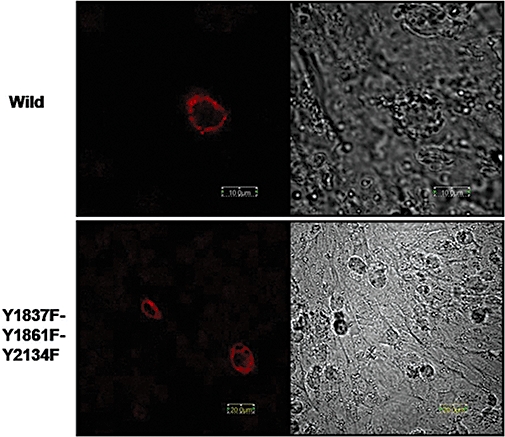
Membrane expression of wild-type (upper) and triple mutant, Y1837F-Y1861F-Y2134F (lower) Ca2+ channels in CHO cells.
Discussion
Excitation–transcription coupling is now recognized as a dynamic process involving ion channel gating and activation of transcription factors. Although many of the quantitative features of this process remain to be elucidated, recent studies suggest that local Ca2+ elevations coupled to high open probability of the channel provide a direct and rapid means for L-type Ca2+ channels to initiate CREB phosphorylation in the nucleus (Deisseroth et al., 1998; Dolmetsch et al., 2001; Barlow et al., 2006). In their recent model, Tsien and colleagues (Wheeler et al., 2008) suggested that Ca2+ increases near the mouth of the channel were sensed by apo-calmodulin resulting in activation of CAMKII that translocates to the nucleus. Interestingly, it is the increase in channel open probability due to depolarization that was necessary for signalling rather than increases in bulk Ca2+ concentrations. In the present study, we tested whether attenuation of the Ca2+ channel function due to tyrosine nitration accompanies loss of transcriptional activation. We have previously shown that tyrosine nitration of the Ca2+ channel occurs during colonic inflammation resulting in decreased Ca2+ channel currents. Our studies have demonstrated that modulation of Ca2+ channels by the intracellular tyrosine kinase, c-Src kinase, is attenuated following colonic inflammation (Kang et al., 2004). Several lines of evidence indicate that the α subunit of Cav1.2b is under the basal regulation of c-Src kinase including evidence of direct association of the α subunit and c-Src in smooth muscle (Wijetunge and Hughes, 1996; Hu et al., 1998; Wijetunge et al., 2000). Src kinase inhibition attenuates Ca2+ currents and tyrosine phosphatase inhibitors or receptor-mediated stimulation of Src kinase enhance Ca2+ currents (Hatakeyama et al., 1996; Wijetunge and Hughes, 1996; Wu et al., 2001; Jin et al., 2002). Colonic inflammation enhanced tyrosine nitration of several proteins, including the Ca2+ channel. We have also previously shown that nitration of tyrosine residues within the C-terminus of the Ca2+ channel prevents protein-protein interaction with Src kinase (Kang et al., 2007), similar to several other studies implicating disabling of the tyrosine phosphorylating regulatory mechanisms by nitration of tyrosine residues of target proteins (Gow et al., 1996; Kong et al., 1996; Greenacre and Ischiropoulos, 2001).
In both hCav1.2b-transfected cells and in smooth muscle tissues, depolarization with high K+ resulted in enhanced expression of nuclear pCREB, similar to that observed in neurons (Deisseroth et al., 1998; Dolmetsch et al., 2001; Wheeler et al., 2008). The activation of pCREB in high K+ can be attributed largely to Ca2+ influx through L-type Ca2+ channels as it was abolished by nifedipine and absent in untransfected cells. In colonic muscle strips, incubating with high K+ in the presence of Ca2+ results in Ca2+-induced contractions which are significantly reduced during inflammation and by pretreatment with peroxynitrite (Ross et al., 2007). Thus, similar to its effects on smooth muscle contraction, peroxynitrite treatment and inflammation also suppressed depolarization-induced CREB phosphorylation in colonic smooth muscle. This effect was largely due to nitration of the Ca2+ channel as nitration of CREB protein was not detected during inflammation. At present, we cannot rule out the possible nitration of other Ca2+-dependent kinase(s) and further studies will be necessary to determine which kinase(s) might be involved in Ca2+-dependent phosphorylation of CREB in smooth muscle. It has been reported that during inflammation, the levels of nitrotyrosine are low and that one to five 3-nitrotyrosine residues per 10 000 were detected (Radi, 2004). This may be attributed to several factors including requirements of consensus sequences in the target protein and the local environment of the tyrosine residue (Abello et al., 2009). In the present studies we only examined Ca2+ influx-dependent CREB phosphorylation; however, in vascular smooth muscle, a role for sarcoplasmic reticular Ca2+ in signalling to CREB has also been demonstrated (Barlow et al., 2006).
The phosphorylation of CREB correlated with activation of the CRE promoter. Both nifedipine and peroxynitrite prevented depolarization-induced activation of CRE. Interestingly, increasing intracellular Ca2+ with ionomycin did not result in CRE activation consistent with a primary role for Ca2+ influx through L-type Ca2+ channels. This is consistent with the findings in neurons where depolarization without Ca2+ influx but with increase in bulk Ca2+ had no effect on signalling to pCREB (Wheeler et al., 2008). The Src kinase inhibitor, PP2, blocked KCl-induced CRE activation suggesting that tyrosine phosphorylation of the Ca2+ channel is an important component for downstream signalling. To further test this, mutation of the tyrosine residues within the C-terminus known to be involved in Src regulation blocked CRE activation. We have identified Y2134 as a potential SH2 binding site (Kang et al., 2007) and mutations of this site, but not of Y1861, blocked CRE activation by depolarization but not by forskolin.
Ca2+ currents are markedly decreased in colonic inflammation, which results in attenuated smooth muscle contraction (Akbarali, 2005). Ross et al. (2007) showed that in the mouse model of colitis induced by TNBS, depolarization induced contractions were attenuated compared with controls and were less sensitive to the src kinase inhibitors. The Ca2+ channels from inflamed colon are nitrated and it is the nitration of C-terminus protein tyrosine residues that prevents Src kinase regulation in basal unstimulated conditions. The addition of a nitro group to protein tyrosine is a selective post-translational modification that alters protein-protein interaction (Pacher et al., 2007; Monteiro et al., 2008). Tyrosine nitration has been demonstrated by immunohistological staining in numerous human diseases and animal models, including inflammatory bowel disease (Middleton et al., 1993; Miller et al., 1993; Singer et al., 1996; Miampamba and Sharkey, 1999). Our findings imply that, similar to excitation–contraction coupling, tyrosine nitration of the colonic smooth muscle Ca2+ channel during inflammation also alters excitation–transcription coupling. These studies also suggest that changes in gene expression in inflammatory bowel diseases may be linked to the altered gating process of the Ca2+ channel through modulation of protein-protein interactions at the C-terminus of the Ca2+ channel.
Acknowledgments
This work was supported by the National Institute of Health [Grant DK046367].
Glossary
Abbreviations:
- AF
atrial fibrillation
- CRE
cyclic AMP response element
- CREB
cyclic AMP response element binding protein
- DAI
disease activity index
- IBMX
isobutylmethylxanthine
- iNOS
inducible nitric oxide synthase
- PP2
4-amino-5-(4-chloro-phenyl)-7-(t-butyl)pyrazolo [3,4-d]pyrimidine
- TNBS
trinitrobenzene sulphonic acid
Conflict of interest
None.
References
- Abello N, Kerstjens HA, Postma DS, Bischoff R. Protein tyrosine nitration: selectivity, physicochemical and biological consequences, denitration, and proteomics methods for the identification of tyrosine-nitrated proteins. J Proteome Res. 2009;8:3222–3238. doi: 10.1021/pr900039c. [DOI] [PubMed] [Google Scholar]
- Akbarali HI. Signal-transduction pathways that regulate smooth muscle function. II. Receptor-ion channel coupling mechanisms in gastrointestinal smooth muscle. Am J Physiol Gastrointest Liver Physiol. 2005;288:G598–602. doi: 10.1152/ajpgi.00402.2004. [DOI] [PubMed] [Google Scholar]
- Akbarali HI, Giles WR. Ca2+ and Ca(2+)-activated Cl- currents in rabbit oesophageal smooth muscle. J Physiol. 1993;460:117–133. doi: 10.1113/jphysiol.1993.sp019462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbarali HI, Pothoulakis C, Castagliuolo I. Altered ion channel activity in murine colonic smooth muscle myocytes in an experimental colitis model. Biochem Biophys Res Commun. 2000;275:637–642. doi: 10.1006/bbrc.2000.3346. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl. 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow CA, Rose P, Pulver-Kaste RA, Lounsbury KM. Excitation-transcription coupling in smooth muscle. J Physiol. 2006;570:59–64. doi: 10.1113/jphysiol.2005.098426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ T, Boknik P, Wohrl S, Wettwer E, Graf EM, Bosch RF, et al. L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation. 2004;110:2651–2657. doi: 10.1161/01.CIR.0000145659.80212.6A. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- Estevez AG, Spear N, Manuel SM, Radi R, Henderson CE, Barbeito L, et al. Nitric oxide and superoxide contribute to motor neuron apoptosis induced by trophic factor deprivation. J Neurosci. 1998;18:923–931. doi: 10.1523/JNEUROSCI.18-03-00923.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow AJ, Duran D, Malcolm S, Ischiropoulos H. Effects of peroxynitrite-induced protein modifications on tyrosine phosphorylation and degradation. FEBS Lett. 1996;385:63–66. doi: 10.1016/0014-5793(96)00347-x. [DOI] [PubMed] [Google Scholar]
- Greenacre SA, Ischiropoulos H. Tyrosine nitration: localisation, quantification, consequences for protein function and signal transduction. Free Radic Res. 2001;34:541–581. doi: 10.1080/10715760100300471. [DOI] [PubMed] [Google Scholar]
- Hatakeyama N, Mukhopadhyay D, Goyal RK, Akbarali HI. Tyrosine kinase-dependent modulation of calcium entry in rabbit colonic muscularis mucosae. Am J Physiol. 1996;270:C1780–C1789. doi: 10.1152/ajpcell.1996.270.6.C1780. [DOI] [PubMed] [Google Scholar]
- Hu XQ, Singh N, Mukhopadhyay D, Akbarali HI. Modulation of voltage-dependent Ca2+ channels in rabbit colonic smooth muscle cells by c-Src and focal adhesion kinase. J Biol Chem. 1998;273:5337–5342. doi: 10.1074/jbc.273.9.5337. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, et al. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- Jin X, Malykhina AP, Lupu F, Akbarali HI. Altered gene expression and increased bursting activity of colonic smooth muscle ATP-sensitive K+ channels in experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2004;287:G274–G285. doi: 10.1152/ajpgi.00472.2003. [DOI] [PubMed] [Google Scholar]
- Jin X, Morsy N, Shoeb F, Zavzavadjian J, Akbarali HI. Coupling of M2 muscarinic receptor to L-type Ca channel via c-src kinase in rabbit colonic circular smooth muscle. Gastroenterology. 2002;123:827–834. doi: 10.1053/gast.2002.35388. [DOI] [PubMed] [Google Scholar]
- Kang M, Morsy N, Jin X, Lupu F, Akbarali HI. Protein and gene expression of Ca2+ channel isoforms in murine colon: effect of inflammation. Pflugers Arch. 2004;449:288–297. doi: 10.1007/s00424-004-1339-5. [DOI] [PubMed] [Google Scholar]
- Kang M, Ross GR, Akbarali HI. COOH-terminal association of human smooth muscle calcium channel Ca(v)1.2b with Src kinase protein binding domains: effect of nitrotyrosylation. Am J Physiol Cell Physiol. 2007;293:C1983–C1990. doi: 10.1152/ajpcell.00308.2007. [DOI] [PubMed] [Google Scholar]
- Kinoshita K, Sato K, Hori M, Ozaki H, Karaki H. Decrease in activity of smooth muscle L-type Ca2+ channels and its reversal by NF-kappaB inhibitors in Crohn's colitis model. Am J Physiol Gastrointest Liver Physiol. 2003;285:G483–93. doi: 10.1152/ajpgi.00038.2003. [DOI] [PubMed] [Google Scholar]
- Kong SK, Yim MB, Stadtman ER, Chock PB. Peroxynitrite disables the tyrosine phosphorylation regulatory mechanism: lymphocyte-specific tyrosine kinase fails to phosphorylate nitrated cdc2(6-20)NH2 peptide. Proc Natl Acad Sci U S A. 1996;93:3377–3382. doi: 10.1073/pnas.93.8.3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Rusch NJ, Striessnig J, Sarna SK. Down-regulation of L-type calcium channels in inflamed circular smooth muscle cells of the canine colon. Gastroenterology. 2001;120:480–489. doi: 10.1053/gast.2001.21167. [DOI] [PubMed] [Google Scholar]
- Miampamba M, Sharkey KA. Temporal distribution of neuronal and inducible nitric oxide synthase and nitrotyrosine during colitis in rats. Neurogastroenterol Motil. 1999;11:193–206. doi: 10.1046/j.1365-2982.1999.00150.x. [DOI] [PubMed] [Google Scholar]
- Middleton SJ, Shorthouse M, Hunter JO. Increased nitric oxide synthesis in ulcerative colitis. Lancet. 1993;341:465–466. doi: 10.1016/0140-6736(93)90211-x. [DOI] [PubMed] [Google Scholar]
- Miller MJ, Sadowska-Krowicka H, Chotinaruemol S, Kakkis JL, Clark DA. Amelioration of chronic ileitis by nitric oxide synthase inhibition. J Pharmacol Exp Ther. 1993;264:11–16. [PubMed] [Google Scholar]
- Monteiro HP, Arai RJ, Travassos LR. Protein tyrosine phosphorylation and protein tyrosine nitration in redox signaling. Antioxid Redox Signal. 2008;10:843–889. doi: 10.1089/ars.2007.1853. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci U S A. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross GR, Kang M, Shirwany N, Malykhina AP, Drozd M, Akbarali HI. Nitrotyrosylation of Ca2+ Channels Prevents c-Src Kinase Regulation of Colonic Smooth Muscle Contractility in Experimental Colitis. J Pharmacol Exp Ther. 2007;322:948–956. doi: 10.1124/jpet.107.123075. [DOI] [PubMed] [Google Scholar]
- Shi XZ, Sarna SK. Transcriptional regulation of inflammatory mediators secreted by human colonic circular smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2005;289:G274–84. doi: 10.1152/ajpgi.00512.2004. [DOI] [PubMed] [Google Scholar]
- Singer II, Kawka DW, Scott S, Weidner JR, Mumford RA, Riehl TE, Stenson WF. Expression of inducible nitric oxide synthase and nitrotyrosine in colonic epithelium in inflammatory bowel disease. Gastroenterology. 1996;111:871–885. doi: 10.1016/s0016-5085(96)70055-0. [DOI] [PubMed] [Google Scholar]
- Wamhoff BR, Bowles DK, McDonald OG, Sinha S, Somlyo AP, Somlyo AV, et al. L-type voltage-gated Ca2+ channels modulate expression of smooth muscle differentiation marker genes via a rho kinase/myocardin/SRF-dependent mechanism. Circ Res. 2004;95:406–414. doi: 10.1161/01.RES.0000138582.36921.9e. [DOI] [PubMed] [Google Scholar]
- Wamhoff BR, Bowles DK, Owens GK. Excitation-transcription coupling in arterial smooth muscle. Circ Res. 2006;98:868–878. doi: 10.1161/01.RES.0000216596.73005.3c. [DOI] [PubMed] [Google Scholar]
- Wheeler DG, Barrett CF, Groth RD, Safa P, Tsien RW. CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J Cell Biol. 2008;183:849–863. doi: 10.1083/jcb.200805048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijetunge S, Hughes AD. Activation of endogenous c-Src or a related tyrosine kinase by intracellular (pY)EEI peptide increases voltage-operated calcium channel currents in rabbit ear artery cells. FEBS Lett. 1996;399:63–66. doi: 10.1016/s0014-5793(96)01177-5. [DOI] [PubMed] [Google Scholar]
- Wijetunge S, Lymn JS, Hughes AD. Effects of protein tyrosine kinase inhibitors on voltage-operated calcium channel currents in vascular smooth muscle cells and pp60(c-src) kinase activity. Br J Pharmacol. 2000;129:1347–1354. doi: 10.1038/sj.bjp.0703186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Davis GE, Meininger GA, Wilson E, Davis MJ. Regulation of the L-type calcium channel by alpha 5beta 1 integrin requires signaling between focal adhesion proteins. J Biol Chem. 2001;276:30285–30292. doi: 10.1074/jbc.M102436200. [DOI] [PubMed] [Google Scholar]