

Abstract
Background and purpose:
Adenosine is a major endogenous regulator of macrophage function, and activates four specific adenosine receptors (A1, A2A, A2B and A3). Here, we have assessed in human lung macrophages the modulation of the expression of adenosine receptor mRNA by lipopolysaccharide (LPS), and the relative contributions of the different adenosine receptors to LPS-induced production of tumour necrosis factor (TNF)-α and chemokines.
Experimental approach:
Lung macrophages isolated from resected lungs were stimulated with LPS and treated with adenosine receptor agonists or/and antagonists. Adenosine receptor expression was assessed with qRT-PCR. Cytokines were measured in lung macrophage supernatants with elisa.
Key results:
LPS increased (about 400-fold) mRNA for A2A adenosine receptors, decreased mRNA for A1 and A2B, but had no effect on A3 adenosine receptor mRNA. The adenosine receptor agonist NECA inhibited TNF-α production concentration dependently, whereas the A1 receptor agonist, CCPA, and the A3 receptor agonist, AB-MECA, inhibited TNF-α production only at concentrations affecting A2A receptors. NECA also inhibited the production of CCL chemokines (CCL2, CCL3, CCL4, CCL5) and CXCL chemokines (CXCL9 and CXCL10), but not that of CXCL1, CXCL8 and CXCL5. Reversal of NECA-induced inhibition of TNF-α and chemokine production by the selective A2A adenosine receptor antagonist ZM 241385, but not the A2B receptor antagonist, MRS 1754, or the A3 receptor antagonist, MRS 1220, indicated involvement of A2A receptors.
Conclusions and implications:
LPS up-regulated A2A adenosine receptor gene transcription, and this receptor subtype mediated inhibition of the LPS-induced production of TNF-α and of a subset of chemokines in human lung macrophages.
Keywords: adenosine, adenosine receptors, lung macrophages, tumour necrosis factor-α, chemokines, lipopolysaccharide
Introduction
Adenosine is an endogenous purine nucleoside that is rapidly released from all cells upon ATP degradation under conditions of stress, hypoxia or chronic inflammation (Spicuzza et al., 2006). Adenosine clearly plays a role in inflammatory lung diseases, such as asthma and chronic obstructive pulmonary disease (COPD) (Varani et al., 2006; Brown et al., 2008; Wilson, 2008). The diverse effects of extracellular adenosine are mediated through activation of four distinct G-protein-coupled receptors: A1, A2A, A2B and A3 (nomenclature follows; Alexander et al., 2008). Adenosine receptors are ubiquitously expressed throughout the body with virtually all the cell types involved in airway inflammation expressing one or more adenosine receptor subtypes (Varani et al., 2006; Brown et al., 2008).
Adenosine receptors are expressed on monocytes and macrophages, and, through activation of these receptors, adenosine modulates monocyte and macrophage functions (Hasko et al., 2007). Because adenosine is released under the same conditions as those causing macrophage activation, adenosine receptors may enable macrophages to fine-tune their responses to stressful conditions, particularly acute or chronic airway inflammation (Hasko et al., 2007). Tumour necrosis factor (TNF)-α, a well-known pro-inflammatory cytokine with a wide range of biological functions, is secreted primarily by monocytes and macrophages. In addition to inhibition of TNF-α production, adenosine can down-regulate interleukin (IL)-12 production and up-regulate IL-10 release from monocytes and macrophages (Hasko et al., 2007).
TNF-α production by monocytes and macrophages can be inhibited by A1, A2A and A3 adenosine receptors, and IL-12 production by A2A and A3 adenosine receptors. The receptor subtypes involved depend on cell origin, species and inflammatory stimulus (Hasko et al., 2007). On human monocytes, activation of A2A receptors is predominantly responsible for inhibition of lipopolysaccharide (LPS)-induced TNF-α production by adenosine receptor agonists (Zhang et al., 2005; Ezeamuzie and Khan, 2007; Hamano et al., 2008). A2A receptors are coupled to Gs proteins, elevate intracellular levels of cAMP, activate the protein kinase A (PKA) pathway involved in the inhibition of LPS-induced TNF-α production in human monocytes and macrophages (Bryn et al., 2006), thereby explaining the A2A receptor-mediated inhibition of TNF-α production (Palmer and Trevethick, 2008). In addition to TNF-α, the production of IL-12 and of the chemokines, CCL-3 and CCL-4, are also inhibited by cAMP-dependent pathways in human blood monocytes and monocyte-derived macrophages (Kimata et al., 1998; Bryn et al., 2006).
Macrophages play a critical role in the pathophysiology of COPD and asthma (Barnes, 2008). However, the role of adenosine receptors in the regulation of TNF-α production by human lung macrophages has not been addressed previously. In addition to the release of inflammatory cytokines, like TNF-α, macrophages orchestrate lung inflammation by releasing chemokines to recruit leukocytes (Barnes, 2008). Therefore, this study was designed to investigate the effect of LPS on the expression of adenosine receptor mRNA, and to differentiate the relative contributions of the different adenosine receptor subtypes to the regulation of LPS-induced production of TNF-α, CC chemokines [CCL2 (MCP-1), CCL3 (MIP-1α), CCL4 (MIP-1β), CCL5 (RANTES)] and CXC chemokines [CXCL1 (GRO-α), CXCL5 (ENA-78), CXCL8 (IL-8), CXCL9 (MIG), CXCL10 (IP-10)], which are involved in the recruitment of neutrophils, monocytes and lymphocytes within the airways. We found that all four adenosine receptors were expressed on lung macrophages, and that A2A adenosine receptor expression was dramatically increased following LPS stimulation. We also demonstrate that LPS-induced TNF-α and chemokine production is inhibited mainly through activation of the A2A receptor subtype, while other adenosine receptor subtypes do not seem to play a role in regulating production of cytokines and chemokines in lung macrophages.
Methods
Isolation and culture of human lung macrophages
The use of resected lung tissues for research purposes was approved by the Regional Ethics Committee for Biomedical Research. Lung tissues were obtained from 11 patients (age: 59 ± 9 years, sex (M : F): 6:5, FEV1: 81 ± 4% predicted, FEV1/FVC ratio: 0.77 ± 0.04, smokers/ex-smokers: 6/5, pack years: 49 ± 8) undergoing surgical resection for lung carcinoma who had not received preoperative anti-cancer chemotherapy or radiotherapy. Lung macrophages were isolated as described previously (Jeyaseelan et al., 2005) with some modifications. Briefly, peripheral tissues far from the tumour were separated from their capsule, washed with sterile culture medium [Roswell Park Memorial Institute (RPMI) 1640 supplemented with 100 µg·mL−1 of streptomycin and 100 U·mL−1 of penicillin], then finely minced with scissors and washed again with RPMI 1640. The collected fluid was centrifuged (10 min, 300× g), and the cell pellet was resuspended in RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum (FCS), L-glutamine (2 mM) and antibiotics (complete medium). The cell suspension was adjusted to 106 viable cells·mL−1, and 1 mL of cell suspension was deposited in each well of 24-well plastic tissue culture plates and left at 37°C in a 5% CO2 humidified atmosphere for at least 1 h. The non-adherent cells were removed by three gentle washings with warm medium, 1 mL of complete medium was then added to each well and the remaining cells were maintained at 37°C and 5% CO2 overnight. The adherent population of cells (160 ± 16 × 103 cells per well) were >95% pure macrophages using light microscope cytological examination after May–Grünewald–Giemsa staining (Coger, Paris, France) and using CD68 immunocytochemistry (monoclonal mouse anti-human CD68 antibody, KP1 clone, Dako, Trappes, France). Cell viability exceeded 90% as assessed by trypan blue dye exclusion.
Treatment of lung macrophages with adenosine receptor agonists or antagonists
Stock solutions of 10 mM adenosine receptor agonists and antagonists were prepared in dimethylsulphoxide (DMSO) except for the A2B receptor antagonist, MRS 1754, solubilized at 100 mM. All subsequent dilutions were prepared in complete medium. The maximal DMSO concentration applied to cells in culture did not exceed 0.1%, and had no major effect on either cell viability or TNF-α production (data not shown) as previously reported (Bshesh et al., 2002). The 24-well plates containing lung macrophages were washed with warm medium, and 1 mL of fresh medium supplemented with 1% FCS was added. Lung macrophages were exposed to adenosine receptor antagonists or their respective vehicles for 15 min before being pre-incubated for 1 h (37°C, 5% CO2) with adenosine receptor agonists or vehicles prior to LPS stimulation (1 µg·mL−1) for 24 h. Antagonist concentrations for ZM 241385, MRS 1754 and MRS 1220 were chosen based on their reported affinities for human recombinant adenosine receptors (Fredholm et al., 2001). All wells were run in duplicate for each series of experiments performed with lung macrophages isolated from a resected lung. After incubation with LPS, lung macrophage culture supernatants were collected and stored at −80°C prior to cytokine quantification. None of the treatments used in this study altered cell viability.
Cytokine assays
Cytokine production was assessed by measuring their concentrations in the lung macrophage supernatants with elisa (Duoset Development System), according to the manufacturer's instructions (R&D Systems Europe, Lille, France). The supernatants were diluted with RPMI as appropriate, and the optical density was determined at 450 nm with an MRX II microplate reader from Dynex Technologies (Saint-Cloud, France). Cytokine concentrations are expressed as pg or ng per 106 lung macrophages. The detection limits of these assays were 8 pg·mL−1 for CCL3, 16 pg·mL−1 for TNF-α, CXCL5, CXCL10, CCL2, CCL4 and CCL5, 32 pg·mL−1 for CXCL1 and CXCL8, and 62 pg·mL−1 for CXCL9.
qRT-PCR analysis
Lung macrophages, treated or not with LPS (1 µg·mL−1) for 24 h, were harvested in RNAlater, and total RNA was extracted with RNeasy Mini Kit (Qiagen, Courtabeuf, France). After DNAse I treatment (DNAse set, Qiagen), 1 µg of total RNA was used to generate single-stranded cDNA (cDNA Archive Kit, Applied Biosystems, Courtabeuf, France). Reactions without reverse transcriptase were systematically run in parallel. Absence of contaminating genomic DNA was assessed by amplifying 24 housekeeping genes (18S, ACTB, AGPAT1, B2M, EEF1A2, GAPDH, GUSB, HDAC1, HMBS, HPRT1, ILF2, PMM1, POLR2H, PPIA, PSMA1, RPL13, RPL37A, SDHA, TAX1BP1, TBP, TPT1, UBC, VIL2, YWHAZ).
A specific TaqMan low-density array, based on pre-designed reagents (Assay-on-Demand, Applied Biosystems), was set up to evaluate adenosine receptor expression by qRT-PCR: A1 receptor (ADORA1): Hs00181231_m1; A2A receptor (ADORA2A): Hs00169123_m1; A2B receptor (ADORA2B): Hs00386497_m1; A3 receptor (ADORA3): Hs00181232_m1. qRT-PCR was carried out with 100 ng of cDNA in 2 µL of final reaction volume in 384-well microfluidic plates pre-loaded with probe and primers on the ABI PRISM 7900 Sequence Detection System using TaqMan Universal PCR Master Mix (Applied Biosystems). Gene expression fold changes were calculated according to the ΔΔCt method (Livak and Schmittgen, 2001). Sample profiles were obtained for the 24 housekeeping genes to determine the best set of genes leading to the most accurate normalization. The mean values of TPT1, PPIA and UBC were selected for further normalization of data. Genes were considered to be significantly expressed, and their transcript was measurable if their corresponding threshold cycle (Ct) value was ≤35.
Statistical analysis
Data are expressed as means ± SEM; n represents the number of patients from whom lung macrophage preparations were obtained. The concentration–effect curves were analysed using non-linear regression GraphPad Prism Version 5.01 (GraphPad Software Inc., San Diego, CA, USA), and sigmoidal curves were plotted to analyse the effects of the agonists (in the absence or presence of antagonists) on cytokine production. Statistical analyses used one-way repeated measures anova followed by Dunnett's post-tests. Significance was defined as P < 0.05.
The potency (pD2) of adenosine receptor agonists was defined as the negative log10 of the agonist concentration achieving 50% of the maximal response (EC50). The dissociation constant (KB) of the antagonist ZM 241385 at its respective adenosine receptor was estimated using the following equation: KB=[B]/[DR − 1], where DR is the dose ratio (EC50 of the agonist in the presence of the antagonist divided by the EC50 of the same agonist in the absence of the antagonist) and [B]= molar antagonist concentration (Arunlakshana and Schild, 1959).
Materials
The following chemicals were used: penicillin–streptomycin stabilized solution, DMSO, l-glutamine, FCS, RNAlater, trypan blue dye and LPS (from Escherichia coli serotype 0111:B4) were purchased from Sigma (St Louis, MO, USA). RPMI 1640 medium, phosphate-buffered saline and BSA were from Eurobio Biotechnology (Les Ulis, France). NECA (5′-N-ethyl-carboxamidoadenosine), CCPA (2-chloro-N6-cyclopentyladenosine), ZM 241385 (4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo-[2,3-a][1,3,5]triazin-5-yl-amino]ethyl)phenol), MRS 1754 (N-(4-cyano-phenyl)-2-[4-(2,6-dioxo-1,3-dipropyl-2,3,4,5,6,7-hexahydro-1H-purin-8-yl)-phenoxy]acetamide) and MRS 1220 (9-chloro-2-(2-furanyl)-5-[(phenylacetyl)amino][1,2,4]-triazolo[1,5-c]quinazoline) were purchased from Tocris Bioscience (Bristol, UK). AB-MECA (4-aminobenzyl-5-N-methylcarboxamidoadenosine) was obtained from Sanofi-Aventis (Montpellier, France). All cell culture plastics were from TPP (Trasadingen, Switzerland).
Results
LPS changes adenosine receptor mRNA expression in human lung macrophages
In unstimulated cells, gene expression results indicated that lung macrophages preferentially expressed A1 and A2B adenosine receptors, and only weakly expressed the other two adenosine receptors. Exposure of lung macrophages to LPS for 24 h evoked about a 400-fold increase of A2A receptor mRNA, and about a 10-fold decrease of A1 and A2B receptor mRNA, but had no effect on A3 receptor mRNA (Table 1). Therefore, the ratios of expression of A1 : A2A : A2B : A3 receptors by unstimulated cells were approximately 11:1:32:1 and, after LPS stimulation, were 1:360:3:1.
Table 1.
Levels of expression of mRNA for adenosine receptor subtypes and their modulation by LPS in human lung macrophages
| Receptor |
Expression levels (2−ΔCt) |
Fold change |
|
|---|---|---|---|
| Control | LPS | LPS versus Control | |
| A1 | 3.8 | 0.3 | −14.0 (−5.9, −100.0) |
| A2A | 0.3 | 100.7 | 359.7 (115.4, 1038.8) |
| A2B | 10.7 | 0.9 | −12.6 (−3.8, −33.3) |
| A3 | 0.3 | 0.3 | −1.2 (−50.0, 7.1) |
Data are mean relative quantifications (min, max) (n= 5 different preparations) expressed as 2−ΔCt, where ΔCt is the difference between target Ct and mean Ct of the pool of reference genes.
Effects of adenosine receptor agonists and antagonists on TNF-α production by LPS-stimulated human lung macrophages
The effects of serial concentrations of the non-selective adenosine receptor agonist NECA, the A1 receptor agonist, CCPA and the A3 receptor agonist, AB-MECA, were investigated on LPS-stimulated TNF-α production by human lung macrophages. LPS stimulation of human lung macrophages induced a mean 20-fold increase of TNF-α production (to 2083 ± 303 pg 10−6 cells). NECA inhibited LPS-stimulated TNF-α production in a concentration-dependent manner (Figure 1), reaching its maximal inhibition of approximately 80% at the highest concentration evaluated (10 µM). NECA had no influence on the low TNF-α production (112 ± 21 pg 10−6 cells) by unstimulated lung macrophages (n= 5).
Figure 1.

Effects of NECA, a non-selective adenosine receptor agonist; CCPA, an A1 receptor agonist; and AB-MECA, an A3 receptor agonist, on the production of TNF-α by LPS-stimulated human lung macrophages. Values are means ± SEM of five experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
Neither CCPA nor AB-MECA inhibited TNF-α production at concentrations up to and including 100 nM (Figure 1). But at higher concentrations, both compounds were able to interact with the other adenosine receptors (Fredholm et al., 2001; Cappellacci et al., 2008), and thus the maximal inhibition of TNF-α production by AB-MECA was 26 ± 6% at 10 µM (n= 5), whereas the maximal effect of CCPA was 56 ± 5% at 10 µM (n= 5).
The A3 adenosine receptor antagonist, MRS 1220, at a selective concentration of 10 nM (Jacobson et al., 1997) did not reverse NECA inhibition of LPS-induced TNF-α production, thereby excluding the participation of the A3 receptor [NECA alone: pD2= 7.3 ± 0.2; NECA + MRS 1220, pD2= 7.1 ± 0.1 (n= 5)].
To elucidate the roles of A2 adenosine receptor subtypes in LPS-induced TNF-α production, selective A2A and A2B receptor antagonists were evaluated for their abilities to block the NECA-induced inhibition. Notably, the A2A adenosine receptor antagonist, ZM 241385 (30 nM) caused a major rightward shift of the NECA concentration–response curve (Figure 2A). The apparent KB value is consistent with the reported affinity of ZM 241385 for the human recombinant A2A receptor (Fredholm et al., 2001). That value and the strong expression of A2A receptors suggest that this adenosine receptor subtype is the predominant receptor responsible for NECA inhibition of LPS-induced TNF-α production. TNF-α production did not rise when the A2A receptors were blocked by ZM 241385 in the presence of LPS, suggesting that endogenous adenosine release did not down-regulate TNF-α production via the A2A receptor under our experimental conditions. On the other hand, pretreatment of human lung macrophages with MRS 1754 (100 nM), an A2B receptor antagonist, failed to prevent NECA inhibition of TNF-α production (Figure 2B). In addition, TNF-α production remained unchanged when the A2B or A3 receptors were blocked by MRS 1754 or MRS 1220 respectively.
Figure 2.

Effects of the selective A2A adenosine receptor antagonist ZM 241385 (A) and A2B receptor antagonist MRS 1754 (B) on the inhibition of TNF-α production by the adenosine receptor agonist NECA. Cells were pretreated with the antagonists or vehicle for 15 min, followed by incubation with NECA for 1 h before being stimulated with LPS (1 µg·mL−1). Values are means ± SEM of five experiments.
Effect of NECA on CCL and CXCL chemokines produced by LPS-stimulated lung macrophages
Screening with human cytokine antibody arrays (RayBio Human Cytokine Antibody Array V, RayBiotech, Le Perray en Yvelines, France, and Proteome Profiler Array, R&D Systems) of the LPS-dependent accumulation of chemokines in human lung macrophage supernatants indicated that the main CC chemokines were CCL2, CCL3, CCL4 and CCL5, and the main CXC chemokines were CXCL1, CXCL5, CXCL8, CXCL9 and CXCL10 (data not shown). Unstimulated lung macrophages produced variable amounts of chemokines with CXCL5 and CXCL8 having the highest basal production. LPS stimulation also induced variable increases of chemokine production, with the highest increases being observed for CXCL10, CCL3 and CCL5 (Table 2). After 24 h of exposure to LPS, CXCL5 and CXCL8 were the predominant CXC chemokines, and CCL3 and CCL4 were the predominant CCL chemokines in lung macrophage supernatants.
Table 2.
The effect of NECA on chemokine production in culture supernatants of human lung macrophages, with or without LPS
| Chemokine (n) |
NECA (M) |
||||||
|---|---|---|---|---|---|---|---|
| Basal | LPS only | LPS+10−9 | LPS+10−8 | LPS+10−7 | LPS+10−6 | LPS+10−5 | |
| CXCL1 (6) | 1.5 ± 0.4 | 22.3 ± 5.2 | 22.7 ± 5.2 | 22.0 ± 5.0 | 22.5 ± 5.6 | 20.9 ± 5.3 | 17.8 ± 4.7** |
| CXCL5 (6) | 25.1 ± 7.8 | 196.8 ± 72.1 | 180.1 ± 53.0 | 189.8 ± 62.4 | 195.4 ± 65.1 | 212.9 ± 75.2 | 190.7 ± 71.4 |
| CXCL8 (6) | 22.4 ± 5.8 | 181.5 ± 43.8 | 186.5 ± 44.6 | 190.8 ± 47.8 | 173.2 ± 54.2 | 170.3 ± 55.0 | 158.1 ± 50.3* |
| CXCL9 (5) | 0.7 ± 0.3 | 6.9 ± 0.4 | 6.8 ± 0.6 | 5.3 ± 0.7* | 4.5 ± 0.4** | 3.9 ± 0.2*** | 3.5 ± 0.2*** |
| CXCL10 (7) | 0.03 ± 0.01 | 5.0 ± 1.4 | 4.7 ± 1.5 | 4.1 ± 1.3 | 3.2 ± 1.0* | 2.7 ± 0.9** | 2.4 ± 0.9*** |
| CCL2 (6) | 2.4 ± 1.1 | 7.0 ± 1.7 | 6.5 ± 1.6 | 5.9 ± 1.6* | 5.8 ± 1.7* | 5.4 ± 1.7** | 4.6 ± 1.7*** |
| CCL3 (6) | 2.2 ± 1.1 | 55.3 ± 11.7 | 54.3 ± 11.2 | 54.0 ± 11.7 | 44.8 ± 9.4* | 39.2 ± 9.3** | 32.5 ± 7.5***. |
| CCL4 (8) | 5.7 ± 1.9 | 72.5 ± 8.0 | 69.0 ± 8.0 | 66.9 ± 6.8 | 59.1 ± 6.5*** | 51.4 ± 5.6*** | 43.4 ± 6.2*** |
| CCL5 (6) | 0.05 ± 0.01 | 1.8 ± 0.7 | 1.3 ± 0.4 | 1.3 ± 0.4 | 1.4 ± 0.5 | 1.2 ± 0.4* | 1.1 ± 0.5* |
Macrophages were treated with medium (basal) only or LPS (1 µg·mL−1) in the absence (LPS only) or presence (LPS +) of a range of concentrations (10−9–10−5 M) of the non-selective adenosine receptor agonist NECA. Supernatants were harvested at 24 h, and analysed for chemokine levels by elisa. Results are expressed in ng 10−6 cells. Number of different preparations of macrophages in parentheses. Asterisks indicate significant effects of NECA on cytokine production, compared to the control experiments (LPS only)
P < 0.05;
P < 0.01;
P < 0.001).
NECA inhibited the LPS-stimulated production of CCL2, CCL3, CCL4, CCL5, CXCL9 and CXCL10 in a concentration-dependent manner (Table 2). Maximal NECA inhibitions (at 10 µM) of LPS-stimulated chemokine productions were 52 ± 8% for CCL2, 43 ± 3% for CCL3, 42 ± 7% for CCL4 and 36 ± 5% for CCL5; and 53 ± 7% for CXCL9 and 63 ± 8% for CXCL10. The pD2 for NECA inhibition of CCL5 and CCL2 productions were 7.4 ± 0.5 and 6.2 ± 0.4 respectively. LPS-induced CXCL1 and CXCL8 productions were barely affected by NECA, which had no effect on CXCL5. NECA did not alter the basal production of CCL2, CCL3 and CCL4 by unstimulated lung macrophages (n= 3–4).
Because A2A adenosine receptors were involved in NECA inhibition of LPS-induced TNF-α production by human lung macrophages, we investigated the possible contribution of these adenosine receptors to the modulation of chemokine production. As shown in Figure 3A–D, respectively, the A2A adenosine receptor antagonist, ZM 241385 (30 nM), shifted to the right the concentration–response curves of NECA as an inhibitor of production of CCL3, CCL4, CXCL9 and CXCL10, whereas the A2B receptor antagonist, MRS 1754 (100 nM), did not change NECA inhibition of chemokine production (data not shown). ZM 241385 also antagonized NECA inhibition of CCL5 and CCL2 productions with respective estimated KB of 0.8 and 2.7 nM. Chemokine production was not increased when the A2A receptors were blocked by ZM 241385 in the presence of LPS. Chemokine production was also unchanged when the A2B adenosine receptors were blocked by MRS 1754.
Figure 3.
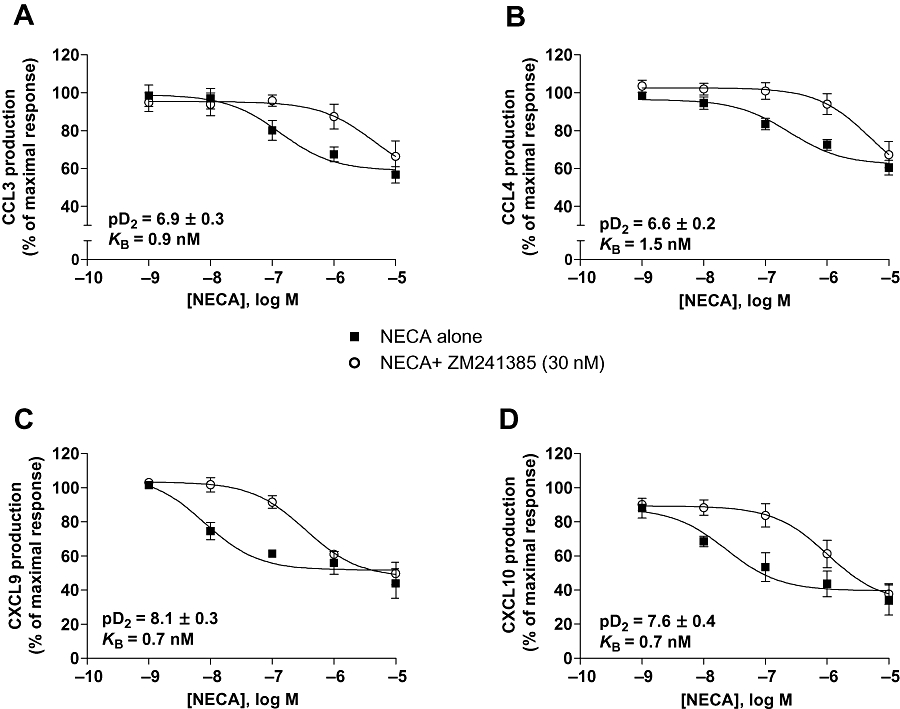
Effects of the selective A2A receptor antagonist ZM 241385 on the inhibition of CCL3 (A), CCL4 (B), CXCL9 (C) or CXCL10 (D) production by the adenosine receptor agonist NECA. Cells were pretreated with the antagonist or vehicle for 15 min, followed by incubation with NECA for 1 h before being stimulated with LPS (1 µg·mL−1). For each chemokine, the pD2 of NECA in the absence of ZM 241385, and the dissociation constant (KB) of the antagonist ZM 241385 are indicated in the graph. Values are means ± SEM of six to eight experiments.
Discussion and conclusions
The results of this study confirmed the expression of mRNA for all adenosine receptors in human lung macrophages, and demonstrated the LPS-induced modulation of their expression. We evaluated, for the first time with this human cell type, the adenosine receptors involved in the inhibition of LPS-induced TNF-α production, and extended these results to chemokines involved in airway inflammation.
All four adenosine receptors are expressed by monocytes and macrophages. However adenosine receptor expression is relatively low in quiescent monocytes, but increases during their differentiation into macrophages and also appears to change upon inflammatory activation (Bours et al., 2006). Immunohistochemical localization of adenosine receptors in human peripheral lung parenchyma revealed the expression of A2 and A3 receptors and, to a lesser degree, A1 receptors on lung macrophages (Varani et al., 2006). But neither the relative expressions of the four adenosine receptor types nor the modulation of their expression by LPS on human lung macrophages had been addressed previously. Our results are the first to demonstrate the expression of all four adenosine receptor mRNA on freshly isolated human lung macrophages with A2B receptor mRNA predominance on unstimulated cells. The very strong induction of A2A receptor mRNA elicited by LPS in lung macrophages is in line with that reported for the mouse myelomonocytic leukemia cell line WEHI-3, mouse intraperitoneal macrophages and human monocyte-derived macrophages, but was much greater than that observed for human blood monocytes (Suzuki et al., 2000) or the human monocyte leukemia cell line THP-1 (Bshesh et al., 2002). Our findings also demonstrated that A2A receptors had the highest mRNA expression followed by those of A2B, A1 and A3 receptors on LPS-stimulated lung macrophages with more than 10-fold decreases of A2B and A1 receptor transcripts in response to LPS. These results contrast with the increase of A2B receptor transcripts elicited by LPS on both WEHI-3 cells and mouse intraperitoneal macrophages (Murphree et al., 2005). Expression of the A3 adenosine receptors was unaffected by LPS, and remained low, as reported for WEHI-3 and THP-1 cells, and mouse intraperitoneal macrophages (Murphree et al., 2005).
The two A2 adenosine receptor subtypes, along with the other adenosine receptor types, have been implicated in the suppression of LPS-induced cytokine production by mouse monocytes/macrophages, depending on the cell origin and species, whereas for human monocytes, A2A receptors appear to be mainly involved (Bours et al., 2006; Hasko et al., 2007; Takahashi et al., 2007; Kreckler et al., 2009).
In this study, the involvement of A2A adenosine receptors in the inhibition of cytokine production by LPS-stimulated human lung macrophages was clearly indicated by three features of our results. First, the mRNA for the A2A receptor was markedly up-regulated by LPS, whereas A1 and A2B receptor mRNAs were clearly decreased, with A3 receptor mRNA at a constantly low level. Second, the A1 and A3 receptor agonists were only mildly inhibitory on TNF-α production in response to LPS. Last, the selective A2A receptor antagonist ZM 241385, but not the A2B receptor antagonist MRS 1754 or the A3 receptor antagonist MRS 1220, reversed the NECA-induced inhibition of LPS-induced TNF-α and chemokine production. Although NECA did not inhibit basal production of cytokines in unstimulated lung macrophages, we cannot suggest a direct relationship between LPS-induced up-regulation of A2A adenosine receptor mRNA and the inhibitory effect of A2A receptor stimulation on LPS-induced cytokine production in the absence of time-course experiments. Pertinently, for these in vitro studies, we selected a clinically relevant concentration of endotoxin (1 µg·mL−1). Endotoxin concentrations as high as 1–100 ng·mL−1 have been detected in bronchoalveolar fluids from patients with acute respiratory distress syndrome or pneumonia (Martin et al., 1997). Because bronchoalveolar lavage in humans has been reported to dilute lung fluids by ∼100-fold, lung fluid endotoxin concentrations in those patients had been estimated to be as high as 0.1–10 µg·mL−1 (Goodman et al., 1998). In addition, those authors estimated endotoxin concentrations in the lung fluids of rabbits with E. coli-induced experimental pneumonia to be ∼1 µg·mL−1, similar to the concentration we have used.
It is widely accepted that activated macrophages play a central role in the regulation of immune and inflammatory activities, and tissue remodelling. Bacterial LPS or cigarette smoke activates lung macrophages to release pro-inflammatory chemokines that recruit and activate blood-derived leukocytes through binding to chemokine receptors. The chemokine receptors CXCR3 and CCR5 are strongly expressed on lung CD8+ T cells and monocytes, and CCL3 and CXCL9 expressions in whole lung tissues from COPD patients were also correlated with disease activity (Freeman et al., 2007).
NECA or adenosine down-regulated the LPS-induced production of CXCR3-activating chemokines (CXCL9 and CXCL10) and CCR5-activating chemokines (CCL3, CCL4 and CCL5) through activation of A2A adenosine receptors, as deduced from the reversal by ZM 241385 of NECA-induced inhibition of their production and the lack of activity of MRS 1754. The production of these chemokines was previously reported to be regulated by cAMP-dependent pathways in monocytes, macrophages, airway epithelial cells or airway smooth muscle cells. (Delgado and Ganea, 2001; Hallsworth et al., 2001; Takayama et al., 2002; Bryn et al., 2006; Ayer et al., 2008; Reddy et al., 2008). The A2A adenosine receptor couples to Gs proteins, and stimulates adenylate cyclase and intracellular cAMP accumulation. (Bshesh et al., 2002; Nemeth et al., 2003; Fotheringham et al., 2004; Palmer and Trevethick, 2008). In human monocytes and monocyte-derived macrophages, cAMP inhibits LPS-induced TNF-α and CCL4 production through the PKA pathway (Bryn et al., 2006). In addition to the PKA pathway, cAMP can activate the exchange protein directly activated by cAMP (Epac) pathway in various cell types including monocytes and macrophages (Bryn et al., 2006; Palmer and Trevethick, 2008). However, the inhibition of LPS-induced cytokine production by cAMP was not regulated by the Epac pathway in human monocytes and macrophages (Bryn et al., 2006). In thioglycollate-elicited mouse peritoneal macrophages, activation of A2A adenosine receptors inhibits LPS-induced TNF-α production at the level of gene transcription through a unique cAMP-dependent, but PKA- and Epac-independent signalling pathway involving protein phosphatase activity without inhibition of nuclear translocation and DNA binding of NF-κB (Kreckler et al., 2009). In contrast, activation of A2A receptors has been shown to inhibit either nuclear translocation of NF-κB or its transcriptional activation in different myeloid, lymphoid and epithelial cell lines (Palmer and Trevethick, 2008). The intracellular pathway mediating the inhibitory effect of A2A receptor activation on cytokine production appears to depend on the cell type and the stimulating agent, and will require specific investigation in LPS-stimulated human lung macrophages. CXCL1, CXCL5 and CXCL8 are mainly involved in the local recruitment of neutrophils (Palmqvist et al., 2007; Viola and Luster, 2008). CXCL8 is not only produced in high quantities, but is also a more effective chemo-attractant than either CXCL1 or CXCL5, and is responsible for almost all neutrophil chemotactic activity in the supernatants of LPS-stimulated alveolar macrophages (Goodman et al., 1998). NECA had only mild or no inhibitory effect on CXCL1, CXCL5 and CXCL8. That finding is consistent with the observation that the production of these chemokines is weakly affected or unaffected by agents that increase cAMP intracellular concentrations (e.g. β2-adrenoceptor agonists, forskolin, phosphodiesterase IV inhibitors) in human monocytes/macrophages or sputum cells (Zhong et al., 1995; Yoshimura et al., 1997; Hallsworth et al., 2001; Pozo et al., 2002; Profita et al., 2003; Reddy et al., 2008). However, A2A adenosine receptors are expressed on neutrophils, and their activation not only inhibits adhesion and recruitment, but also inhibits degranulation of activated neutrophils (Spicuzza et al., 2006).
Taken together, our results suggest that Toll-like receptor 4 stimulation of human lung macrophages initiates a down-regulatory pathway to control inflammation by the induction of the anti-inflammatory A2A adenosine receptor. These receptors are fairly resistant to adenosine-induced desensitization (Zezula and Freissmuth, 2008), and may act as a regulatory mechanism to orchestrate the production of cytokines, like TNF-α, and of a subset of chemokines involved in the lung recruitment of inflammatory cells.
Acknowledgments
We thank Patricia Tchepikoff for qRT-PCR technical assistance, and Janet Jacobson for writing and editorial assistance.
Glossary
Abbreviations:
- A1, A2A, A2B, A3
adenosine receptor genes (ADORA1, ADORA2A, ADORA2B, ADORA3)
- AB-MECA
4-aminobenzyl-5-N-methylcarboxamidoadenosine
- CCPA
2-chloro-N6-cyclopentyladenosine
- COPD
chronic obstructive pulmonary disease
- DMSO
dimethylsulphoxide
- FCS
heat-inactivated fetal calf serum
- IL
interleukin
- LPS
lipopolysaccharide
- MRS 1220
9-chloro-2-(2-furanyl)-5-[(phenylacetyl)amino][1,2,4]-triazolo[1,5-c]quinazoline
- MRS 1754
N-(4-cyano-phenyl)-2-[4-(2,6-dioxo-1,3-dipropyl-2,3,4,5,6,7-hexahydro-1H-purin-8-yl)-phenoxy]acetamide
- NECA
5′-N-ethyl-carboxamidoadenosine
- PBS
phosphate-buffered saline
- PKA
protein kinase A
- RPMI
Roswell Park Memorial Institute
- TNF
tumour necrosis factor
- ZM 241385
4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo-[2,3-a][1,3,5]triazin-5-yl-amino]ethyl)phenol
Conflicts of interest
None.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol Chemother. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayer LM, Wilson SM, Traves SL, Proud D, Giembycz MA. 4,5-Dihydro-1H-imidazol-2-yl)-[4-(4-isopropoxy-benzyl)-phenyl]-amine (RO1138452) is a selective, pseudo-irreversible orthosteric antagonist at the prostacyclin (IP)-receptor expressed by human airway epithelial cells: IP-receptor-mediated inhibition of CXCL9 and CXCL10 release. J Pharmacol Exp Ther. 2008;324:815–826. doi: 10.1124/jpet.107.129312. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118:3546–3556. doi: 10.1172/JCI36130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. 2006;112:358–404. doi: 10.1016/j.pharmthera.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Brown RA, Clarke GW, Ledbetter CL, Hurle MJ, Denyer JC, Simcock DE, et al. Elevated expression of adenosine A1 receptor in bronchial biopsy specimens from asthmatic subjects. Eur Respir J. 2008;31:311–319. doi: 10.1183/09031936.00003707. [DOI] [PubMed] [Google Scholar]
- Bryn T, Mahic M, Enserink JM, Schwede F, Aandahl EM, Tasken K. The cyclic AMP-Epac1-Rap1 pathway is dissociated from regulation of effector functions in monocytes but acquires immunoregulatory function in mature macrophages. J Immunol. 2006;176:7361–7370. doi: 10.4049/jimmunol.176.12.7361. [DOI] [PubMed] [Google Scholar]
- Bshesh K, Zhao B, Spight D, Biaggioni I, Feokistov I, Denenberg A, et al. The A2A receptor mediates an endogenous regulatory pathway of cytokine expression in THP-1 cells. J Leukoc Biol. 2002;72:1027–1036. [PubMed] [Google Scholar]
- Cappellacci L, Franchetti P, Vita P, Petrelli R, Lavecchia A, Costa B, et al. 5′-Carbamoyl derivatives of 2′-C-methyl-purine nucleosides as selective A1 adenosine receptor agonists: affinity, efficacy, and selectivity for A1 receptor from different species. Bioorg Med Chem. 2008;16:336–353. doi: 10.1016/j.bmc.2007.09.035. [DOI] [PubMed] [Google Scholar]
- Delgado M, Ganea D. Inhibition of endotoxin-induced macrophage chemokine production by VIP and PACAP in vitro and in vivo. Arch Physiol Biochem. 2001;109:377–382. doi: 10.1076/apab.109.4.377.4237. [DOI] [PubMed] [Google Scholar]
- Ezeamuzie CI, Khan I. The role of adenosine A(2) receptors in the regulation of TNF-alpha production and PGE(2) release in mouse peritoneal macrophages. Int Immunopharmacol. 2007;7:483–490. doi: 10.1016/j.intimp.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Fotheringham JA, Mayne MB, Grant JA, Geiger JD. Activation of adenosine receptors inhibits tumor necrosis factor-alpha release by decreasing TNF-alpha mRNA stability and p38 activity. Eur J Pharmacol. 2004;497:87–95. doi: 10.1016/j.ejphar.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Ap IJ, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Freeman CM, Curtis JL, Chensue SW. CC chemokine receptor 5 and CXC chemokine receptor 6 expression by lung CD8+ cells correlates with chronic obstructive pulmonary disease severity. Am J Pathol. 2007;171:767–776. doi: 10.2353/ajpath.2007.061177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman RB, Strieter RM, Frevert CW, Cummings CJ, Tekamp-Olson P, Kunkel SL, et al. Quantitative comparison of C-X-C chemokines produced by endotoxin-stimulated human alveolar macrophages. Am J Physiol. 1998;275:L87–L95. doi: 10.1152/ajplung.1998.275.1.L87. 1 Pt 1. [DOI] [PubMed] [Google Scholar]
- Hallsworth MP, Twort CH, Lee TH, Hirst SJ. Beta(2)-adrenoceptor agonists inhibit release of eosinophil-activating cytokines from human airway smooth muscle cells. Br J Pharmacol. 2001;132:729–741. doi: 10.1038/sj.bjp.0703866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamano R, Takahashi HK, Iwagaki H, Kanke T, Liu K, Yoshino T, et al. Stimulation of adenosine A2A receptor inhibits LPS-induced expression of intercellular adhesion molecule 1 and production of TNF-alpha in human peripheral blood mononuclear cells. Shock. 2008;29:154–159. doi: 10.1097/shk.0b013e31812385da. [DOI] [PubMed] [Google Scholar]
- Hasko G, Pacher P, Deitch EA, Vizi ES. Shaping of monocyte and macrophage function by adenosine receptors. Pharmacol Ther. 2007;113:264–275. doi: 10.1016/j.pharmthera.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Park KS, Jiang JL, Kim YC, Olah ME, Stiles GL, et al. Pharmacological characterization of novel A3 adenosine receptor-selective antagonists. Neuropharmacology. 1997;36:1157–1165. doi: 10.1016/s0028-3908(97)00104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyaseelan S, Manzer R, Young SK, Yamamoto M, Akira S, Mason RJ, et al. Toll-IL-1 receptor domain-containing adaptor protein is critical for early lung immune responses against Escherichia coli lipopolysaccharide and viable Escherichia coli. J Immunol. 2005;175:7484–7495. doi: 10.4049/jimmunol.175.11.7484. [DOI] [PubMed] [Google Scholar]
- Kimata M, Shichijo M, Daikoku M, Inagaki N, Mori H, Nagai H. Pharmacological modulation of LPS-induced MIP-1 alpha production by peripheral blood mononuclear cells. Pharmacology. 1998;56:230–236. doi: 10.1159/000028202. [DOI] [PubMed] [Google Scholar]
- Kreckler LM, Gizewski E, Wan TC, Auchampach JA. Adenosine suppresses LPS-induced TNF-α production by murine macrophages through a protein kinase A- and Epac-independent signaling pathway. J Pharmacol Exp Ther. 2009;331:1051–1061. doi: 10.1124/jpet.109.157651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martin TR, Rubenfeld GD, Ruzinski JT, Goodman RB, Steinberg KP, Leturcq DJ, et al. Relationship between soluble CD14, lipopolysaccharide binding protein, and the alveolar inflammatory response in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 1997;155:937–944. doi: 10.1164/ajrccm.155.3.9117029. [DOI] [PubMed] [Google Scholar]
- Murphree LJ, Sullivan GW, Marshall MA, Linden J. Lipopolysaccharide rapidly modifies adenosine receptor transcripts in murine and human macrophages: role of NF-kappaB in A(2A) adenosine receptor induction. Biochem J. 2005;391:575–580. doi: 10.1042/BJ20050888. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth ZH, Leibovich SJ, Deitch EA, Sperlagh B, Virag L, Vizi ES, et al. Adenosine stimulates CREB activation in macrophages via a p38 MAPK-mediated mechanism. Biochem Biophys Res Commun. 2003;312:883–888. doi: 10.1016/j.bbrc.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Palmer TM, Trevethick MA. Suppression of inflammatory and immune responses by the A(2A) adenosine receptor: an introduction. Br J Pharmacol. 2008;153(Suppl 1):S27–S34. doi: 10.1038/sj.bjp.0707524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmqvist C, Wardlaw AJ, Bradding P. Chemokines and their receptors as potential targets for the treatment of asthma. Br J Pharmacol. 2007;151:725–736. doi: 10.1038/sj.bjp.0707263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo D, Guerrero JM, Calvo JR. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit LPS-stimulated MIP-1alpha production and mRNA expression. Cytokine. 2002;18:35–42. doi: 10.1006/cyto.2002.1024. [DOI] [PubMed] [Google Scholar]
- Profita M, Chiappara G, Mirabella F, Di Giorgi R, Chimenti L, Costanzo G, et al. Effect of cilomilast (Ariflo) on TNF-alpha, IL-8, and GM-CSF release by airway cells of patients with COPD. Thorax. 2003;58:573–579. doi: 10.1136/thorax.58.7.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PJ, Aksoy MO, Yang Y, Li XX, Ji R, Kelsen SG. Inhibition by salmeterol and cilomilast of fluticasone-enhanced IP-10 release in airway epithelial cells. COPD. 2008;5:5–11. doi: 10.1080/15412550701817573. [DOI] [PubMed] [Google Scholar]
- Spicuzza L, Di Maria G, Polosa R. Adenosine in the airways: implications and applications. Eur J Pharmacol. 2006;533:77–88. doi: 10.1016/j.ejphar.2005.12.056. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Hashimoto S, Toyoda N, Nagai S, Yamazaki N, Dong HY, et al. Comprehensive gene expression profile of LPS-stimulated human monocytes by SAGE. Blood. 2000;96:2584–2591. [PubMed] [Google Scholar]
- Takahashi HK, Iwagaki H, Hamano R, Wake H, Kanke T, Liu K, et al. Effects of adenosine on adhesion molecule expression and cytokine production in human PBMC depend on the receptor subtype activated. Br J Pharmacol. 2007;150:816–822. doi: 10.1038/sj.bjp.0707126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K, Garcia-Cardena G, Sukhova GK, Comander J, Gimbrone MA, Jr, Libby P. Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. J Biol Chem. 2002;277:44147–44154. doi: 10.1074/jbc.M204810200. [DOI] [PubMed] [Google Scholar]
- Varani K, Caramori G, Vincenzi F, Adcock I, Casolari P, Leung E, et al. Alteration of adenosine receptors in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173:398–406. doi: 10.1164/rccm.200506-869OC. [DOI] [PubMed] [Google Scholar]
- Viola A, Luster AD. Chemokines and their receptors: drug targets in immunity and inflammation. Annu Rev Pharmacol Toxicol. 2008;48:171–197. doi: 10.1146/annurev.pharmtox.48.121806.154841. [DOI] [PubMed] [Google Scholar]
- Wilson CN. Adenosine receptors and asthma in humans. Br J Pharmacol. 2008;155:475–486. doi: 10.1038/bjp.2008.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Kurita C, Nagao T, Usami E, Nakao T, Watanabe S, et al. Effects of cAMP-phosphodiesterase isozyme inhibitor on cytokine production by lipopolysaccharide-stimulated human peripheral blood mononuclear cells. Gen Pharmacol. 1997;29:633–638. doi: 10.1016/s0306-3623(96)00580-0. [DOI] [PubMed] [Google Scholar]
- Zezula J, Freissmuth M. The A(2A)-adenosine receptor: a GPCR with unique features? Br J Pharmacol. 2008;153(Suppl 1):S184–S190. doi: 10.1038/sj.bjp.0707674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JG, Hepburn L, Cruz G, Borman RA, Clark KL. The role of adenosine A2A and A2B receptors in the regulation of TNF-alpha production by human monocytes. Biochem Pharmacol. 2005;69:883–889. doi: 10.1016/j.bcp.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Zhong WW, Burke PA, Drotar ME, Chavali SR, Forse RA. Effects of prostaglandin E2, cholera toxin and 8-bromo-cyclic AMP on lipopolysaccharide-induced gene expression of cytokines in human macrophages. Immunology. 1995;84:446–452. [PMC free article] [PubMed] [Google Scholar]