

Abstract
Background and purpose:
Hydrogen sulphide is an important mediator of gastric mucosal defence. The use of non-steroidal anti-inflammatory drugs continues to be limited by their toxicity, particularly in the upper gastrointestinal tract. We evaluated the gastrointestinal safety and anti-inflammatory efficacy of a novel hydrogen sulphide-releasing derivative of naproxen, ATB-346 [2-(6-methoxy-napthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester].
Experimental approach:
The ability of ATB-346 versus naproxen to cause gastric damage was evaluated in healthy rats and in rats with compromised gastric mucosal defence. Effects on the small intestine and on the healing of ulcers were also assessed. The ability of ATB-346 to inhibit cyclooxygenase-1 and 2 and to reduce inflammation in vivo was also evaluated.
Key results:
ATB-346 suppressed gastric prostaglandin E2 synthesis as effectively as naproxen, but produced negligible damage in the stomach and intestine. In situations in which the gastric mucosa was rendered significantly more susceptible to naproxen-induced damage (e.g. ablation of sensory afferent nerves, inhibition of endogenous nitric oxide or hydrogen sulphide synthesis, co-administration with aspirin, antagonism of KIR6.x channels), ATB-346 did not cause significant damage. Unlike naproxen and celecoxib, ATB-346 accelerated healing of pre-existing gastric ulcers. In a mouse airpouch model, ATB-346 suppressed cyclooxygenase-2 activity and inhibited leukocyte infiltration more effectively than naproxen. ATB-346 was as effective as naproxen in adjuvant-induced arthritis in rats, with a more rapid onset of activity. Unlike naproxen, ATB-346 did not elevate blood pressure in hypertensive rats.
Conclusions and implications:
ATB-346 exhibits anti-inflammatory properties similar to naproxen, but with substantially reduced gastrointestinal toxicity.
Keywords: hydrogen sulphide, non-steroidal anti-inflammatory drug, ulcer, inflammation, gastrointestinal, blood pressure, arthritis, naproxen, cyclooxygenase, mucosal defence
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) remain the most commonly used drugs for treating the symptoms of osteoarthritis and several other inflammatory diseases, despite their significant gastrointestinal (GI) and cardiovascular toxicity (Grosser et al., 2006; Wallace, 2008). The gastric toxicity of NSAIDs is linked closely to the ability of these drugs to inhibit prostaglandin (PG) synthesis, with both COX-1 and COX-2 contributing significantly to mucosal defence and repair (Wallace et al., 2000; 2008;). On the other hand, damage to the small intestine induced by NSAIDs appears to be related more to their topical irritant actions, particularly when the drugs are excreted in bile, than to the suppression of PG synthesis (Wallace, 2008).
In addition to PG, several other mediators contribute significantly to GI mucosal defence and repair. For example, nitric oxide (NO) modulates many of the same elements of mucosal defence (e.g. blood flow, mucus and bicarbonate secretion) as do PGs (Brown et al., 1993; Wallace, 2008), and can promote the healing of ulcers (Elliott et al., 1995). These actions of NO were exploited in the design of NO-releasing NSAIDs, which exhibit comparable anti-inflammatory effects to the parent NSAIDs with reduced detrimental effects in the GI and cardiovascular systems (Wallace and Del Soldato, 2003; Wallace et al., 2009a).
Like NO, hydrogen sulphide (H2S) is a gaseous mediator with physiological and pathophysiological roles in many organs (Wang, 2002; Fiorucci et al., 2006; Li and Moore, 2008). H2S has been shown to be produced in the GI tract (Fiorucci et al., 2005; Linden et al., 2008; Wallace et al., 2009b) and to contribute to gastric mucosal defence (Fiorucci et al., 2005) and the healing of gastric ulcers (Wallace et al., 2007b). In the intestine, H2S modulates epithelial secretion (Schicho et al., 2006) and promotes resolution of colitis (Wallace et al., 2009b). Like NO, H2S inhibits leukocyte adherence to the vascular endothelium (Zanardo et al., 2006) and appears to play an important role in the regulation of systemic blood pressure (Yang et al., 2008). Also similar to NO, H2S has been exploited in the design of novel NSAIDs. An H2S-releasing derivative of diclofenac has been shown to be more effective than the parent drug in reducing inflammation, while producing significantly less damage in the GI tract (Li et al., 2007; Wallace et al., 2007a).
In the present study, we have evaluated the anti-inflammatory and GI-damaging effects of a novel H2S-releasing derivative of naproxen, ATB-346 [2-(6-methoxy-napthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester]. Naproxen is among the most commonly used NSAIDs, in part because of evidence that its use may be associated with less cardiovascular toxicity than selective COX-2 inhibitors and other NSAIDs (Kearney et al., 2006). This study included examination of the gastric safety of ATB-346 in circumstances in which mucosal defence was significantly impaired, in an attempt to model more closely the clinical scenario in which NSAIDs are most frequently used (i.e. patients with co-morbidities that elevate susceptibility to NSAID-induced ulceration). Our results demonstrate that ATB-346 is at least as effective as naproxen as an anti-inflammatory drug, while exhibiting a substantial reduction in toxic effects in the GI tract.
Methods
Animals
Animal studies were conducted in accordance with the guidelines established by the Canadian Council of Animal Care, with all protocols being approved by our institutional animal care committee. Male, Wistar rats (200–225 g) and male, C57BL6J mice (25–27 g) were obtained from Charles River Laboratories (Montreal, QC, Canada) and were fed standard laboratory chow and water ad libitum. The animals were housed in pairs and kept in a room having controlled temperature (22 ± 1°C), humidity (65–70%) and light cycle (12 h light/12 h dark).
Zymosan airpouch model
Mice were deprived of food, but not water, for 18–20 h prior to experiments. The airpouch was induced as described previously (Edwards et al., 1981; Wallace et al., 1999), with slight modifications. Briefly, 3 mL of air was injected subcutaneously into the back of the mice. Additional injections of 1.5 mL of air were performed 3 and 6 days after the first injection. Twenty-four hours after the last injection of air, 1 mL of either saline or a 1% solution of zymosan was injected into the pouch. All of the injections were performed under isoflurane anaesthesia. One hour prior to zymosan injection into the airpouch, mice were treated orally with naproxen or ATB-346 (each at 30 µmol·kg−1) or vehicle (1% carboxymethylcellulose). Each group consisted of five to six mice. An additional group of mice was treated with another derivative of naproxen, ATB-345 [2-(6-methoxy-napthalen-2-yl)-propionic acid 4-(5-thioxo-5H-[1,2]dithiol-3-yl)-phenyl ester] (30 µmol·kg−1), which has a different H2S-releasing moiety to that in ATB-346 (Figure 1). Six hours after zymosan injection, the mice were anaesthetized with sodium pentobarbital (60 mg·kg−1, i.p.), and blood was drawn from the inferior vena cava for measurement of whole blood thromboxane B2 (TXB2) synthesis (by elisa), as an index of COX-1 activity (Wallace et al., 1999). Immediately thereafter, 1 mL of heparin-treated saline was injected into the pouch. The airpouch was carefully opened by a small incision. The exudate was collected, the volume measured and an aliquot used to quantify leukocyte numbers using a Sysmex KX-21N haematology analyser (Sysmex Canada, Mississauga, ON, Canada). The exudate was centrifuged at 1000×g for 10 min. The supernatant was collected and stored at −80°C for measurement of immunoreactive PGE2, as an index of COX-2 activity (Wallace et al., 1999).
Figure 1.
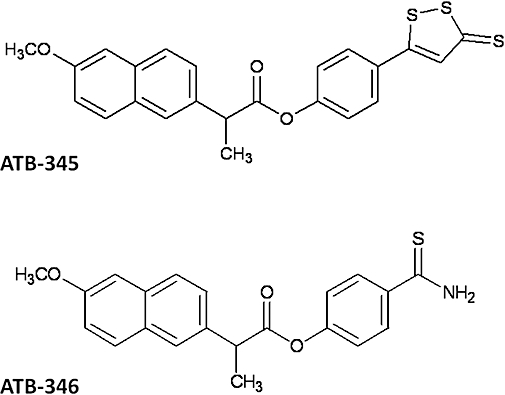
Chemical structures of two hydrogen sulphide-releasing derivatives of naproxen (ATB-345 and ATB-346). ATB-345, 2-(6-methoxy-napthalen-2-yl)-propionic acid 4-(5-thioxo-5H-[1,2]dithiol-3-yl)-phenyl ester; ATB-346, 2-(6-methoxy-napthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester.
Adjuvant arthritis
Polyarthritis was induced in rats via an injection into the base of the tail of 100 µL of Freund's Complete Adjuvant containing 0.6 mg of Mycobacterium butirricum (Cicala et al., 2000). The volume of both hindpaws of each rat was blindly measured using a hydroplethysmometer (Ugo Basile, Comerio, Italy) prior to the injection of the adjuvant, and on days 7, 10, 14 and 21 after adjuvant administration. Groups of six to seven rats each were treated twice-daily, orally, with naproxen (4 µmol·kg−1), ATB-346 (4 µmol·kg−1) or vehicle (1% carboxymethylcellulose) on days 7 to 21. At the end of the experiment, the stomach and small intestine were blindly examined for evidence of ulceration and bleeding.
Acute gastric damage
Rats were fasted for 16–18 h, then were randomized to groups (n≥ 5) and given vehicle or one of the test drugs orally. Naproxen and ATB-346 were tested at doses of 30, 60, 120 and 2740 µmol·kg−1. Three hours later, the rats were killed by an overdose of pentobarbital sodium and the stomach was excised and examined. To assess gastric mucosal damage, the lengths (in mm) of all lesions were measured with a digital calliper, and a ‘gastric damage score’ was calculated for each stomach by summing these values (Wallace et al., 2000). This assessment was performed by an individual unaware of the treatments the rats had received. Samples of the gastric tissue were excised and processed, as described previously (Wallace et al., 2000), for measurement of immunoreactive PGE2.
ATB-346 consists of a molecule of naproxen linked via an ester bond to 4-hydroxythiobenzamide (TBZ) (Figure 1). To determine if co-administration of unconjugated naproxen and TBZ would protect the gastric mucosa from damage, the following experiment was performed. Groups of five rats each, deprived of food for the previous 16–18 h, were given naproxen (50 µmol·kg−1) orally together with an equal volume of vehicle, or vehicle containing TBZ at 50, 100 or 150 µmol·kg−1. Another group of rats received ATB-346 at 50 µmol·kg−1. Three hours later, gastric damage was blindly scored as described above.
Intestinal damage
Groups of six rats (not fasted) were treated twice-daily for 5 days with naproxen (90 µmol·kg−1), ATB-346 (90 µmol·kg−1) or vehicle. Prior to the first administration of test drug or vehicle, a blood sample was drawn from the tail vein for determination of haematocrit (Reuter et al., 1997). Six hours after the final administration of test drug or vehicle, another blood sample was drawn for determination of haematocrit, and the rats were then killed by an overdose of sodium pentobarbital. The small intestine was excised, and the extent of haemorrhagic damage to the small intestine was blindly quantified by measuring the lengths of lesions in mm and then summing these to give a damage score for each rat (Reuter et al., 1997).
Impaired gastric mucosal defence
Experiments were performed as described above to compare the gastric damaging effects of naproxen to those of ATB-346. In these experiments, a number of other procedures were employed to increase the gastric mucosal susceptibility to damage. These included administration of the test drugs to rats that had been treated with aspirin (10 mg·kg−1 i.p.), NG-nitro-L-arginine methylester (L-NAME; 15 mg·kg−1 i.p.), β-cyanoalanine (BCA; 50 mg·kg−1 i.p.) or glibenclamide (10 mg·kg−1 i.p.). Aspirin was given immediately before administration of the test drugs/vehicle, while the other agents were given 30 min before administration of the test drugs/vehicle. In the experiments in which aspirin was administered, one group of rats was cotreated with celecoxib (30 µmol·kg−1 p.o.).
The test drugs or vehicle were also administered to rats in which sensory afferent neurones had been ablated by prior treatment with capsaicin, as described previously (McCafferty et al., 1997).
Gastric ulcer healing
Ulcers were induced using a previously described model (Okabe et al., 1971; Dudar et al., 2008), with slight modifications. In brief, mice were anaesthetized with isoflurane, the abdomen was opened with a midline incision and the stomach was exposed to permit application of glacial acetic acid (200 µL, 20% vol·vol−1) to the serosal surface of the corpus region. The acetic acid was applied using the barrel of a 1 mL syringe, such that the contact area was 28 mm2. One minute later, the acetic acid was removed by aspiration, and the area of contact was washed three times with 200 µL of saline. The peritoneum and skin were then closed with sutures. Three days after application of acetic acid to the stomach, five mice were killed by an overdose of sodium pentobarbital. The stomach was removed and opened by an incision along the greater curvature, then pinned out, mucosal side up, on a paraffin block. A 25 mm2 grid was placed alongside the ulcer and the stomach was photographed. Planimetric analysis, using the grid as reference, allowed for determination of the area of the ulcer (Dudar et al., 2008). The remaining mice were randomized to one of four groups, and they began receiving one of the following twice-daily, orally: naproxen (60 µmol·kg−1), ATB-346 (60 µmol·kg−1), celecoxib (30 µmol·kg−1) or vehicle. After 4 days of drug/vehicle administration, the mice were killed and the areas of the gastric ulcers were blindly measured, as described above.
Arterial blood pressure
Effects on systemic arterial blood pressure were assessed in healthy, untreated rats and in rats given drinking water supplemented with L-NAME (400 mg·L−1) for the previous 2 weeks to induce hypertension (Muscaráet al., 2000b). The rats were anaesthetized with sodium pentobarbital and a carotid artery was cannulated for measurement of systemic blood pressure. After a stable blood pressure had been recorded for at least 20 min, the rats received an i.p. injection of naproxen (90 µmol·kg−1), ATB-346 (90 µmol·kg−1) or vehicle (1% carboxymethylcellulose). Also, for comparison, we examined the effects of a second NSAID, diclofenac (60 µmol·kg−1), and an H2S-releasing derivative of diclofenac, ATB-337 [(2-(2,6-dichloro-phenylamino)-phenyl)-acetic acid 4-(3-thioxo-3H-[1,2]dithiol-4-yl)-phenyl ester] (60 µmol·kg−1), which has been described previously (Li et al., 2007; Wallace et al., 2007a). Blood pressure was recorded for 60 min following the injections.
Statistical analyses
All data are presented as the mean ± SEM. Comparisons of two groups of data to one another were performed using Student's t-test (with Bonferroni correction in cases where heterogeneity of variance existed). Comparisons among more than two groups of data were performed using anova followed either by the Dunnett's Multiple Comparison test (parametric) or Mann–Whitney test (non-parametric), as appropriate. An associated probability (P-value) of less than 5% was considered significant.
Materials
ATB-346, ATB-345 and ATB-337 were synthesized by Antibe Therapeutics Inc. (Toronto, ON, Canada). Aspirin, diclofenac sodium, sodium naproxen, L-NAME, BCA and glibenclamide were purchased from Sigma-Aldrich (St. Louis, MO, USA). elisa kits for measuring TXB2 and PGE2 were obtained from Cayman Chemicals (Ann Arbor, MI, USA). Celecoxib was obtained from American Custom Chemicals Ltd. (San Diego, CA, USA). TBZ was purchased from SynChem Inc. (Des Plaines, IL, USA).
Drug and molecular target nomenclature conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2008).
Results
Inhibition of COX-1 and COX-2 activity
The airpouch model permits the evaluation of effects of a drug on systemic COX-1 activity, via measurement of whole blood TXB2 synthesis, and COX-2 activity, via measurement of PGE2 levels in the airpouch exudates (Wallace et al., 1999). Administration of zymosan into the airpouch resulted in a profound influx of leukocytes (Figure 2A) and a 15-fold increase in exudate concentrations of PGE2 (Figure 2B). Exudate leukocyte and PGE2 levels were significantly reduced by naproxen and by the two H2S-releasing naproxen derivatives. In the case of PGE2 levels, significantly greater inhibition was observed with ATB-346 than with naproxen or ATB-345. Whole blood TXB2 synthesis was almost completely suppressed by naproxen and ATB-346, but only 60% inhibition was observed in mice treated with ATB-345 (Figure 2C). Despite marked suppression of systemic COX-1 activity, neither of the H2S-releasing donors caused gastric damage, while haemorrhagic erosions were evident in the mice treated with naproxen (Figure 2D).
Figure 2.
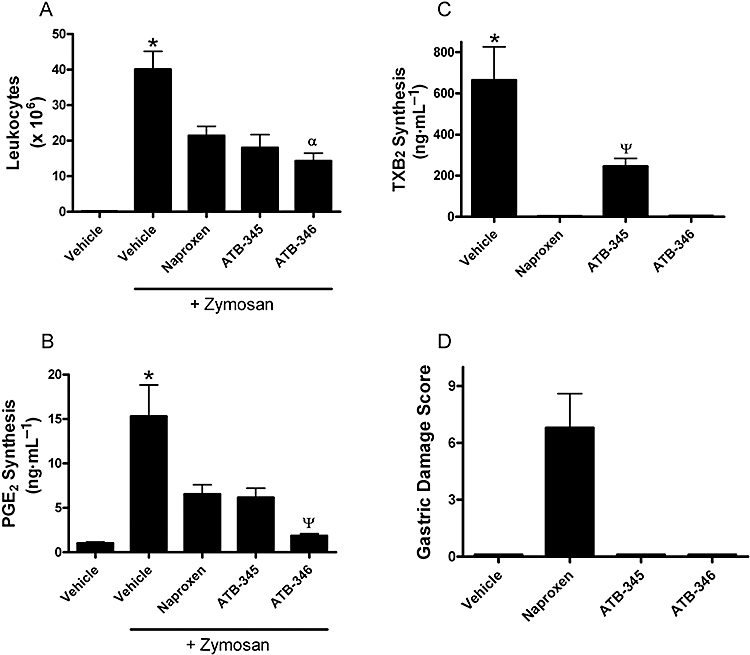
Effects of naproxen and two hydrogen sulphide-releasing naproxen derivatives (ATB-345 and ATB-346) in a model of zymosan-induced inflammation in the mouse. Samples of inflammatory exudates and of blood were collected 6 h after injection of zymosan into a preformed airpouch on the back of the mice. The test drugs were administered orally at a dose of 30 µmol·kg−1 1 h prior to zymosan administration. The test drugs significantly reduced infiltration of leukocytes into the airpouch (A), PGE2 levels in the exudates, which are indicative of cyclooxygenase-2 activity (B), and whole blood thromboxane synthesis, which is indicative of cyclooxygenase-1 activity (C). Gastric damage was observed in the mice treated with naproxen, but not in the other groups (D). *P < 0.05 versus all other groups. αP < 0.05 versus the naproxen-treated group; ΨP < 0.05 versus the other groups treated with one of the test drugs (anova and Dunnett's Multiple Comparison test). Each group consisted of five to six mice. ATB-345, 2-(6-methoxy-napthalen-2-yl)-propionic acid 4-(5-thioxo-5H-[1,2]dithiol-3-yl)-phenyl ester; ATB-346, 2-(6-methoxy-napthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester; PG, prostaglandin; TXB2, thromboxane B2.
Anti-inflammatory effects on adjuvant-Induced arthritis
A pronounced increase in paw volume was observed during days 14–21 after injection of Freund's Complete Adjuvant into base of the tail in rats (Figure 3). Twice-daily oral treatment with naproxen (4 µmol·kg−1) resulted in a significant reduction of paw volume on day 21, but not on day 14. In contrast, treatment with ATB-346 produced a significant reduction of paw volume on days 14 and 21. The paw volume on these days did not differ significantly from those on day 0 (i.e. prior to adjuvant administration). Rats treated with naproxen did not exhibit any gastric damage at the conclusion of the study, but blood was evident in the small intestine. Rats treated with ATB-346 did not exhibit any signs of damage or bleeding in the stomach or small intestine.
Figure 3.
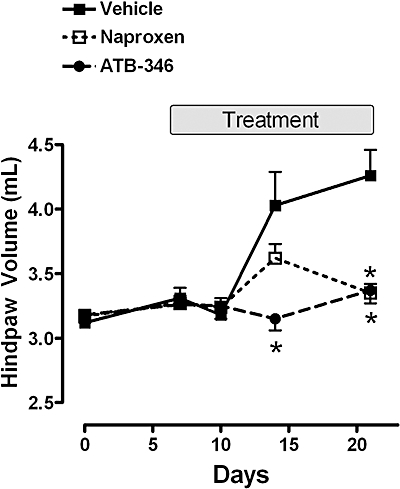
The reduction of adjuvant arthritis-associated paw swelling by orally administered naproxen and ATB-346 (both at 4 µmol·kg−1 twice-daily). Freund's complete adjuvant was administered on day 0, after the initial paw volume measurement. Results are shown as the summed volumes of the two hindpaws. Treatment with the test drugs or vehicle was carried out from days 7 to 21 after adjuvant administration. Naproxen significantly reduced paw oedema at day 21 (*P < 0.05 vs. the vehicle-treated group), but not at day 14. ATB-346 significantly reduced paw oedema at days 14 and 21 (*P < 0.05 vs. the vehicle-treated group). Each group consisted of six to seven rats (data were compared using anova and Dunnett's Multiple Comparison test). ATB-346, 2-(6-methoxy-napthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester.
Gastric damage and PG synthesis
To evaluate more fully the effects of ATB-346 on the gastric mucosa, a dose–response study was performed in rats. Naproxen given orally at doses of 30, 60 or 120 µmol·kg−1 produced haemorrhagic erosions in the stomach that increased in severity with the dose (Figure 4). With all three doses, gastric PGE2 synthesis was inhibited by naproxen (by 88%, 93% and 99% respectively). When ATB-346 was given at the same doses, gastric damage did not develop (this was confirmed by blind histological evaluation). Moreover, the inhibition of PGE2 synthesis by ATB-346 at doses of 30, 60 or 120 µmol·kg−1 (87%, 95% and 99% respectively) did not differ significantly from that observed with naproxen. We then examined the effects of a very high dose of naproxen and ATB-346 (2.74 mmol·kg−1). Naproxen induced the formation of extensive and severe haemorrhagic erosion (Figure 4). At this very high dose, ATB-346 also induced the formation of some haemorrhagic erosion, but the extent was markedly less (approximately the same as that produced by naproxen at 1/100th the dose).
Figure 4.
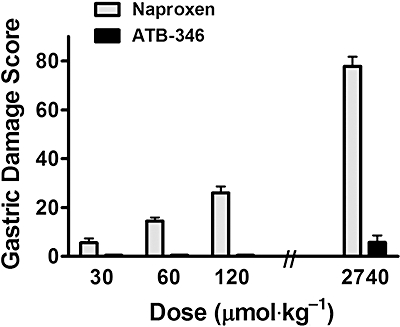
Oral administration of naproxen caused haemorrhagic damage in the stomach that increased in severity in a dose-dependent manner. In contrast, ATB-346 administration caused markedly less gastric damage at all doses tested; the stomach appeared normal with doses of 30 to 120 µmol·kg−1). At the dose of 2740 µmol·kg−1, there was significantly more damage with naproxen than with ATB-346 (P < 0.001; Student's t-test). Each group consisted of at least five rats. ATB-346, 2-(6-methoxy-napthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester.
The gastric-sparing property of ATB-346 was not evident when the two components of this drug (naproxen and TBZ) were administered as separate entities. Naproxen given orally at 50 µmol·kg−1 resulted in a gastric damage score of 15 ± 5, while with an equimolar dose of ATB-346 the gastric damage score was 0.2 ± 0.2. Co-administering the molar equivalent amounts of naproxen and TBZ resulted in a gastric damage score of 14 ± 4. Increasing the amount of TBZ administered to 100 or 150 µmol·kg−1 (with 50 µmol·kg−1 naproxen) did not confer any protective effect (gastric damage scores of 13 ± 3 and 16 ± 5 respectively).
Impaired gastric mucosal defence
Co-administration of naproxen with aspirin, the latter at a dose that by itself produced negligible gastric damage (10 mg·kg−1), resulted in more than a doubling of the gastric damage score as compared to that with naproxen alone (Figure 5A). Similarly, a selective COX-2 inhibitor, celecoxib, caused extensive gastric damage when co-administered with aspirin (but not when given alone). In contrast, the combination of ATB-346 and aspirin did not elicit gastric damage.
Figure 5.
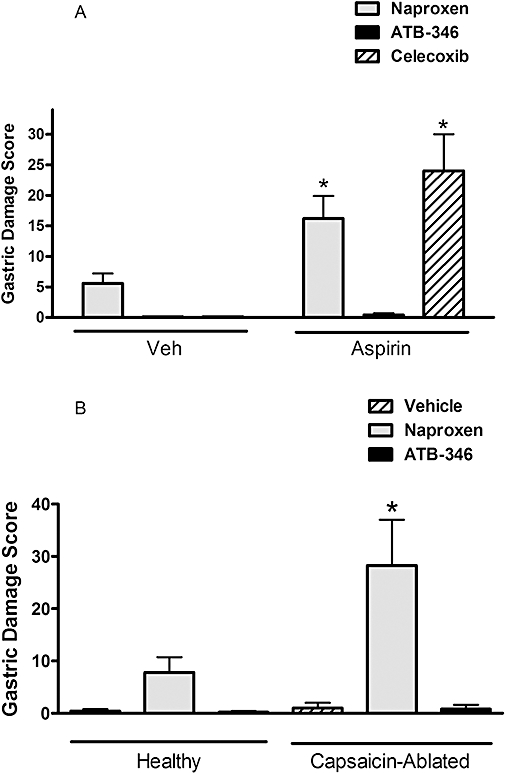
ATB-346 spares the stomach of injury in circumstances in which gastric mucosal defence is impaired. (A) Administration of aspirin at a dose that itself did not cause detectable gastric damage (10 mg·kg−1) resulted in a significant (*P < 0.05; anova and Dunnett's Multiple Comparison test) increase in the severity of gastric damage when co-administered with naproxen (60 µmol·kg−1) or celecoxib (30 µmol·kg−1), but not with ATB-346 (60 µmol·kg−1). (B) Abalation of sensory afferent nerves by neonatal capsaicin treatment resulted in a significant increase (*P < 0.05; Student's t-test) in the severity of naproxen-induced gastric damage, but an equimolar dose (60 µmol·kg−1) of ATB-346 did not cause significant gastric damage in capsaicin-ablated rats. Each group consisted of five to six rats. ATB-346, 2-(6-methoxy-napthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester; Veh, vehicle.
Ablation of gastric sensory afferent nerves with capsaicin has been shown to markedly increase the susceptibility of the stomach to injury induced by a variety of agents (Holzer et al., 1987). In the present study, naproxen-induced gastric damage was approximately three times more severe in rats in which capsaicin-induced ablation had been performed, while ATB-346 still did not induce significant gastric damage (Figure 5B).
Gastric mucosal defence can also be impaired by inhibition of synthesis of NO (Tepperman and Soper, 1993) or H2S (Fiorucci et al., 2005). In rats pretreated with the non-selective NO synthase inhibitor, L-NAME, naproxen produced significantly more severe gastric damage than that produced in rats pretreated with vehicle (Figure 6A). Similarly, administration of BCA, an inhibitor of one of the key enzymes for H2S synthesis in the stomach (Martin et al., 2009) resulted in a significant increase in the severity of naproxen-induced gastric damage. However, no gastric damage was observed in rats treated with ATB-346 following L-NAME or BCA administration (Figure 6B). Several effects of H2S have been suggested to occur through activation of ATP-sensitive potassium channels (KIR6.x), which can be blocked by glibenclamide (Wang, 2002; Distrutti et al., 2006a). Glibenclamide pretreatment itself did not produce any gastric damage, but subsequent administration of naproxen resulted in damage more than twice as extensive as that observed in vehicle pretreated rats (Figure 6C). Glibenclamide pretreatment had no effect on the gastric-sparing properties of ATB-346.
Figure 6.
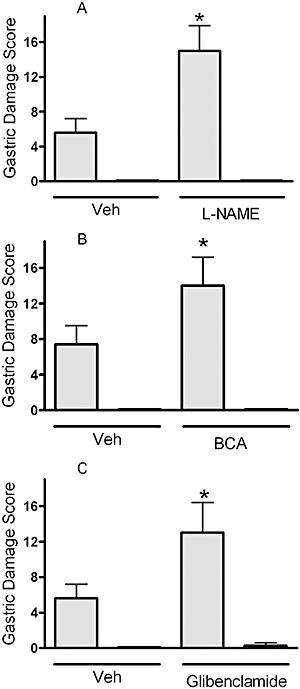
ATB-346 protected the stomach from injury in circumstances in which gastric mucosal defence was impaired. Administration of L-NAME (15 mg·kg−1; A), an inhibitor of nitric oxide synthase, or BCA (50 mg·kg−1; B), an inhibitor of hydrogen sulphide synthesis, significantly increased the severity of gastric damage induced by naproxen (shaded columns; 60 µmol·kg−1; *P < 0.05; Student's t-test), but significant damage was not observed when the rats were treated with an equimolar dose of ATB-346 (solid columns). Pretreatment with glibenclamide (10 mg·kg−1; C), an antagonist of ATP-sensitive potassium (KIR6.x) channels, also significantly increased the severity of naproxen-induced gastric damage, but rats cotreated with ATB-346 did not develop significant gastric injury. Each group consisted of five to six rats. ATB-346, 2-(6-methoxy-napthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester; BCA, β-cyanoalanine; L-NAME, NG-nitro-L-arginine methylester; Veh, vehicle.
Small intestinal injury and bleeding
Twice-daily oral administration of naproxen to rats for 5 days resulted in extensive damage in the small intestine (Figure 7A). Ulcers were mainly concentrated in the jejunum. Blood was evident in the lumen, and consistent with this observation, there was a significant decrease in haematocrit at the end of the study as compared with the pretreatment value (Figure 7B). In contrast, very little damage was observed in the intestine of rats treated with the same dose of ATB-346 (five of the six rats did not exhibit any damage), and there was no change in haematocrit over the course of the 5 days of treatment.
Figure 7.
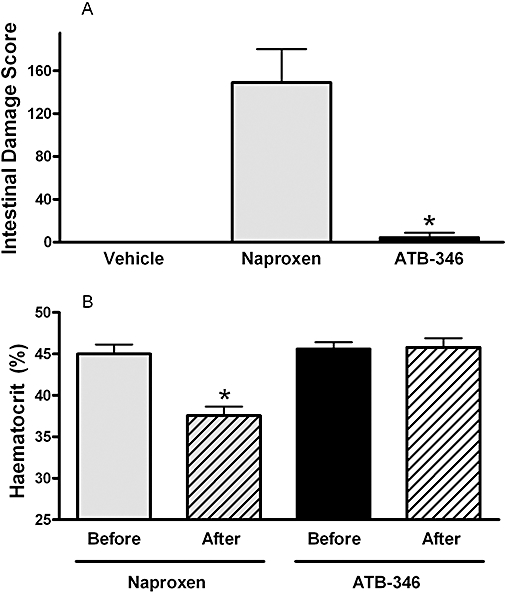
ATB-346 protected the small intestine from damage and bleeding. Twice-daily oral administration of naproxen (90 µmol·kg−1) for 5 days resulted in extensive ulceration and bleeding in the small intestine (A). The latter was evident upon examination of the intestine at the end of the study, and the severity of the bleeding is further exemplified by the significant decrease in haematocrit in naproxen-treated rats (B). Treatment with an equimolar dose of ATB-346 resulted in markedly less intestinal damage (*P < 0.05; anova and Mann–Whitney test) and did not significantly affect the haematocrit, as compared with that measured before drug administration. Each group consisted of six rats. ATB-346, 2-(6-methoxy-napthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester.
Healing of gastric ulcers
In addition to inducing erosions and ulcers, NSAIDs can interfere with the healing of pre-existing ulcers (Stadler et al., 1991). Using a well-characterized rodent model (Okabe et al., 1971; Ma et al., 2002; Dudar et al., 2008), we compared the effects of ATB-346 on gastric ulcer healing with those of naproxen and celecoxib. Within 3 days of serosal application of acetic acid, an ulcer with an average area of ∼25 mm2 had formed on the mucosal surface of the mouse stomach, penetrating through the muscularis mucosae into the submucosa. Over a 4 day period of twice-daily treatment with vehicle, the area of the ulcer was reduced by approximately 46% (Figure 8). In mice treated with naproxen or celecoxib, the extent of healing was significantly less than that in vehicle-treated mice (32% and 27% respectively). In contrast, treatment with ATB-346 significantly enhanced gastric ulcer healing (61%).
Figure 8.
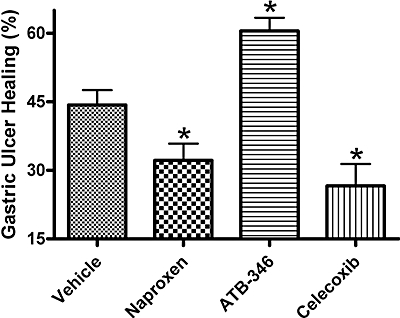
Effects of naproxen, ATB-346 and celecoxib on healing of gastric ulcers in mice. Mice were treated twice-daily with one of the test drugs, or vehicle (orally), from days 3 to 7 post ulcer induction. The extent of healing with each treatment is shown. While naproxen (60 µmol·kg−1) and celecoxib (30 µmol·kg−1) significantly impaired ulcer healing as compared with the vehicle-treated group, ATB-346 (60 µmol·kg−1) significantly enhanced ulcer healing. Each group consisted of six to seven mice (*P < 0.05 vs. the vehicle-treated group; anova and Dunnett's Multiple Comparison test). ATB-346, 2-(6-methoxy-napthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester.
Effects on systemic blood pressure
Intraperitoneal injection of ATB-346 or naproxen to healthy, untreated rats did not elicit any significant change in systemic arterial blood pressure over the 60 min that followed. In both cases, the mean arterial blood pressure remained in the 95–110 mmHg range. The mean arterial blood pressure in the rats that had received L-NAME in their drinking water for the previous 2 weeks was 153 ± 5 mmHg (n= 32). During the hour after i.p. administration of naproxen at a dose of 90 µmol·kg−1, there was an increase above basal blood pressure of ∼12 mmHg (Figure 9). In contrast, the increase in blood pressure observed following administration of an equimolar dose of ATB-346 (∼4 mmHg) did not differ significantly from that observed following administration of vehicle (∼3 mmHg). Similar results were obtained when another NSAID (diclofenac; 60 µmol·kg−1) was compared with an equimolar dose of a H2S-releasing diclofenac derivative, ATB-337 (Figure 9).
Figure 9.
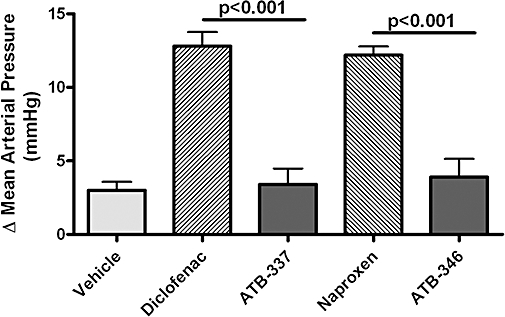
Unlike conventional NSAIDs (diclofenac and naproxen at 60 and 90 µmol·kg−1 respectively), equimolar doses of hydrogen sulphide-releasing derivatives of these drugs (ATB-337 and ATB-346 respectively) did not significantly elevate mean arterial blood pressure in rats with hypertension induced by addition of L-NAME to the drinking water (400 mg·L−1). The basal blood pressure (after 2 weeks of consumption of the supplemented drinking water) was 153 ± 5 mmHg (n= 32), with no significant differences in basal blood pressure among the treatment groups. Results are shown as the change in mean arterial blood pressure over the course of 1 h after an i.p. bolus injection of the test drug or vehicle. Each group consisted of five to seven rats (anova and Dunnett's Multiple Comparison test). ATB-337, (2-(2,6-dichloro-phenylamino)-phenyl)-acetic acid 4-(3-thioxo-3H-[1,2]dithiol-4-yl)-phenyl ester; ATB-346, 2-(6-methoxy-napthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester; L-NAME, NG-nitro-L-arginine methylester; NSAIDs, non-steroidal anti-inflammatory drugs.
Discussion
Considerable resources have been invested in the past four decades in the development of effective anti-inflammatory and analgesic drugs that do not cause the GI ulceration and bleeding associated with the use of conventional NSAIDs. Selective COX-2 inhibitors arrived on the market in the mid 1990s with a promise of reduced GI toxicity. These drugs do produce less GI ulceration and bleeding than traditional NSAIDs in some groups of patients and in healthy animals (Masferrer et al., 1994; Silverstein et al., 2000), but when given concomitantly with aspirin or in other circumstances in which mucosal defence is impaired, the safety advantage over conventional NSAIDs is negligible (Maricic et al., 1999; Silverstein et al., 2000; Wallace et al., 2000; Fiorucci et al., 2003; Lanas and Scheiman, 2007). Selective COX-2 inhibitors have also been found to interfere with healing of pre-existing ulcers in rodent studies (Mizuno et al., 1997; Ma et al., 2002). These findings suggest that preclinical studies of novel anti-inflammatory drugs would be more useful if they included examination of the effects of those drugs in models in which gastric mucosal defence is impaired and in models of pre-existing injury. Thus, in the present study we evaluated ATB-346 in healthy animals and in several models characterized by impaired mucosal defence, as well as in a model of gastric ulcer healing.
In the present study, we examined the effects of a novel H2S-releasing derivative of one of the most commonly used NSAIDs, naproxen. ATB-346 exhibited anti-inflammatory activity comparable, or even superior, to equimolar doses of naproxen in two models (mouse airpouch and rat adjuvant arthritis). In healthy rats, ATB-346 caused negligible gastric damage even when given at an extremely high dose (2.7 mmol·kg−1). On a kg−1 basis, this represents a dose >100 times the usual human dose of naproxen. The small amount of haemorrhagic damage observed in rats treated with the high dose of ATB-346 was equivalent to that observed with naproxen at 1/100th that dose. We also observed a marked gastric-sparing effect of ATB-346 in several models in which gastric mucosal defence was impaired. These included co-administration of aspirin, pretreatment with inhibitors of NO or H2S synthesis, co-administration of a KIR6.x antagonist and prior ablation of sensory afferent nerves, all of which significantly increased the severity of naproxen-induced gastric damage. Moreover, while naproxen and celecoxib significantly inhibited gastric ulcer healing in mice, there was a significant enhancement of healing in mice treated with ATB-346. When given repeatedly over several days, naproxen elicited extensive ulceration and bleeding in the small intestine. In contrast, ATB-346 had no effect on haematocrit and produced very little mucosal damage.
Several lines of evidence suggest that H2S is a potent mediator of GI mucosal defence, and that the GI-sparing effects of ATB-346 are attributable to its ability to release H2S. First, H2S donors have been shown to protect the gastric mucosa from NSAID-induced damage (Fiorucci et al., 2005; Wallace, 2009). Second, inhibition of endogenous H2S synthesis results in a significant increase in severity of NSAID-induced gastric damage, as seen in the present study and previously (Fiorucci et al., 2005). Third, the conjugation of different H2S donors to different NSAIDs has been shown to consistently result in a GI-sparing effect (Li et al., 2007; Wallace et al., 2007a; 2008;). While ATB-346 did not produce significant gastric damage in various models of impaired mucosal defence, co-administration of the two moieties of this compound (naproxen and TBZ) resulted in gastric damage comparable in severity to that seen with naproxen alone. The explanation for this finding may lie in the different levels of release of H2S from TBZ versus that from ATB-346. Release of H2S from ATB-346 incubated in buffer or in rat liver homogenates was found to be about six times greater than that from an equimolar concentration of TBZ (Wallace et al., 2008). Similar results have been reported with an H2S-releasing derivative of mesalamine (Distrutti et al., 2006b).
ATB-346 did not produce significant GI injury despite inhibiting systemic COX-1 activity and mucosal PG synthesis as effectively as naproxen. The finding that this compound also did not induce damage when mucosal NO or H2S synthesis was suppressed (by L-NAME and BCA respectively) suggests that stimulation of endogenous release of those mediators did not account for its GI safety. Many effects of H2S have been reported to occur via stimulation of ATP-sensitive potassium (KIR6.x) channels. However, ATB-346 still did not elicit gastric damage when given to rats pretreated with a KIR6.x antagonist (glibenclamide) at a dose that significantly exacerbated naproxen-induced damage. Thus, activation of these potassium channels by ATB-346 does not appear to be the prime mechanism underlying its GI safety. Li et al. (2007) demonstrated that a H2S-releasing derivative of diclofenac suppressed endotoxin-induced generation of several pro-inflammatory cytokines (interleukin-1β, tumour necrosis factor-α) and provided evidence that this may be mediated via inhibition of activation of the transcription factor NFκB. Inhibitors of NFκB activation have been shown to reduce the severity of NSAID-induced gastric damage (Brand et al., 1999). Inhibition by H2S of NSAID-induced leukocyte adherence in the GI microcirculation is another possible mechanism underlying the GI safety of ATB-346. Leukocyte adherence is an early, key event in the pathogenesis of mucosal injury associated with NSAIDs (Wallace, 1993). H2S is a potent inhibitor of leukocyte adherence induced by NSAIDs and other agonists (Zanardo et al., 2006; Andruski et al., 2008). While NSAIDs have been shown to trigger leukocyte adherence and up-regulation of endothelial (ICAM-1) and leukocyte (LFA-1) adhesion molecules, these events were prevented by co-administration of an H2S donor with the NSAID (Fiorucci et al., 2005), and did not occur following administration of an H2S-releasing derivative of diclofenac (Wallace et al., 2007a). H2S can also prevent the decrease in gastric blood flow that is seen following NSAID administration (Fiorucci et al., 2005), which could contribute to the gastric safety of ATB-346. However, both the inhibition of leukocyte adherence and the vasodilator effects of H2S have been reported to be inhibited by glibenclamide (Wang, 2002; Fiorucci et al., 2005; Zanardo et al., 2006), while the gastric safety of ATB-346 in the present study was unaffected by glibenclamide.
The withdrawal of rofecoxib (Vioxx®) from worldwide markets in 2004 raised awareness of the cardiovascular toxicity of coxibs and of traditional NSAIDs (Grosser et al., 2006; Kearney et al., 2006). The underlying mechanism for this toxicity has not been identified and is the subject of considerable debate (Grosser et al., 2006; Mitchell and Warner, 2006). Sustained suppression of platelet thromboxane synthesis is generally regarded as beneficial in terms of cardiovascular health during NSAID therapy, counteracting the detrimental effects of suppression of endothelial prostacyclin synthesis (Grosser et al., 2006). The long-lasting suppression of platelet thromboxane synthesis by naproxen, versus most other NSAIDs, has been suggested to account for the relative cardiovascular safety of naproxen (Kearney et al., 2006). Both traditional NSAIDs and selective COX-2 inhibitors cause small but clinically significant increases in systemic blood pressure, which can result in significant increases in the risk of myocardial infarction and stroke (Singh et al., 2003). While a detailed study of the cardiovascular safety of ATB-346 has not been performed, it is noteworthy that this compound suppressed systemic COX-1 activity (i.e. platelet thromboxane synthesis) as effectively as naproxen. Moreover, bolus injection of a high dose of ATB-346 to hypertensive rats did not significantly affect mean arterial blood pressure, while a significant increase was seen with an equimolar dose of naproxen. H2S has recently been shown to contribute to regulation of blood pressure in mice (Yang et al., 2008). We did not observe any effect of ATB-346 on blood pressure in normotensive rats, consistent with the findings of Li et al. (2007), who studied an H2S-releasing diclofenac derivative. The effectiveness of coupling of an endogenous vasodilator with an NSAID to reduce the detrimental effects of the NSAID on blood pressure has been demonstrated previously. NO-releasing NSAIDs have been shown to have beneficial effects in hypertensive animals and humans (Muscaráet al., 1998; 2000a; Karlsson et al., 2009; Wallace et al., 2009a; White et al., 2009).
In summary, the present study provides evidence for the remarkable GI-sparing properties of an H2S-releasing derivative of naproxen. The H2S-releasing moiety contained within ATB-346, TBZ, has not been previously utilized for this purpose. In healthy animals, ATB-346 was in the order of 100-fold safer than naproxen, in terms of eliciting gastric damage, but in two models of inflammation it exerted effects comparable to or superior to those of naproxen. Importantly, and in contrast to selective COX-2 inhibitors, ATB-346 did not cause significant gastric damage when given to rats in which mucosal defence was significantly compromised, and it enhanced, rather than inhibited, healing of pre-existing gastric ulcers. Finally, the lack of effect of ATB-346 on blood pressure in hypertensive rats suggests that this compound may also exhibit a better cardiovascular profile than conventional NSAIDs. H2S-releasing NSAIDs appear to represent a very promising alternative to existing therapies for the treatment of inflammation and pain.
Glossary
Abbreviations:
- GI
gastrointestinal
- H2S
hydrogen sulphide
- NO
nitric oxide
- NSAIDs
non-steroidal anti-inflammatory drugs
- PG
prostaglandin
Conflicts of interest
Each of the authors of this paper holds shares in Antibe Therapeutics Inc., a company developing H2S-releasing drugs, including those described in this paper.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andruski B, McCafferty DM, Ignacy T, Millen B, McDougall JJ. Leukocyte trafficking and pain behavioral responses to a hydrogen sulphide donor in acute monoarthritis. Am J Physiol. 2008;295:R814–R820. doi: 10.1152/ajpregu.90524.2008. [DOI] [PubMed] [Google Scholar]
- Brand SJ, Morise Z, Tagerud S, Mazzola L, Granger DN, Grisham MB. Role of the proteasome in rat indomethacin-induced gastropathy. Gastroenterology. 1999;116:865–873. doi: 10.1016/s0016-5085(99)70069-7. [DOI] [PubMed] [Google Scholar]
- Brown JF, Keates AC, Hanson PJ, Whittle BJ. Nitric oxide generators and cGMP stimulate mucus secretion by rat gastric mucosal cells. Am J Physiol. 1993;265:G418–G422. doi: 10.1152/ajpgi.1993.265.3.G418. [DOI] [PubMed] [Google Scholar]
- Cicala C, Ianaro A, Fiorucci S, Calignano M, Buci M, Gerli R, et al. NO-naproxen modulates inflammation, nociception and downregulates T cell response in rat Freund's adjuvant arthritis. Br J Pharmacol. 2000;130:1399–1405. doi: 10.1038/sj.bjp.0703449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distrutti E, Sediari L, Mencarelli A, Renga B, Orlandi S, Antonelli E, et al. Evidence that hydrogen sulphide exerts antinociceptive effects in the gastrointestinal tract by activating KATP channels. J Pharmacol Exp Ther. 2006a;316:325–335. doi: 10.1124/jpet.105.091595. [DOI] [PubMed] [Google Scholar]
- Distrutti E, Sediari L, Mencarelli A, Renga B, Orlandi S, Russo G, et al. 5-Amino-2-hydroxybenzoic acid 4-(5-thioxo-5H-[1,2]dithiol-3yl)-phenyl ester (ATB-429), a hydrogen sulfide-releasing derivative of mesalamine, exerts antinociceptive effects in a model of postinflammatory hypersensitivity. J Pharmacol Exp Ther. 2006b;319:447–458. doi: 10.1124/jpet.106.106435. [DOI] [PubMed] [Google Scholar]
- Dudar GK, D'Andrea LD, Di Stasi R, Pedone C, Wallace JL. A vascular endothelial growth factor mimetic accelerates gastric ulcer healing in an iNOS-dependent manner. Am J Physiol. 2008;295:G374–G381. doi: 10.1152/ajpgi.90325.2008. [DOI] [PubMed] [Google Scholar]
- Edwards JW, Sedgwick AD, Willoughby DA. The formulation of a structure with features of synovial lining by subcutaneous injection of air: an in vivo tissue culture system. J Pathol. 1981;134:147–156. doi: 10.1002/path.1711340205. [DOI] [PubMed] [Google Scholar]
- Elliott SN, McKnight W, Cirino G, Wallace JL. A nitric oxide-releasing nonsteroidal anti-inflammatory drug accelerates gastric ulcer healing in rats. Gastroenterology. 1995;109:524–530. doi: 10.1016/0016-5085(95)90341-0. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Santucci L, Wallace JL, Sardina M, Romano M, del Soldato P, et al. Interaction of a selective cyclooxygenase-2 inhibitor with aspirin and NO-releasing aspirin in the human gastric mucosa. Proc Natl Acad Sci USA. 2003;100:10937–10941. doi: 10.1073/pnas.1933204100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorucci S, Antonelli E, Distrutti E, Rizzo G, Mencarelli A, Orlandi S, et al. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology. 2005;129:1210–1224. doi: 10.1053/j.gastro.2005.07.060. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Distrutti E, Cirino G, Wallace JL. The emerging roles of hydrogen sulfide in the gastrointestinal tract and liver. Gastroenterology. 2006;131:259–271. doi: 10.1053/j.gastro.2006.02.033. [DOI] [PubMed] [Google Scholar]
- Grosser T, Fries S, Fitzgerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4–15. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer P, Schluet W, Lippe IT, Sametz W. Involvement of capsaicin-sensitive sensory neurons in gastrointestinal function. Acta Physiol Hung. 1987;69:403–411. [PubMed] [Google Scholar]
- Karlsson J, Pivodic A, Aguirre D, Schnitzer TJ. Efficacy, safety, and tolerability of the cyclooxygenase-inhibiting nitric oxide donator naproxcinod in treating osteoarthritis of the hip or knee. J Rheumatol. 2009;36:1290–1297. doi: 10.3899/jrheum.081011. [DOI] [PubMed] [Google Scholar]
- Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. Br Med J. 2006;332:1302–1308. doi: 10.1136/bmj.332.7553.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanas A, Scheiman J. Low-dose aspirin and upper gastrointestinal damage: epidemiology, prevention and treatment. Curr Med Res Opin. 2007;23:163–173. doi: 10.1185/030079907X162656. [DOI] [PubMed] [Google Scholar]
- Li L, Moore PK. Putative biological roles of hydrogen sulfide in health and disease: a breath of not so fresh air? Trends Pharmacol Sci. 2008;29:84–90. doi: 10.1016/j.tips.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Li L, Rossoni G, Sparatore A, Lee LC, Del Soldato P, Moore PK. Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radic Biol Med. 2007;42:706–719. doi: 10.1016/j.freeradbiomed.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Linden DR, Sha L, Mazzone A, Stoltz GJ, Bernard CE, Furne JK, et al. Production of the gaseous signal molecule hydrogen sulfide in mouse tissues. J Neurochem. 2008;106:1577–1585. doi: 10.1111/j.1471-4159.2008.05502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, del Soldato P, Wallace JL. Divergent effects of new cyclooxygenase inhibitors on gastric ulcer healing: shifting the angiogenic balance. Proc Natl Acad Sci USA. 2002;99:13243–13247. doi: 10.1073/pnas.202392199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCafferty DM, Wallace JL, Sharkey KA. Effects of chemical sympathectomy and sensory nerve ablation on experimental colitis in the rat. Am J Physiol. 1997;272:G272–G280. doi: 10.1152/ajpgi.1997.272.2.G272. [DOI] [PubMed] [Google Scholar]
- Maricic N, Ehrlich K, Gretzer B, Schuligoi R, Respondek M, Peskar BM. Selective cyclo-oxygenase-2 inhibitors aggravate ischaemia-reperfusion injury in the rat stomach. Br J Pharmacol. 1999;128:1659–1666. doi: 10.1038/sj.bjp.0702966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GR, McKnight GW, Dicay MS, Coffin CS, Ferraz JG, Wallace JL. Hydrogen sulphide synthesis in the rat and mouse gastrointestinal tract. Dig Liver Dis. 2009 doi: 10.1016/j.dld.2009.05.016. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, et al. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci USA. 1994;91:3228–3232. doi: 10.1073/pnas.91.8.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JA, Warner TD. COX isoforms in the cardiovascular system: understanding the activities of non-steroidal anti-inflammatory drugs. Nat Rev Drug Discov. 2006;5:75–86. doi: 10.1038/nrd1929. [DOI] [PubMed] [Google Scholar]
- Mizuno H, Sakamoto C, Matsuda K, Wada K, Uchida T, Noguchi H, et al. Induction of cyclooxygenase 2 in gastric mucosal lesions and its inhibition by the specific antagonist delays healing in mice. Gastroenterology. 1997;112:387–397. doi: 10.1053/gast.1997.v112.pm9024292. [DOI] [PubMed] [Google Scholar]
- Muscará MN, McKnight W, Del Soldato P, Wallace JL. Effect of a nitric oxide-releasing naproxen derivative on hypertension and gastric damage induced by chronic nitric oxide inhibition in the rat. Life Sci. 1998;62:PL235–PL240. doi: 10.1016/s0024-3205(98)00072-1. [DOI] [PubMed] [Google Scholar]
- Muscará MN, McKnight W, Lovren F, Triggle CR, Cirino G, Wallace JL. Antihypertensive properties of a nitric oxide-releasing naproxen derivative in two-kidney, one-clip rats. Am J Physiol. 2000a;279:H528–H535. doi: 10.1152/ajpheart.2000.279.2.H528. [DOI] [PubMed] [Google Scholar]
- Muscará MN, Vergnolle N, Lovren F, Triggle CR, Elliott SN, Asfaha S, et al. Selective cyclo-oxygenase-2 inhibition with celecoxib elevates blood pressure and promotes leukocyte adherence. Br J Pharmacol. 2000b;129:1423–1430. doi: 10.1038/sj.bjp.0703232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Roth JL, Pfeiffer CJ. A method for experimental, penetrating gastric and duodenal ulcers in rats. Observations on normal healing. Am J Dig Dis. 1971;16:277–284. doi: 10.1007/BF02235252. [DOI] [PubMed] [Google Scholar]
- Reuter BK, Davies NM, Wallace JL. Nonsteroidal anti-inflammatory drug-enteropathy in rats: role of permeability, bacteria and enterohepatic circulation. Gastroenterology. 1997;112:109–117. doi: 10.1016/s0016-5085(97)70225-7. [DOI] [PubMed] [Google Scholar]
- Schicho R, Krueger D, Zeller F, Von Weyhern CW, Frieling T, Kimura H, et al. Hydrogen sulfide is a novel prosecretory neuromodulator in the guinea-pig and human colon. Gastroenterology. 2006;131:1542–1552. doi: 10.1053/j.gastro.2006.08.035. [DOI] [PubMed] [Google Scholar]
- Silverstein FE, Faich G, Goldstein JL, Simon LS, Pincus T, Whelton A, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: a randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA. 2000;284:1247–1255. doi: 10.1001/jama.284.10.1247. [DOI] [PubMed] [Google Scholar]
- Singh G, Miller JD, Huse DM, Pettitt D, D'Agostino RB, Russell MW. Consequences of increased systolic blood pressure in patients with osteoarthritis and rheumatoid arthritis. J Rheumatol. 2003;30:714–719. [PubMed] [Google Scholar]
- Stadler P, Armstrong D, Margalith D, Saraga E, Stolte M, Lualdi P, et al. Diclofenac delays healing of gastroduodenal mucosal lesions. Double-blind, placebo-controlled endoscopic study in healthy volunteers. Dig Dis Sci. 1991;36:594–600. doi: 10.1007/BF01297025. [DOI] [PubMed] [Google Scholar]
- Tepperman BL, Soper BD. Interaction of nitric oxide and salivary gland epidermal growth factor in the modulation of rat gastric mucosal integrity. Br J Pharmacol. 1993;110:229–234. doi: 10.1111/j.1476-5381.1993.tb13797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JL. Gastric ulceration – critical events at the neutrophil-endothelium interface. Can J Physiol Pharmacol. 1993;71:98–102. doi: 10.1139/y93-014. [DOI] [PubMed] [Google Scholar]
- Wallace JL. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol Sci. 2007;28:501–505. doi: 10.1016/j.tips.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Wallace JL. Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn't the stomach digest itself? Physiol Rev. 2008;88:1547–1565. doi: 10.1152/physrev.00004.2008. [DOI] [PubMed] [Google Scholar]
- Wallace JL. Physiological and pathophysiological roles of hydrogen sulfide in the gastrointestinal tract. Antioxid Redox Signal. 2009 doi: 10.1089/ars.2009.2900. in press. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Del Soldato P. The therapeutic potential of NO-NSAIDs. Fundam Clin Pharmacol. 2003;17:11–20. doi: 10.1046/j.1472-8206.2003.00125.x. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Chapman K, McKnight W. Limited anti-inflammatory efficacy of cyclo-oxygenase-2 inhibition in carrageenan airpouch inflammation. Br J Pharmacol. 1999;126:1200–1204. doi: 10.1038/sj.bjp.0702420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JL, McKnight W, Reuter BK, Vergnolle N. NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology. 2000;119:706–714. doi: 10.1053/gast.2000.16510. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Caliendo G, Santagada V, Cirino G, Fiorucci S. Gastrointestinal safety and anti-inflammatory effects of a hydrogen sulfide-releasing diclofenac derivative in the rat. Gastroenterology. 2007a;132:261–271. doi: 10.1053/j.gastro.2006.11.042. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Dicay M, McKnight W, Martin GR. Hydrogen sulfide enhances ulcer healing in rats. FASEB J. 2007b;21:4070–4076. doi: 10.1096/fj.07-8669com. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Cirino G, Santagada V, Caliendo G. Hydrogen sulfide derivatives of nonsteroidal anti-inflammatory drugs. 2008. United States Patent Application No. WO/2008/009127.
- Wallace JL, Viappiani S, Bolla M. Cyclooxygenase-inhibiting nitric oxide donators for osteoarthritis. Trends Pharmacol Sci. 2009a;30:112–117. doi: 10.1016/j.tips.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Vong L, McKnight W, Dicay M, Martin GR. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology. 2009b;137:569–578. doi: 10.1053/j.gastro.2009.04.012. [DOI] [PubMed] [Google Scholar]
- Wang R. Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 2002;16:1792–17987. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- White WB, Schnitzer TJ, Fleming R, Duquesroix B, Beekman M. Effects of the cyclooxygenase inhibiting nitric oxide donator naproxcinod versus naproxen on systemic blood pressure in patients with osteoarthritis. Am J Cardiol. 2009;104:840–845. doi: 10.1016/j.amjcard.2009.05.014. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006;20:2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]